

Abstract
Tankyrase 1 is a poly(ADP-ribose) polymerase (PARP) that participates in a broad range of cellular activities due to interaction with multiple binding partners. Tankyrase 1 recognizes a linear six-amino-acid degenerate motif and, hence, has hundreds of potential target proteins. Binding of partner proteins to tankyrase 1 usually results in their poly(ADP-ribosyl)ation (PARsylation) and can lead to ubiquitylation and proteasomal degradation. However, it is not known how tankyrase 1 PARP activity is regulated. Here we identify GDP-mannose 4,6-dehydratase (GMD) as a binding partner of tankyrase 1. GMD is a cytosolic protein required for the first step of fucose synthesis. We show that GMD is complexed to tankyrase 1 in the cytosol throughout interphase, but its association with tankyrase 1 is reduced upon entry into mitosis, when tankyrase 1 binds to its other partners TRF1 (at telomeres) and NuMA (at spindle poles). In contrast to other binding partners, GMD is not PARsylated by tankyrase 1. Indeed, we show that GMD inhibits tankyrase 1 PARP activity in vitro, dependent on the GMD tankyrase 1 binding motif. In vivo, depletion of GMD led to degradation of tankyrase 1, dependent on the catalytic PARP activity of tankyrase 1. We speculate that association of tankyrase 1 with GMD in the cytosol sequesters tankyrase 1 in an inactive stable form that can be tapped by other target proteins as needed.
INTRODUCTION
Tankyrase 1 is a poly(ADP)ribose polymerase (PARP) that, like other PARPs, uses NAD+ as a substrate to generate poly(ADP-ribose) (PAR) onto protein acceptors (31). Poly(ADP-ribosyl)ation (PARsylation) is a dramatic posttranslational modification that can alter protein function. Tankyrase 1 (and its closely related homolog tankyrase 2) is distinguished by an ever-increasing number and remarkably diverse group of binding partners (13, 16). Tankyrases recognize a linear six-amino-acid RXXPDG (or degenerate) motif (TNKS-binding motif) