

Abstract
Leishmania, an obligate intracellular parasite, binds several receptors to trigger engulfment by phagocytes, leading to cutaneous or visceral disease. These receptors include complement receptor 3 (CR3), used by promastigotes, and the Fc receptor (FcR), used by amastigotes. The mechanisms mediating uptake are not well understood. Here we show that Abl family kinases mediate both phagocytosis and the uptake of Leishmania amazonensis by macrophages (Mϕs). Imatinib, an Abl/Arg kinase inhibitor, decreases opsonized polystyrene bead phagocytosis and Leishmania uptake. Interestingly, phagocytosis of IgG-coated beads is decreased in Arg-deficient Mϕs, while that of C3bi-coated beads is unaffected. Conversely, uptake of C3bi-coated beads is decreased in Abl-deficient Mϕs, but that of IgG-coated beads is unaffected. Consistent with these results, Abl-deficient Mϕs are inefficient at C3bi-opsonized promastigote uptake, and Arg-deficient Mϕs are defective in IgG1-opsonized amastigote uptake. Finally, genetic loss of Abl or Arg reduces infection severity in murine cutaneous leishmaniasis, and imatinib treatment results in smaller lesions with fewer parasites than in controls. Our studies are the first to demonstrate that efficient phagocytosis and maximal Leishmania infection require Abl family kinases. These results highlight Abl family kinase-mediated signaling pathways as potential therapeutic targets for leishmaniasis.
INTRODUCTION
Leishmania parasites cause cutaneous or visceral disease in 1 to 2 million people a year in the developing world (17). Leishmania undergoes two life cycle stages: (i) the promastigote, found in the sand fly, and (ii) the amastigote, found in mammalian hosts. When an infected sand fly bites a host, the injected promastigotes must be engulfed by phagocytes to establish infection. The promastigotes then differentiate into amastigotes within the phagolysosome. If the amastigote finds itself outside a cell, it must be reengulfed for continued infection (23).
Several Mϕ surface proteins permit Leishmania uptake. Promastigote internalization is mediated by the fibronectin receptor (integrin α5β1) (2), the mannose-fucose receptor (63, 64), and complement receptors CR1 (10) and CR3 (38). Promastigotes may interact directly with CR3 (49), but binding is facilitated by opsonization with C3bi, a complement component (22, 37, 40, 45). Both CR3 and the Fc receptor (FcR) mediate amastigote uptake (16); interactions with the latter are facilitated by IgG opsonization (35). The FcR subclass FcγR, which mediates IgG-mediated phagocytosis (33), is most likely responsible for amastigote uptake by Mϕs. Indeed, internalization of IgG-opsonized amastigotes via FcRγI and -III sustains infection in murine cutaneous leishmaniasis (8, 24, 65). Adhesion of Leishmania to any of these receptors causes an actin-rich phagocytic cup to form and engulf the parasite (30). Our study explores the requirement for actin regulatory proteins in efficient Leishmania internalization.
The Abl family kinases Abl and Arg translate signals from adhesion and growth factor receptors into cytoskeletal structural changes (1, 43). Integrin engagement stimulates Abl family kinases to interact with and phosphorylate activators of the actin-polymerizing Arp2/3 complex in fibroblasts (5, 27, 28, 29, 34, 36, 61), causing dynamic cell edge protrusions resembling phagocytic intermediates. Interestingly, Abl family kinases have been implicated in endocytosis (21, 58, 59), macropinocytosis (13), and autophagy (66). They are also required for infectivity of viruses such as polyomavirus (56) and poxviruses (46, 47). Their precise roles in phagocytosis are less clear, although they are necessary for internalizing HIV (18) and bacteria such as Shigella (5), Salmonella (31) and mycobacteria (41). RNA interference (RNAi) screens have implicated Abl family kinases in the uptake of Pseudomonas (44) and Chlamydia (14). Imatinib, an Abl/Arg inhibitor, decreases uptake of zymosan particles (a yeast cell wall derivative) (11). However, roles for Abl and Arg in the uptake of Leishmania or other parasites by Mϕs have not been explored.
We report that Abl and Arg play complementary, nonredundant roles in the phagocytosis of opsonized beads and Leishmania. Genetic loss of Abl function significantly reduces complement-mediated phagocytosis, while Arg activity is required for efficient immunoglobulin-mediated phagocytosis. Consistent with this result, Abl permits uptake of complement-opsonized L. amazonensis promastigotes, likely through CR3, while Arg mediates uptake of IgG-opsonized amastigotes, most likely via FcγRIII. In addition, using imatinib or mice lacking either Abl or Arg, we show that Abl family kinases facilitate infection in murine cutaneous leishmaniasis. Our results implicate cytoskeletal-based cell invasion pathways as promising drug targets to combat leishmaniasis.
MATERIALS AND METHODS
Mice.
C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). Abl and Arg knockout mice (abl−/− and arg−/−) are in a mixed C57BL/6 × 129Sv/J genetic background as described previously (25). Mice lacking Abl in hematopoetic and endothelial cells, some in an arg−/− background (double knockout [dKO]), were generated by crossing mice carrying floxed Abl alleles (36) with mice carrying the Tie2-Cre allele (26) (generously provided by Richard Flavell, Yale Immunobiology). The arg−/− and ablflox/flox/Tie2-Cre+ mice used in in vivo infection experiments (as well as dKO mice) were all backcrossed to C57BL/6 at least 4 times during generation. All experiments, particularly the in vivo infection experiments using genetically manipulated mice, were performed with wild-type littermates (WTLM) to control for genetic background. The Institutional Animal Care and Use Committee at Yale University approved all experimental protocols.
Cell culture.
For opsonized bead experiments using imatinib and CR3/FcR blocking experiments, RAW 264.7 cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% heat-inactivated, endotoxin-free fetal bovine serum (FBS) (Invitrogen, Grand Island, NY). For other experiments, cells were harvested from the tibias and femurs of WTLM, abl−/−, arg−/−, and arg−/− ablflox/flox/Tie2-Cre+ mice and differentiated into bone-marrow-derived (BM) primary Mϕs (WT or WTLM, abl−/−, arg−/−, or dKO) over 7 days by growing them in a mixture of DMEM, 10% FBS, and 20% supernatant from L929 cells. Differentiation into BM Mϕs was confirmed by fluorescence-activated cell sorter (FACS) using antibodies to F4/80 and C11b (eBioscience, San Diego, CA); over 98% of differentiated cells were positive for both markers.
Parasite culture.
Leishmania amazonensis promastigotes (strain MHOM/BR/767/LTB0016) were grown at 24°C in Schneider's Drosophila medium supplemented with 15% heat-inactivated, endotoxin-free FBS and 10 μg/ml gentamicin (24). For Mϕ invasion, promastigotes were incubated at stationary phase for 7 days to maximize infective metacyclic promastigotes, which we defined as those isolated after purification at 3,000 × g for 30 min through a step Percoll gradient (Sigma, St. Louis, MO). For experiments with L. amazonensis amastigotes, strain IFLA/BR/67/PH8 (kindly provided by Norma Andrews, University of Maryland) was grown axenically at 32°C in M199 (Invitrogen) at pH 4.5 supplemented with 20% FBS, 1% penicillin-streptomycin, 0.1% hemin (25 mg/ml in 50% triethanolamine), 10 mM adenine, 5 mM l-glutamine, 0.25% glucose, 0.5% Trypticase, and 40 mM sodium succinate (20). Where indicated, promastigotes and amastigotes were incubated in medium containing dimethyl sulfoxide (DMSO) or 3.3 μM imatinib (LC Laboratories, Woburn, MA) to monitor for effects on parasite growth. Both strains were passaged through mice to maintain virulence.
Immunoblotting.
WT, abl−/−, arg−/−, or dKO Mϕs were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100) (27), and the protein concentration was assessed using bicinchoninic acid (BCA) (Thermo Scientific, Waltham, MA). Lysates were run on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Abl was visualized with mouse anti-24-11 (Santa Cruz, Santa Cruz, CA) at 1:1,000. Arg was visualized with a rabbit polyclonal antibody, as previously described (25) at 1:250. Immunoblotting with mouse antiactin (Millipore, Billerica, MA) at 1:10,000 was performed as a loading control.
To measure relative Abl or Arg activation after receptor engagement, phorbol-12-myristate-13-acetate (PMA; Sigma)-activated, macrophage colony-stimulating factor (M-CSF)-starved WT, abl−/−, arg−/−, or dKO Mϕs were added to dishes that were coated with C3bi or rabbit IgG1 (Sigma) for 15 min and lysed with a buffer of 20 mM Tris, 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100 that contained protease and phosphatase inhibitors (61). Lysates of WT Mϕs allowed to spread on uncoated dishes were run to confirm that Crk was phosphorylated in response to CR3 or FcR engagement. Equivalent micrograms of protein were loaded onto 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were probed with anti-phosphorylated Crk (anti-pCrk; Cell Signaling, Beverly, MA) at 1:1500 and then stripped and reprobed for Crk (Santa Cruz) at 1:500 to confirm protein levels. Relative levels of pCrk, normalized to Crk, were compared among samples using Image J analysis software. Each experiment was performed 5 times. Statistical significance was assessed with one-way analyses of variance (ANOVA).
Phagocytosis assays.
Primary Mϕs or RAW 264.7 cells, as indicated, were incubated overnight in M-CSF-starved media or serum-free media, respectively. Experiments were performed at ∼50% confluence. For drug experiments, Mϕs were preincubated in 3.3 μM imatinib or DMSO (Sigma) for 2 h. Where indicated, RAW cells were incubated in M1/70 supernatant (rat isotype IgG2b; Developmental Studies Hybridoma Bank, University of Iowa) at 1:10 for 20 min to block CR3 or F16/32 (also rat IgG2b; eBioscience) at 1:50 to block FcγRII and -III. An irrelevant antibody of the same isotype also did not affect uptake. For C3bi experiments, 2-μm-diameter latex yellow-green beads (Sigma) were coated with human IgM (Sigma) at 1:100 for 1 h at 37°C incubated in freshly isolated mouse serum diluted 1:1 in phosphate-buffered saline (PBS) for 1 h at 37°C. For IgG experiments, beads were incubated with rabbit IgG (Sigma) diluted 1:250 for 1 h at 37°C. Prior to C3bi bead uptake, Mϕs were activated with PMA (Sigma) for 1 h. Mϕs were incubated with a ratio of 10 to 15 particles per cell for 30 min at 37°C. Rates of uptake were compared between opsonized and unopsonized beads to monitor for opsonization effectiveness. Opsonization was confirmed by FACS analysis using a goat polyclonal antibody to C3b (Pierce, Rockford, IL) and an A568-conjugated donkey anti-goat secondary antibody (Invitrogen) or an A488-conjugated goat anti-rabbit secondary antibody for IgG (Invitrogen). After uptake, coverslips were rinsed, fixed with 3% formaldehyde for 15 min, and blocked with 2% bovine serum albumin (BSA) without permeabilization. Coverslips with C3bi-coated beads were incubated with rabbit anti-human IgM (Sigma) at 1:250. All coverslips were labeled with A594-conjugated goat anti-rabbit secondary antibody (Invitrogen) at 1:250. Coverslips were permeabilized with 0.25% Triton for 10 min, and stained with DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen). This process, termed a “two-color immunofluorescence assay” (62), allowed us to distinguish the beads that were internalized from those that remained outside cells. Fluorescence was visualized on a Nikon Eclipse TE2000-5 inverted fluorescence microscope with a 40× Nikon objective (1.0 aperture) by an observer blinded to the experimental treatment. At least 10 randomly selected fields were visualized for a total of at least 100 Mϕs and over 100 beads per experimental determination. The mean phagocytic index (PI), the number of particles internalized per 100 cells, was calculated for each experiment. The phagocytic index for control cells was taken as the maximum value (100%) for each experiment; the phagocytic index for the Abl kinase-deficient cells was determined and compared to that of the control. Means ± standard errors (SE) were calculated, and each experiment was performed at least three times. One-sample Student's t tests were performed to assess statistical significance. The representative images shown in Fig. 1 and 2 were taken with a Qimaging cooled charge-coupled device (CCD) camera (mono, 12 bit) at room temperature, acquired with Nikon Imaging software, and processed in Adobe Photoshop.
Fig 1.

Abl family kinases are required for optimal phagocytosis. (A and B) Imatinib decreases C3bi- and IgG-opsonized bead uptake. BM Mϕs or RAW 264.7 cells were treated with 3.3 μM imatinib or DMSO for 2 h. Ten C3bi- or IgG-opsonized beads were incubated per Mϕ for 30 min at 37°C. Two-color IF distinguished between intracellular (green) and extracellular (orange) beads. Nuclei are labeled with DAPI. (A) Image of C3bi-opsonized bead uptake by BM Mϕs treated with DMSO (top) or imatinib (bottom). The left panels show representative fields; the right panels show enlarged areas (boxed). Left scale bar, 20 μm; right scale bar, 5 μm. (B) Imatinib inhibits phagocytosis. Bars show the mean phagocytic index (PI) ± standard error (SE) for RAW 264.7 cells treated with 3.3 μM imatinib normalized to the DMSO-treated PI (100%) for each experiment. **, P = 0.0063for C3bi-coated bead uptake by DMSO versus imatinib-treated cells, and P = 0.0093 for IgG-coated bead uptake by DMSO versus imatinib-treated cells, by one-sample t test (n = 3 experiments). (C) Imatinib does not affect adhesion. Bars show percentages of adhered beads per 100 imatinib-treated RAW 264.7 cells compared to DMSO-treated cells from the experiment shown in panel B. (D) The relative decrease in PI in imatinib-treated Mϕs does not change even after 3 h of incubation. Mϕs were treated with DMSO or 3.3 μM imatinib prior to incubation with zymosan particles for the time indicated. The PI is shown for each time point of imatinib-treated Mϕs, normalized to DMSO-treated Mϕs at the first time point. Shown is one representative experiment of two experiments. (E) Immunoblots of Arg and Abl expression in BM Mϕs isolated from WT, arg−/−, abl−/−, or arg−/− ablflox/flox/Tie2-Cre+ (dKO) mice. dKO Mϕs express low levels of a truncated, kinase-inactive form of Abl (indicated by arrow) that migrates 10 kDa faster than full-length Abl on immunoblots. Actin is a loading control. (F) Mϕs lacking Arg with decreased Abl levels (dKO) have phagocytic defects. The graph shows the normalized mean PI ± SE for dKO Mϕs compared to WTLM Mϕs (abbreviated “WT”). *, P = 0.035 for C3bi-coated bead uptake by WTLM versus dKO Mϕs, and P = 0.019 for IgG-coated bead uptake by WTLM versus dKO Mϕs, by one-sample t test.(n = 3 experiments)
Fig 2.

Abl facilitates complement-mediated phagocytosis, while Arg governs immunoglobulin-mediated phagocytosis. (A and B) abl−/− Mϕs exhibit defects in complement-mediated phagocytosis. (A) Images of C3bi-opsonized bead uptake by WTLM (top, labeled “WT”) and abl−/− (bottom) Mϕs. Left panels, wide-field images; right panels, enlarged areas (boxed). Mϕs were incubated with beads and processed as in Fig. 1E. Scale bar, 20 μm in the left panels and 7 μm in the right panels. (B) Graph showing the mean PI ± SE for C3bi-opsonized beads by abl−/− and arg−/− Mϕs, normalized to WTLM (100%). *, P = 0.032 for WTLM versus abl−/− Mϕs by one-sample t test (n = 3 experiments). (C) Abl-YFP expression corrects C3bi-mediated phagocytic defects of dKO Mϕs. Shown is the mean PI ± SE for C3bi-opsonized bead uptake by Abl-YFP, Arg-YFP, and YFP-complemented Mϕs, normalized to WTLM Mϕs. *, P < 0.05, and **, P < 0.01, by one-way ANOVA (n = 3 experiments). (D and E) arg−/− Mϕs have defects in immunoglobulin-mediated phagocytosis. (D) Image of IgG-opsonized bead uptake by WTLM (top) and arg−/− (bottom) Mϕs. Coverslip processing, panels, and scale bars are as in panel A. (E) Graph showing mean PI ± SE for IgG-opsonized bead uptake by abl−/− and arg−/− Mϕs, normalized to WTLM Mϕs. *, P = 0.028 for WTLM versus arg−/− Mϕs by one-sample t test (n = 4 experiments). (F) Arg-YFP expression corrects IgG phagocytic defects in dKO Mϕs. Shown is the mean PI ± SE for IgG-coated bead uptake by Abl-YFP-, Arg-YFP-, and YFP-complemented Mϕs, normalized to WTLM Mϕs. *, P < 0.05, and **, P < 0.01 by one-way ANOVA (n = 3 experiments).
Zymosan particles (Sigma) were used to determine kinetics of the effect of imatinib treatment. Primary Mϕs were incubated with 3.3 μM imatinib or DMSO and then incubated with Oregon green-labeled zymosan at a ratio of 5 particles per Mϕ for up to 3 h prior to fixation and processing for two-color immunofluorescence (IF). Fixed coverslips were incubated with rabbit antizymosan (Sigma) and A594-conjugated goat anti-rabbit secondary antibody (Invitrogen), both at 1:250, prior to visualization and quantification of the PI.
For imaging and quantification of phagocytic cups in WT, abl−/−, arg−/−, or dKO Mϕs, 4-μm-diameter Far-red beads (Bangs Laboratories, Fishers, IL) were opsonized, then added to cells for 10 min. Cells were fixed, and phagocytic cups were labeled with Oregon Green phalloidin (1:100; Invitrogen). For the z-stack merges shown, phagocytic cups were imaged with a Nikon Ti-E Eclipse inverted spinning-disk confocal microscope with a 1-μm step size and photographed with a 14-bit Hamamatsu (C9100-50) electron-multiplied, charge-coupled device (EMCCD). Images were converted to TIFF format, and the middle three layers of the phagocytic cup were merged using ImageJ. For quantification, 50 cups per category from multiple experiments were measured by an observer blinded to genotype using ImageJ and categories were compared using one-way ANOVA. Relative intensity of phagocytic cups was also measured to control for differences in cell thickness and did not decrease in knockout macrophages.
Leishmania uptake assays.
Primary mouse bone marrow-derived Mϕs were employed for all assays except the phagocytic receptor blocking experiments, which employed RAW cells. For imatinib assays, Mϕs were incubated with 3.3 μM imatinib for 2 h prior to the assay. For C3bi opsonization experiments, metacyclic promastigotes were incubated with freshly isolated mouse serum for 1 h, and Mϕs were activated with PMA for 1 h prior to experiments. Amastigotes were opsonized with anti-P8–proteoglycolipid complex (monoclonal antibody IgG1) (42) for 1 h. Opsonization was confirmed as above. Where indicated, RAW cells were incubated for 20 min in M1/70 supernatant (Developmental Studies Hybridoma Bank) at 1:10 to block CR3 or F16/32 (eBioscience) at 1:50 to block FcγRII and -III. Coverslips containing primary Mϕs or RAW cells were incubated with metacyclic promastigotes at a ratio of 10:1 or with amastigotes at a ratio of 2:1 for 90 min (unopsonized) or for 20 min (opsonized), unless otherwise indicated, and washed prior to fixation with 3% paraformaldehyde. External promastigotes were labeled with mouse anti-gp46 (48), while external amastigotes were labeled with mouse anti-P8. All coverslips were labeled with A594-conjugated goat anti-mouse secondary antibody (Invitrogen). After permeabilization, promastigotes were relabeled with mouse anti-gp46, and amastigotes were relabeled with mouse anti-P8 and A488-conjugated goat anti-mouse secondary antibody (Invitrogen); coverslips were costained with DAPI. Rates of uptake at 20 min were compared between opsonized and unopsonized parasites to monitor opsonization efficiency. For all experiments, at least 10 randomly selected fields were visualized containing at least 100 Mϕs and at least 100 parasites per experimental determination. All experiments were performed 3 times unless otherwise indicated.
Complementation experiments.
Primary BM Mϕs were isolated from dKO mice and nucleofected (Lonza, Basel, Switzerland) with plasmids containing Arg conjugated with yellow fluorescent protein (Arg-YFP), Abl-YFP, or YFP alone (36, 60). WT Mϕs were also transfected with YFP as a control for nucleofection. All Mϕs were immediately plated and selected in G418 (Invitrogen) for 5 days prior to experiments. Phagocytosis assays with C3bi- or IgG-coated beads were performed as described above.
Murine infections.
For all mice, 1 × 106 metacyclic promastigotes suspended in PBS were injected subcutaneously in the dorsal right hind foot. For imatinib- or DMSO-treated C57B/6 mice, between 4 and 8 mice per group were infected per experiment and three independent experiments were performed. For imatinib treatment, mice were given 200 mg/kg/day of imatinib (LC Laboratories) in their drinking water or DMSO (the diluent) starting 4 days prior to infection and continuing throughout the experiment. Water intake was monitored by bottle weight to ensure appropriate dosing. For arg−/− mice and their littermate controls, between 4 and 5 mice per group were infected per experiment, and two independent experiments were performed. For ablflox/flox/Tie2-Cre+ and their littermate controls, between 4 and 8 mice per group were infected per each of two duplicate experiments. Lesion size was monitored weekly with a dial gauge caliper. The ratio of the infected to the uninfected hind foot was calculated (7). Both the control and Abl family kinase-deficient groups were compared to their respective degree of swelling at week 0 by ANOVA. Parasite burdens within lesions were assessed by limiting dilution upon sacrifice between weeks 12 and 20 (7).
Spleens from infected DMSO- versus imatinib-treated mice were harvested for cytokine profiling (53–55). Briefly, spleen cells were prepared in RPMI 1640 (Invitrogen) supplemented with 10% heat-inactivated FBS,5 × 10−5 M 2-mercaptoethanol (Sigma), and 100 μg of penicillin-streptomycin sulfate/ml. Cells were plated and stimulated with promastigote lysates (equivalent to 2 × 106 to 2 × 107 parasites per well, as indicated) or concanavalin A (ConA) (5 μg/ml; Sigma). Supernatants were harvested after 72 h of incubation and stored at −70°C until the levels of tumor necrosis factor (TNF-α), interleukin-4 (IL-4), IL-10, IL-17, or gamma interferon (IFN-γ) were measured with specific enzyme-linked immunosorbent assays (ELISAs) (BD Biosciences). Background cytokine levels were determined by using supernatants from unstimulated cell populations.
RESULTS
Abl family kinases are required for efficient phagocytosis.
To test if Abl and Arg might facilitate phagocytosis, we determined if imatinib affected C3bi or polyclonal IgG-opsonized bead uptake. We used two-color immunofluorescence (IF) to distinguish internalized from adherent beads and measured the phagocytic index (PI) after 30 min of incubation in the presence of imatinib or DMSO (Fig. 1A). Treatment of RAW 264.7 cells with 3.3 μM imatinib decreased the PI for C3bi- and IgG-coated beads by 53.0% ± 4.2% and 70.8% ± 6.9%, respectively, relative to that for controls (Fig. 1B). Similar results were observed for primary bone-marrow-derived (BM) Mϕs (data not shown). Opsonized beads bound to imatinib-treated RAW 264.7 cells at levels indistinguishable from those in controls (Fig. 1C), indicating that decreased invasion did not simply result from reduced adhesion. Internalization defects also persisted over long incubations (up to 3 h), demonstrating that imatinib-treated Mϕs did not overcome phagocytic defects even if significant time elapsed (Fig. 1D).
Imatinib also inhibits other kinases, such as c-KIT receptor tyrosine kinase and platelet-derived growth factor receptor (PDGFR) (3), both of which, like Abl family kinases, can direct actin cytoskeleton remodeling. To specifically test the requirement for Abl and Arg in phagocytosis, we isolated BM Mϕs from mice lacking Abl (abl−/−) or Arg (arg−/−) and wild type littermates (WTLM). Mϕs cannot be recovered from mice with germ line deletions of both Abl and Arg, which die at midgestation (25). Instead, we employed a conditional knockout strategy using a hematospecific/endothelial-specific Cre allele (Tie2-Cre) (26) to inactivate a floxed abl allele in an arg−/− genetic background (arg−/− ablflox/flox/Tie2-Cre+, henceforth termed dKO). dKO Mϕs lack Arg and full-length Abl, although they express low levels of a truncated form of Abl lacking exon 5 (Fig. 1E). This isoform, also observed after brain-specific recombination of the floxable abl allele (36), migrates 10 kDa faster than Abl on immunoblots and lacks kinase activity. When dKO Mϕs were incubated with C3bi- or IgG-coated beads, the PI was reduced by 34.7% ± 6.7% and 37.8% ± 5.3%, respectively (Fig. 1F). This reduction was smaller than that observed following treatment with imatinib, possibly due to inhibition of other kinases involved in phagocytosis (3). In combination, however, these results demonstrate that Abl and Arg are necessary for optimal complement- or immunoglobulin-mediated phagocytosis.
Abl promotes complement-mediated phagocytosis, while Arg mediates immunoglobulin-mediated phagocytosis.
To delineate the specific contributions of Abl or Arg to phagocytosis, we examined bead uptake by Mϕs isolated from abl−/−, arg−/−, or WTLM mice. Surprisingly, abl−/− Mϕs were less able to phagocytose C3bi-coated beads; their PI decreased by 65.2% ± 11.9% from WTLM Mϕs (Fig. 2A and B). However, abl−/− Mϕs phagocytosed IgG-coated beads at efficiencies that were indistinguishable from those of WTLM Mϕs (Fig. 2E). Defects in complement-mediated phagocytosis were specifically due to loss of Abl, as dKO Mϕs reexpressing Abl-YFP phagocytosed C3bi-coated beads efficiently (Fig. 2C). Interestingly, arg−/− Mϕs took up C3bi-coated beads as well as WTLM Mϕs (Fig. 2B), but had a significantly reduced ability to engulf IgG-coated beads (by 36.8% ± 9.2%) (Fig. 2D and E). Complementation of dKO Mϕs with Arg-YFP restored efficient phagocytosis of IgG-coated beads (Fig. 2F). Collectively, these data indicate that Abl mediates complement-mediated phagocytosis, while Arg governs immunoglobulin-mediated phagocytosis.
Having established that Abl contributes to complement-mediated phagocytosis and Arg contributes to immunoglobulin-mediated phagocytosis, we tested whether the activities of these kinases were differentially responsive to activation of their respective receptors. The Crk adaptor protein is required for efficient phagocytosis and is a substrate of both Abl and Arg (1). Reduced Crk phosphorylation has been observed in cells lacking Abl and Arg during Shigella uptake (5). We thus used Crk phosphorylation in WT, dKO, abl−/−, and arg−/− Mϕs to measure the relative contributions of each kinase to Crk phosphorylation following engagement of the distinct uptake receptors. Initially, we demonstrated that CR3 or FcR engagement stimulates Crk phosphorylation in WT Mϕs (Fig. 3). We found that during FcR engagement, Crk phosphorylation is reduced in arg−/− and dKO Mϕs by 70.6% ± 11.9% and 54.2% ± 12.7%, respectively, compared to that in WT Mϕs (Fig. 3A and B). However, IgG-induced Crk phosphorylation is unaltered in abl−/− Mϕs relative to that in the WT Mϕs (Fig. 3A and B). We also found that Crk phosphorylation during CR3 engagement is reduced in abl−/− and dKO Mϕs by 49.3% ± 13.5% and 65.0 ± 9.1%, respectively, compared to that in WT Mϕs; C3bi-mediated Crk phosphorylation is unaltered in arg−/− Mϕs (Fig. 3C and D). Together, these data indicate that Abl and Arg contribute differentially to Crk phosphorylation in response to signaling from CR3 and the FcR.
Fig 3.
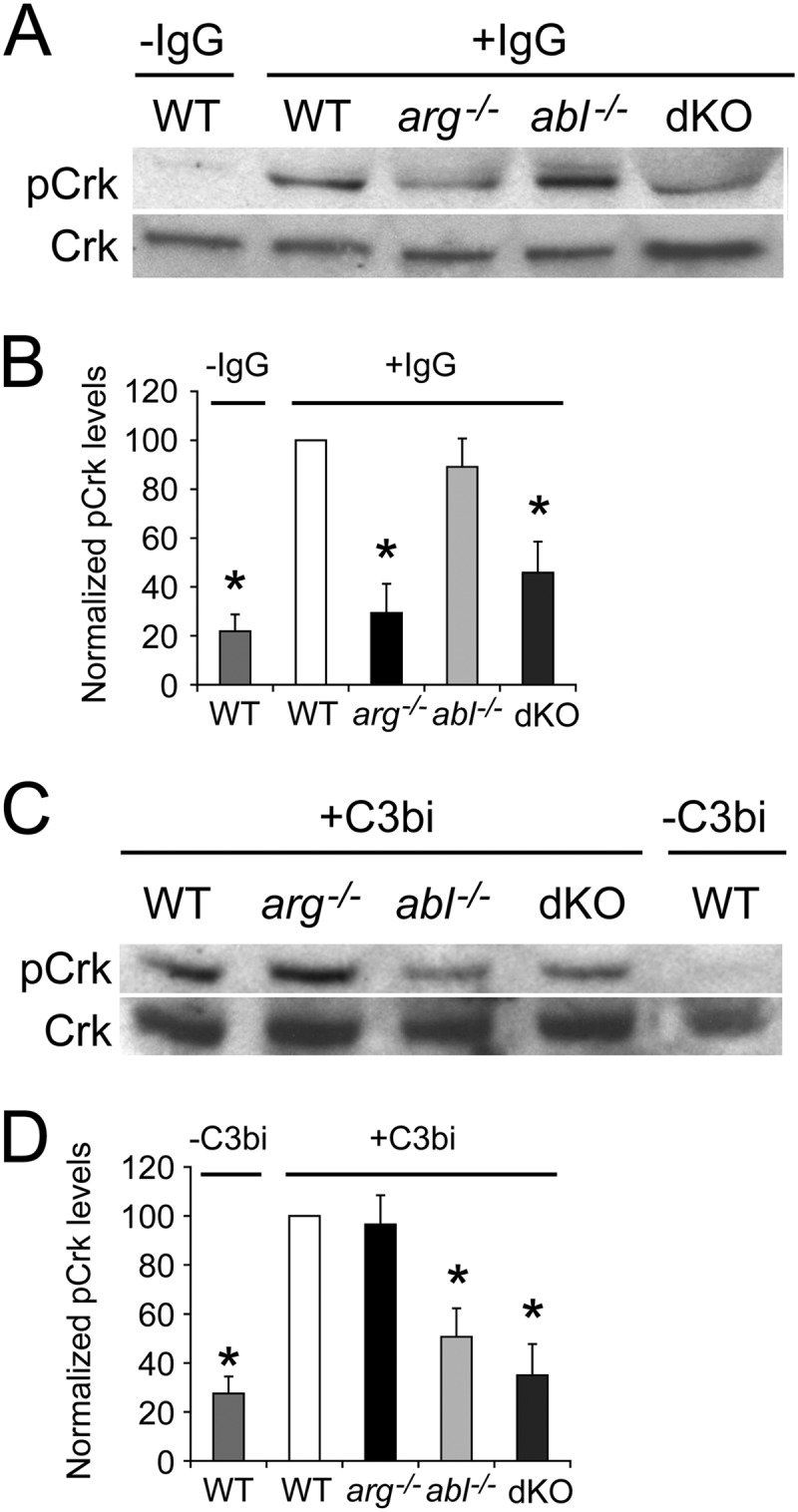
Arg and Abl stimulate Crk phosphorylation following engagement of the FcR and CR3 receptors, respectively. (A and B) Phosphorylation of the Abl/Arg substrate CrkII (pCrk) induced upon FcR engagement is decreased in arg−/− and dKO Mϕs compared to WT or abl−/− Mϕs. M-CSF-starved-Mϕs were adhered to uncoated plates (−IgG) or plates coated with mouse IgG1 (+IgG) for 15 min before lysis and processing for immunoblotting with an antibody to pCrk. (A) Representative immunoblot of pCrk (top) and total Crk (bottom) in WT Mϕs (± IgG) and IgG-stimulated arg−/−, abl−/− and dKO Mϕs. (B) Graph showing relative levels of pCrk, normalized to Crk levels, among WT (±IgG), arg−/−, abl−/−, and dKO Mϕs. Relative levels of pCrk for each category have been normalized to the level in IgG-treated WT Mϕs. *, P < 0.05 by ANOVA when pCrk levels in starred categories are compared to IgG-stimulated WT or abl−/− Mϕs (n = 5 experiments). (C and D) CR3 engagement-induced Crk phosphorylation is decreased in abl−/− and dKO Mϕs compared to WT or arg−/− Mϕs. Mϕs were added to uncoated plates (−C3bi) or C3bi-coated plates (+C3bi) for 15 min before processing as described above. (C) Representative immunoblot of pCrk (top) and Crk (bottom) in C3bi-stimulated WT, arg−/−, abl−/−, and dKO Mϕs and unstimulated WT Mϕs. (D) Graph showing relative levels of pCrk, normalized to Crk, among unstimulated WT Mϕs and C3bi-stimulated WT, arg−/−, abl−/−, and dKO Mϕs. *, P < 0.05 by ANOVA if pCrk levels in starred categories are compared to levels in either WT or arg−/− Mϕs (n = 5 experiments).
To determine if the incipient phagocytic cups in abl−/− and arg−/− Mϕs differed from those in control cells during phagocytosis, we stained F-actin with phalloidin in actively phagocytosing Mϕs. We found that F-actin-rich phagocytic cups are larger in arg−/− and dKO Mϕs undergoing IgG-mediated phagocytosis (Fig. 4A and B) than those in WT Mϕs. The phagocytic cups in arg−/− Mϕs also contained multiple ring-like structures not seen in WT Mϕs (Fig. 4A). When we examined abl−/− and dKO Mϕs undergoing complement-mediated phagocytosis, we found that abl−/− and dKO Mϕs had larger phagocytic cups that were less tightly organized around the particle being internalized than those of the WT Mϕs (Fig. 4A and C).
Fig 4.

Loss of Arg or Abl yields large F-actin-rich phagocytic cups during immunoglobulin-mediated or complement-mediated phagocytosis. (A) Three examples of WT and arg−/− Mϕs undergoing immunoglobulin-mediated phagocytosis (+IgG), as well as WT and abl−/− Mϕs undergoing complement-mediated phagocytosis (+C3bi). Mϕs were incubated with red beads and fixed/permeabilized. F-actin was labeled with green phalloidin. Left panel, actin alone; right panel, merged actin and bead. Scale bar, 5 μm. (B) Phagocytic cups in cells undergoing IgG-mediated phagocytosis are larger in arg−/− and dKO Mϕs than in controls. The graph shows the average area of phagocytic cups (normalized to WT) in WT, abl−/−, arg−/−, and dKO Mϕs. *, P < 0.05 for arg−/− or dKO Mϕs compared to WT or abl−/− Mϕs by one-way ANOVA. The difference between WT and abl−/− Mϕs is not statistically significant. (C) Phagocytic cups in cells undergoing complement-mediated phagocytosis are larger in abl−/− and dKO Mϕs than in controls. Normalization performed as for panel B. *, P < 0.05 for abl−/− or dKO Mϕs compared to WT and arg−/− Mϕs by one-way ANOVA.
Abl mediates promastigote engulfment.
CR3 helps mediate promastigote uptake, while the FcR has been implicated in amastigote uptake. Thus, we hypothesized that Mϕs would require Abl for efficient promastigote uptake, whereas Arg would be needed for amastigote uptake. We first determined the time course for engulfment of C3bi-opsonized L. amazonensis promastigotes, as occurs during infection (39), compared to promastigotes that had been grown in culture. Cultured promastigotes required 90 min to near maximal levels of cell entry (Fig. 5A). Opsonization of promastigotes with C3bi increased uptake efficiency significantly at early time points, but the effects of opsonization decreased over time (Fig. 5B). Treatment of Mϕs with 3.3 μM imatinib decreased internalization of cultured promastigotes by 54.8% ± 8.6%, that of C3bi-opsonized promastigotes by 53.3% ± 5.6%, and that of IgG1-opsonized promastigotes by 48.7% ± 8.8% (Fig. 5C). These uptake defects persisted over 3 h (Fig. 5D). However, 3.3 μM imatinib did not affect the growth of promastigotes in culture (Fig. 5E), indicating that it does not act directly on the parasites.
Fig 5.

Abl mediates promastigote engulfment. (A) Time course for cultured promastigote uptake. Promastigotes were incubated with primary Mϕs for the time indicated, and the PI was calculated. Shown is the absolute PI for a representative experiment of two experiments. (B) The effect of C3bi opsonization on uptake decreases over time. Shown is the PI ± SE of cultured and opsonized promastigotes over time, normalized to the PI of cultured promastigotes at 20 min, from a representative experiment of 2. (C) Imatinib decreases uptake of L. amazonensis promastigotes. Mϕs were treated with 3.3 μM imatinib or DMSO. Promastigotes were grown in culture (Promastigotes) or preincubated with mouse serum (+C3bi) or mouse IgG1 anti-gp46 (+IgG) and incubated with Mϕs for 20 min (+C3bi, +IgG) or 90 min (Promastigotes). An antibody to gp46 differentiated internalized (green) from external (orange) promastigotes. The graph shows the average PI ± SE for imatinib-treated Mϕs for cultured promastigotes, C3bi-opsonized promastigotes, and IgG-opsonized promastigotes, each normalized to their respective DMSO-treated Mϕs for 3 experiments. *, P = 0.024 for uptake by DMSO- versus imatinib-treated Mϕs for cultured promastigotes, P = 0.011 for C3bi-opsonized promastigotes, and P = 0.032 for IgG-opsonized promastigotes by one-sample t test. (D) Imatinib continues to decrease parasite uptake even after 3 h. The graph shows the average PI ± SE for imatinib-treated Mϕs, normalized to their respective DMSO-treated Mϕs. *, P < 0.05 by one-tailed t test (n = 3 experiments). (E) Imatinib is not toxic to promastigotes. Shown is a representative experiment of two experiments following promastigotes per ml of triplicate culture in control, DMSO-treated, or 3.3 μM imatinib-treated media over 3 days. (F) C3bi-opsonized promastigote uptake is decreased in Mϕs lacking Abl and Arg. WTLM versus dKO Mϕs were incubated with C3bi-opsonized promastigotes for 20 min. The graph shows the mean PI ± SE by dKO Mϕs, normalized to WTLM Mϕs (labeled WT). **, P = 0.0068 for WTLM versus dKO Mϕs by one-sample t test. (G) Abl mediates C3bi-opsonized promastigote uptake. The graph shows the mean PI ± SE by abl−/− and arg−/− Mϕs, normalized to WTLM Mϕs (labeled WT). **, P = 0.0046 for WTLM versus abl−/− Mϕs by one-sample t test (n = 3 experiments). (H) A CR3-blocking antibody (M1/70) decreases promastigote uptake. RAW 264.7 cells were preincubated with M1/70 or F16/32 (an FcR blocking antibody; both rat IgG2) prior to incubation with C3bi-opsonized promastigotes. The graph shows the mean PI + SE for untreated, M1/70-treated, or F16/32-treated RAW 264.7 cells. **, P < 0.01 for M1/70-treated cells by one-way ANOVA (n = 3 experiments).
Mϕs lacking both Arg and Abl have defects in C3bi-opsonized promastigote uptake, which decreased by 32.9% + 2.7% (Fig. 5F). As C3bi-opsonized promastigotes should interact with CR3, we tested whether abl−/− Mϕs were less efficient at engulfing opsonized promastigotes. Indeed, uptake of C3bi-opsonized promastigotes decreased in abl−/− Mϕs by 42.5% ± 2.9% (Fig. 5G). However, arg−/− Mϕs had no defects in C3bi-opsonized promastigote uptake (Fig. 5G). An antibody to CR3 prevented opsonized promastigote uptake (Fig. 5H), indicating that uptake of C3bi-opsonized promastigotes occurred through CR3, as previously shown (38).
Arg mediates amastigote engulfment.
We next examined internalization of amastigotes by Mϕs. Amastigotes isolated from infected animals are coated with IgG (8), making this condition the most physiologically relevant. Similar to C3bi-opsonized promastigotes, IgG1-opsonized L. amazonensis amastigotes were readily engulfed by Mϕs in 20 min (data not shown). A concentration of 3.3 μM imatinib did not affect the growth of amastigotes in culture (Fig. 6A). Nevertheless, imatinib decreased unopsonized amastigote uptake by 45.9% ± 2.2%, C3bi-opsonized amastigote uptake by 50.3% ± 5.8%, and IgG1-opsonized amastigote uptake by 47.1% ± 3.7% (Fig. 6B). Both dKO and arg−/− Mϕs were less efficient at engulfing IgG1-opsonized amastigotes: the PI decreased by 44.5% ± 8.9% and 46.2% ± 8.8%, respectively, from that of the WT (Fig. 6C and D). abl−/− Mϕs had no defects in the uptake of IgG1-opsonized amastigotes (Fig. 6D). An antibody to FcRγIII prevented IgG1-opsonized amastigote uptake (Fig. 6E), demonstrating that uptake of IgG1-opsonized amastigotes occurred through FcRγIII, as previously shown (65).
Fig 6.

Arg mediates amastigote engulfment. (A) Imatinib is not toxic to amastigotes. Shown is a representative experiment of two experiments following the number of amastigotes per ml in control, DMSO-treated, or imatinib-treated medium over 6 days. (B) Imatinib decreases amastigote uptake. Mϕs were pretreated with 3.3 μM imatinib or DMSO. Mouse anti-P8 IgG1 or freshly isolated mouse serum was used to opsonize amastigote surfaces with IgG1 or C3bi. Amastigotes were incubated with Mϕs for 90 min (unopsonized) or 20 min (opsonized). Two-color IF with anti-P8 distinguished internalized from external amastigotes. The graph shows the mean PI ± SE for imatinib-treated Mϕs, normalized to DMSO-treated Mϕs, of opsonized (+ C3bi, + IgG) and unopsonized (Amastigotes) L. amazonensis parasites. P = 0.0024 (**) for DMSO- versus imatinib-treated Mϕs for unopsonized amastigotes, P = 0.013 (*) for C3bi-opsonized amastigotes, and P = 0.0063 (**) for IgG-opsonized amastigotes by one-sample t test. (C) IgG1-opsonized amastigote uptake decreases in Mϕs lacking Abl and Arg. WTLM versus dKO Mϕs were incubated with IgG1-opsonized amastigotes for 20 min. The graph shows the mean PI ± SE by dKO Mϕs, normalized to WTLM Mϕs (labeled WT). *, P = 0.038 for WTLM versus dKO Mϕs by one-sample t test. (D) Arg mediates IgG-opsonized amastigote uptake. Bars show the mean PI ± SE of amastigotes by abl−/− and arg−/− Mϕs, normalized to WTLM (labeled “WT”). *, P = 0.034 for WTLM versus arg−/− Mϕs by one-sample t test. (E) An FcRγIII-blocking antibody (F16/32) decreases amastigote uptake. RAW 264.7 cells were preincubated with F16/32 or M1/70 prior to incubation with IgG1-opsonized amastigotes. The graph shows the mean PI + SE for untreated, M1/70-treated, or F16/32-treated RAW 264.7 cells. **, P < 0.01 for F16/32-treated cells by one-way ANOVA (n = 3 experiments).
In summary, and consistent with the opsonized bead data, C3bi-opsonized Leishmania promastigotes trigger Abl-mediated engulfment, most likely by binding CR3, whereas IgG1-opsonized amastigotes likely bind FcγRIII and initiate Arg-mediated engulfment.
Reduction of Abl family kinase function lessens lesion severity in murine cutaneous leishmaniasis.
Since engulfment by phagocytes is required for parasite survival, we hypothesized that drug inhibition or genetic loss of Abl or Arg might reduce Leishmania pathogenesis in vivo. Therefore, we used L. amazonensis in the C57BL/6 mouse model of cutaneous leishmaniasis. Mice were treated with 200 mg/kg of body weight/day of oral imatinib or DMSO (the diluent) starting 4 days before inoculation and continuing for the experimental duration. Footpad swelling (i.e., lesion development) was followed over time. Imatinib-treated mice reproducibly had smaller lesions that developed later than those of controls (Fig. 7A). We hypothesized that the smaller lesions observed in imatinib-treated mice would contain fewer parasites, consistent with the Mϕ internalization defects. Quantification of parasite burden by limiting dilution (7) revealed a 6- to 16-fold decrease in parasites isolated from imatinib-treated lesions relative to controls (Fig. 7D).
Fig 7.

Abl family kinases permit efficient infection in a mouse model of cutaneous leishmaniasis. (A) Imatinib-treated mice have smaller lesions than untreated mice. Four to eight C57BL/6 mice per experiment were injected with 1 × 106 L. amazonensis promastigotes in the right hind foot and treated with 200 mg/kg/day of imatinib or DMSO in their drinking water, starting 4 days before infection and continuing until sacrifice. Three separate experiments were performed; shown is a representative experiment containing 8 mice per group. Bars represent the increase in foot size compared to the uninfected foot (normalized to 1) ± SE. *, P < 0.05 by ANOVA. (B) arg−/− mice and (C) ablflox/flox/Tie2-Cre+ mice develop smaller lesions than WTLM when infected with 1 × 106 L. amazonensis promastigotes. Shown in panel B is a cohort of 5 mice per group in a representative experiment performed in duplicate using 4 to 5 mice per group. Shown in panel C is a cohort of 4 mice per group in a representative, duplicated experiment with 4 to 8 mice in each group. *, P < 0.05 by ANOVA. (D) Lesions in imatinib-treated mice contain fewer parasites than lesions in DMSO-treated mice (quantified by limiting dilution). Plotted is the parasite concentration in thousands at the termination of the experiment shown in panel A. *, P = 0.017 for DMSO versus imatinib-treated parasite burdens by two-sample t test. (E and F) arg−/− mice (E) and ablflox/flox/Tie2-Cre+ mice (labeled “abl−/−”) (F) have a lower parasite burden than WTLM. Shown is the final amount of L. amazonensis isolated from the infected foot from the WTLM versus the arg−/− experiment (E) and WTLM versus the ablflox/flox/Tie2-Cre+ experiment (F) from panels B and C, respectively. For panel E, P = 0.033 (*) for WTLM versus arg−/− parasite burdens, and for panel F, P = 0.017 (*) for WTLM versus ablflox/flox/Tie2-Cre+ parasite burdens by two-sample t test.
Mice lacking Abl have immunological defects, including defects in T cell development and function (15, 19, 52, 67), and defective immune responses could alter the pathogenesis of cutaneous leishmaniasis. Thus, we isolated spleens from infected DMSO- and imatinib-treated mice and profiled cytokine secretion after stimulation with high (1 × 107) or low (2 × 106) doses of parasite lysates (Table 1). Overall, there were no significant differences in cytokine (TNF, IL-13, IL-10, IL-4, and IL-17) response to L. amazonensis between splenic cells isolated from DMSO- and imatinib-treated mice (Table 1), with the exception of IFN-γ, which decreased in imatinib-treated mice at higher antigen stimulation. However, resolution of infection is typically associated with increased IFN-γ (53).
Table 1.
Cytokine profiles of DMSO- versus imatinib-treated infected splenic cellsa
| Cytokine | Treatment | Cytokine secretion (pg) after: |
|
|---|---|---|---|
| High stimulation | Low stimulation | ||
| TNF-α | DMSO | <1.6 | <1.6 |
| Imatinib | <1.6 | <1.6 | |
| IFN-γ | DMSO | 388 ± 97.1 | 317 ± 30.8 |
| Imatinib | 135 ± 18.6* | 133 ± 88.8 (NS) | |
| IL-4 | DMSO | <12.5 | <12.5 |
| Imatinib | <12.5 | <12.5 | |
| IL-13 | DMSO | 564 ± 147 | 243 ± 103. |
| Imatinib | 306 ± 130 (NS) | 293 ± 69.8. | |
| IL-10 | DMSO | <100 | <100 |
| Imatinib | <100 | <100 | |
| IL-17 | DMSO | <25 | <25 |
| Imatinib | <25 | <25 | |
Spleens from four L. amazonensis-infected DMSO-treated mice and four imatinib-treated mice were isolated and dissociated. A total of 5 × 106 cells were cultured for 72 h with media (unstimulated), ConA (positive control), or lysates of 1 × 107 parasites (high stimulation) or 2 × 106 parasites (low stimulation). Cytokine ELISAs were performed on harvested supernatants. *, P = 0.04 by two-sample t test. NS, not significant. The lone statistically significant effect is the opposite of what would be expected if IFN-γ mediated the smaller lesions in imatinib-treated mice. Shown is a representative experiment of two experiments.
As imatinib inhibits other kinases, we wanted to address if selective genetic loss of Abl or Arg affected Leishmania pathogenesis. Since our mice were not on a pure genetic background, we infected cohorts of arg−/− or ablflox/flox/Tie2-Cre+ mice and their WTLM with L. amazonensis. arg−/− mice infected with 1 × 106 L. amazonensis parasites developed lesions more slowly than WTLM (Fig. 7B). Infected ablflox/flox/Tie2-Cre+ mice also developed lesions more slowly than WTLM (Fig. 7C). Again, fewer parasites were recovered from infected arg−/− (Fig. 7E) or ablflox/flox/Tie2-Cre+ (Fig. 7F) mice compared to WTLM. These results indicate that Abl and Arg are important for the pathogenesis of cutaneous leishmaniasis.
DISCUSSION
The results provided here are the first to directly demonstrate a role for Abl family kinases in Leishmania infection. We find that Abl regulates complement-mediated phagocytosis, while Arg governs immunoglobulin-mediated phagocytosis. We extend these studies to L. amazonensis uptake by Mϕs and show that Abl mediates promastigote engulfment, whereas Arg mediates amastigote engulfment. Finally, we demonstrate a role for Abl family kinases in cutaneous leishmaniasis and show that the Abl/Arg inhibitor imatinib decreases lesion development and parasite burden in the mouse model.
Previous work has suggested roles for Abl family kinases in phagocytosis (5, 11, 14, 31, 41, 44), but Abl and Arg have never been linked to specific phagocytic receptors. Our work delineates specific yet complementary roles for Abl and Arg in complement-mediated and immunoglobulin-mediated phagocytosis, respectively. First, we observe selective defects in bead uptake, as abl−/− Mϕs have defects in the uptake of complement-coated beads and promastigotes, and arg−/− Mϕs have defects in the uptake of immunoglobulin-coated beads and amastigotes. Second, we find that Abl and Arg both differentially phosphorylate their downstream mediator Crk during CR3 and FcR engagement, respectively. To our knowledge, differential responsiveness of Abl versus that of Arg kinases to distinct upstream receptors has not previously been shown. Finally, we find that phagocytic cups are larger and deformed in abl−/− Mϕs undergoing complement-mediated phagocytosis and in arg−/− Mϕs undergoing IgG-mediated phagocytosis. Thus, abl−/− or arg−/− Mϕs with phagocytic defects appear to form unproductive phagocytic cups that are unable to appropriately internalize beads or parasites. Although they are similar in overall structure, Abl and Arg differ in sequence, particularly at their C-terminal regions, as well as in their subcellular localizations (1). We would anticipate that such differences may confer distinct roles for these kinases during phagocytosis, as has been delineated in other aspects of cell biology (43). For example, Arg, but not Abl, localizes to and is required for invadopodia function in breast cancer cells (32). An alternate hypothesis is that Abl and Arg activate additional downstream targets (besides Crk) that differ between complement- or immunoglobulin-mediated phagocytosis, such as the hematopoietic cortactin homologue HS1 (19). Clearly, Abl/Arg-independent mechanisms also contribute to phagocytosis, since even Mϕs that lack both Abl family kinases still are capable of undergoing reduced levels of phagocytosis. In addition, the difference in phagocytic inhibition seen between abl−/− and dKO Mϕs suggests that the residual “exon 5-less” Abl isoform may support some phagocytic activity, perhaps by providing a scaffolding function, as has been demonstrated for Abl family kinases in other systems (1).
Studies of receptors required for Leishmania uptake have revealed that engagement with several distinct receptors can initiate internalization. For example, promastigotes are known to bind CR3 both directly (49, 50, 57) and after fixing C3bi (37, 40, 45). Consistent with this literature, we also show that antibodies that block CR3 prevent C3bi-opsonized promastigote uptake. C3bi-opsonized promastigotes are not taken up as efficiently by abl−/− Mϕs as by WT or arg−/− Mϕs, indicating that Abl is responsible for efficient complement-mediated phagocytosis. Similarly, our IgG1-opsonized amastigote studies implicate Arg in the control of immunoglobulin-mediated phagocytosis—specifically through FcγRIII, as an FcγRIII blocking antibody decreased amastigote uptake. Therefore, we hypothesize that Arg acts downstream of FcγRIII to mediate amastigote uptake. However, our current studies do not rule out the possibility that Arg can mediate phagocytosis through other FcRs as well.
Our mouse studies indicate that Leishmania survival, replication, and pathogenesis during cutaneous leishmaniasis depend on the Abl/Arg kinases. We propose that the reduced lesion sizes seen in Abl family kinase-deficient mice result at least in part from uptake deficiencies in Mϕs. Consistent with this possibility, fewer parasites were isolated from lesions in imatinib-treated, arg−/− or ablflox/flox/Tie2-Cre+ mice than from controls. Lesion size differences in cutaneous leishmaniasis result from both disparities in parasite invasion and the subsequent burden and from differences in the hosts' inflammatory/immune responses (54). For example, inhibition of phosphoinositide 3-kinase γ, signaling through which has been linked to Abl family kinases (1), affects both the uptake of L. mexicana and the host's inflammatory response to infection, which results in less severe disease in the mouse model (9). We did not observe significant differences in cytokine secretion between stimulated splenic cells isolated from imatinib and DMSO-treated mice, with the exception of IFN-γ, which decreased in imatinib-treated splenic cells at higher antigen stimulation. However, IFN-γ secretion typically increases when infection is resolving (53). Thus, the differences we see in lesion size are not likely to be explained by defective immune responses to Leishmania infection in imatinib-treated mice.
In addition, the proto-oncogene Friend leukemia integration 1 transcription factor (FLI-1), or transcription factor ERGB, regulates wound healing and has been proposed as therapy for systemic sclerosis (12). Polymorphisms in this factor's gene govern susceptibility to cutaneous leishmaniasis (6, 51), and Abl facilitates FLI-1 activation (4). However, decreased FLI-1 activity and subsequent effects on wound healing should not lower parasite burdens (51). Thus, it seems unlikely that differences in FLI-1 activation are solely responsible for the differences we see in lesion size in Abl family kinase-deficient mice.
In conclusion, we have shown that Abl and Arg regulate distinct phagocytic mechanisms. Abl family kinases also govern the uptake of Leishmania and its subsequent pathogenesis. Our results strongly suggest that improving our knowledge of cell entry pathways can provide new lines of therapy for prophylaxis and/or treatment of leishmaniasis. Of note, imatinib is a clinically approved chemotherapeutic agent with a relatively benign side effect profile. In contrast, therapies for leishmaniasis used in the developing world, such as pentavalent antimony compounds and miltefosine, are plagued with multiple toxic side effects, and drug resistance is emerging (17). Future studies will continue to explore the use of imatinib and/or other inhibitors of cell entry for the prophylaxis or treatment of leishmaniasis.
ACKNOWLEDGMENTS
We thank Norma Andrews for providing L. amazonensis strain IFLA/BR/67/PH8 and Richard Flavell for providing Tie2-Cre+ mice. The M1/70 antibody was obtained from the Developmental Studies Hybridoma Bank, developed under the NICHD and maintained by the University of Iowa, Iowa City, IA. We thank all of the members of the McMahon-Pratt and Koleske laboratories, as well as Hanspeter Niederstrasser, for technical assistance, helpful discussions, and critical comments on the manuscript. We also thank the Calderwood laboratory for helpful discussions and use of their Amaxa nucleofector device.
D.M.W. was supported by T32 AI007210, a Pediatric Infectious Diseases Society Fellowship (sponsored by Wyeth Pharmaceuticals), and NRSA F32 AI094905. Work was supported by PHS grants CA133346, NS39475 (A.J.K.), and AI093775 (D.M.P.).
Footnotes
Published ahead of print 4 June 2012
REFERENCES
- 1. Bradley WD, Koleske AJ. 2009. Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J. Cell Sci. 122:3441–3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brittingham A, Chen G, McGwire BS, Chang KP, Mosser DM. 1999. Interaction of Leishmania gp63 with cellular receptors for fibronectin. Infect. Immun. 67:4477–4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buchdunger E, et al. 2000. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther. 295:139–145 [PubMed] [Google Scholar]
- 4. Bujor AM, Asano Y, Haines P, Lafyatis R, Trojanowska M. 2011. The c-Abl tyrosine kinase controls protein kinase Cdelta-induced Fli-1 phosphorylation in human dermal fibroblasts. Arthritis Rheum. 63:1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burton EA, Plattner R, Pendergast AM. 2003. Abl tyrosine kinases are required for infection by Shigella flexneri. EMBO J. 22:5471–5479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castellucci L, et al. 2011. FLI1 polymorphism affects susceptibility to cutaneous leishmaniasis in Brazil. Genes Immun. 12:589–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Champsi J, McMahon-Pratt D. 1988. Membrane glycoprotein M-2 protects against Leishmania amazonensis infection. Infect. Immun. 56:3272–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colmenares M, Constant SL, Kima PE, McMahon-Pratt D. 2002. Leishmania pifanoi pathogenesis: selective lack of a local cutaneous response in the absence of circulating antibody. Infect. Immun. 70:6597–6605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cummings HE, et al. 2012. Critical role for phosphoinositide 3-kinase gamma in parasite invasion and disease progression of cutaneous leishmaniasis. Proc. Natl. Acad. Sci. U. S. A. 109:1251–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Da Silva RP, Hall BF, Joiner KA, Sacks DL. 1989. CR1, the C3b receptor, mediates binding of infective Leishmania major metacyclic promastigotes to human macrophages. J. Immunol. 143:617–622 [PubMed] [Google Scholar]
- 11. Dewar AL, Doherty KV, Hughes TP, Lyons AB. 2005. Imatinib inhibits the functional capacity of cultured human monocytes. Immunol. Cell Biol. 83:48–56 [DOI] [PubMed] [Google Scholar]
- 12. Distler JH, et al. 2007. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. 56:311–322 [DOI] [PubMed] [Google Scholar]
- 13. Dubielecka PM, et al. 2010. Abi1/Hssh3bp1 pY213 links Abl kinase signaling to p85 regulatory subunit of PI-3 kinase in regulation of macropinocytosis in LNCaP cells. FEBS Lett. 584:3279–3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Elwell CA, Ceesay A, Kim JH, Kalman D, Engel JN. 2008. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 4:e1000021 doi:10.1371/journal.ppat.1000021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gu JJ, Zhang N, He YW, Koleske AJ, Pendergast AM. 2007. Defective T cell development and function in the absence of Abelson kinases. J. Immunol. 179:7334–7343 [DOI] [PubMed] [Google Scholar]
- 16. Guy RA, Belosevic M. 1993. Comparison of receptors required for entry of Leishmania major amastigotes into macrophages. Infect. Immun. 61:1553–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haldar A. K., Sen P, Roy S. 2011. Use of antimony in the treatment of leishmaniasis: current status and future directions. Mol. Biol. Int. 2011:571242 doi:10.4061/2011/571242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harmon B, Campbell N, Ratner L. 2010. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 6:e1000956 doi:10.1371/journal.ppat.1000956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang Y, et al. 2008. The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse. Blood 112:111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huynh C, Sacks DL, Andrews NW. 2006. A Leishmania amazonensis ZIP family iron transporter is essential for parasite replication within macrophage phagolysosomes. J. Exp. Med. 203:2363–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jacob M, et al. 2009. Endogenous cAbl regulates receptor endocytosis. Cell. Signal. 21:1308–1316 [DOI] [PubMed] [Google Scholar]
- 22. Johnson E, Hetland G. 1988. A sensitive method to detect synthesis of the functional classical, alternative and terminal pathway of complement by cells cultured in vitro. Scand. J. Clin. Lab. Invest. 48:223–231 [DOI] [PubMed] [Google Scholar]
- 23. Kane MM, Mosser DM. 2000. Leishmania parasites and their ploys to disrupt macrophage activation. Curr. Opin. Hematol. 7:26–31 [DOI] [PubMed] [Google Scholar]
- 24. Kima PE, et al. 2000. Internalization of Leishmania mexicana complex amastigotes via the Fc receptor is required to sustain infection in murine cutaneous leishmaniasis. J. Exp. Med. 191:1063–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koleske AJ, et al. 1998. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron 21:1259–1272 [DOI] [PubMed] [Google Scholar]
- 26. Koni PA, et al. 2001. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J. Exp. Med. 193:741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lapetina S, Mader CC, Machida K, Mayer BJ, Koleske AJ. 2009. Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. J. Cell Biol. 185:503–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lewis JM, Baskaran R, Taagepera S, Schwartz MA, Wang JY. 1996. Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. Proc. Natl. Acad. Sci. U. S. A. 93:15174–15179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li R, Pendergast AM. 2011. Arg kinase regulates epithelial cell polarity by targeting beta1-integrin and small GTPase pathways. Curr. Biol. 21:1534–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lodge R, Descoteaux A. 2008. Leishmania invasion and phagosome biogenesis. Subcell. Biochem. 47:174–181 [DOI] [PubMed] [Google Scholar]
- 31. Ly KT, Casanova JE. 2009. Abelson tyrosine kinase facilitates Salmonella enterica serovar Typhimurium entry into epithelial cells. Infect. Immun. 77:60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mader CC, et al. 2011. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 71:1730–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McKenzie SE, Schreiber AD. 1998. Fc gamma receptors in phagocytes. Curr. Opin. Hematol. 5:16–21 [DOI] [PubMed] [Google Scholar]
- 34. Miller MM, et al. 2010. Regulation of actin polymerization and adhesion-dependent cell edge protrusion by the Abl-related gene (Arg) tyrosine kinase and N-WASp. Biochemistry 49:2227–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morehead J, Coppens I, Andrews NW. 2002. Opsonization modulates Rac-1 activation during cell entry by Leishmania amazonensis. Infect. Immun. 70:4571–4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moresco EM, Donaldson S, Williamson A, Koleske AJ. 2005. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J. Neurosci. 25:6105–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mosser DM, Edelson PJ. 1984. Activation of the alternative complement pathway by Leishmania promastigotes: parasite lysis and attachment to macrophages. J. Immunol. 132:1501–1505 [PubMed] [Google Scholar]
- 38. Mosser DM, Edelson PJ. 1985. The mouse macrophage receptor for C3bi (CR3) is a major mechanism in the phagocytosis of Leishmania promastigotes. J. Immunol. 135:2785–2789 [PubMed] [Google Scholar]
- 39. Mosser DM, Rosenthal LA. 1993. Leishmania-macrophage interactions: multiple receptors, multiple ligands and diverse cellular responses. Semin. Cell Biol. 4:315–322 [DOI] [PubMed] [Google Scholar]
- 40. Mosser DM, Springer TA, Diamond MS. 1992. Leishmania promastigotes require opsonic complement to bind to the human leukocyte integrin Mac-1 (CD11b/CD18). J. Cell Biol. 116:511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Napier RJ, et al. 2011. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe 10:475–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pan AA, McMahon-Pratt D. 1988. Monoclonal antibodies specific for the amastigote stage of Leishmania pifanoi. I. Characterization of antigens associated with stage- and species-specific determinants. J. Immunol. 140:2406–2414 [PubMed] [Google Scholar]
- 43. Peacock JG, Couch BA, Koleske AJ. 2010. The Abl and Arg non-receptor tyrosine kinases regulate different zones of stress fiber, focal adhesion, and contractile network localization in spreading fibroblasts. Cytoskeleton (Hoboken) 67:666–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pielage JF, Powell KR, Kalman D, Engel JN. 2008. RNAi screen reveals an Abl kinase-dependent host cell pathway involved in Pseudomonas aeruginosa internalization. PLoS Pathog. 4:e1000031 doi:10.1371/journal.ppat.1000031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Puentes SM, Sacks DL, da Silva RP, Joiner KA. 1988. Complement binding by two developmental stages of Leishmania major promastigotes varying in expression of a surface lipophosphoglycan. J. Exp. Med. 167:887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reeves PM, et al. 2005. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat. Med. 11:731–739 [DOI] [PubMed] [Google Scholar]
- 47. Reeves PM, et al. 2011. Variola and monkeypox viruses utilize conserved mechanisms of virion motility and release that depend on Abl and Src family tyrosine kinases. J. Virol. 85:21–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rivas L, Kahl L, Manson K, McMahon-Pratt D. 1991. Biochemical characterization of the protective membrane glycoprotein GP46/M-2 of Leishmania amazonensis. Mol. Biochem. Parasitol. 47:235–243 [DOI] [PubMed] [Google Scholar]
- 49. Russell DG. 1987. The macrophage-attachment glycoprotein gp63 is the predominant C3-acceptor site on Leishmania mexicana promastigotes. Eur. J. Biochem. 164:213–221 [DOI] [PubMed] [Google Scholar]
- 50. Russell DG, Wright SD. 1988. Complement receptor type 3 (CR3) binds to an Arg-Gly-Asp-containing region of the major surface glycoprotein, gp63, of Leishmania promastigotes. J. Exp. Med. 168:279–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sakthianandeswaren A, et al. 2010. Fine mapping of Leishmania major susceptibility locus lmr2 and evidence of a role for Fli1 in disease and wound healing. Infect. Immun. 78:2734–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Silberman I, et al. 2008. T cell survival and function requires the c-Abl tyrosine kinase. Cell Cycle 7:3847–3857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Soong L, et al. 1997. Role of CD4+ T cells in pathogenesis associated with Leishmania amazonensis infection. J. Immunol. 158:5374–5383 [PubMed] [Google Scholar]
- 54. Soong L, Duboise SM, Kima P, McMahon-Pratt D. 1995. Leishmania pifanoi amastigote antigens protect mice against cutaneous leishmaniasis. Infect. Immun. 63:3559–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soong L, et al. 1996. Disruption of CD40-CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity 4:263–273 [DOI] [PubMed] [Google Scholar]
- 56. Swimm AI, et al. 2010. Abl family tyrosine kinases regulate sialylated ganglioside receptors for polyomavirus. J. Virol. 84:4243–4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Talamas-Rohana P, Wright SD, Lennartz MR, Russell DG. 1990. Lipophosphoglycan from Leishmania mexicana promastigotes binds to members of the CR3, p150,95 and LFA-1 family of leukocyte integrins. J. Immunol. 144:4817–4824 [PubMed] [Google Scholar]
- 58. Tanos B, Pendergast AM. 2006. Abl tyrosine kinase regulates endocytosis of the epidermal growth factor receptor. J. Biol. Chem. 281:32714–32723 [DOI] [PubMed] [Google Scholar]
- 59. Tanos BE, Pendergast AM. 2007. Abi-1 forms an epidermal growth factor-inducible complex with Cbl: role in receptor endocytosis. Cell. Signal. 19:1602–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang Y, Miller AL, Mooseker MS, Koleske AJ. 2001. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc. Natl. Acad. Sci. U. S. A. 98:14865–14870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Warren MS, et al. 2012. Integrin β1 signals through Arg to regulate postnatal dendritic arborization, synapse density, and behavior. J. Neurosci. 32:2824–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wetzel DM, Hakansson S, Hu K, Roos D, Sibley LD. 2003. Actin filament polymerization regulates gliding motility by apicomplexan parasites. Mol. Biol. Cell 14:396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilson ME, Pearson RD. 1986. Evidence that Leishmania donovani utilizes a mannose receptor on human mononuclear phagocytes to establish intracellular parasitism. J. Immunol. 136:4681–4688 [PubMed] [Google Scholar]
- 64. Wilson ME, Pearson RD. 1988. Roles of CR3 and mannose receptors in the attachment and ingestion of Leishmania donovani by human mononuclear phagocytes. Infect. Immun. 56:363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Woelbing F, et al. 2006. Uptake of Leishmania major by dendritic cells is mediated by Fcgamma receptors and facilitates acquisition of protective immunity. J. Exp. Med. 203:177–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yogalingam G, Pendergast AM. 2008. Abl kinases regulate autophagy by promoting the trafficking and function of lysosomal components. J. Biol. Chem. 283:35941–35953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zipfel PA, Zhang W, Quiroz M, Pendergast AM. 2004. Requirement for Abl kinases in T cell receptor signaling. Curr. Biol. 14:1222–1231 [DOI] [PubMed] [Google Scholar]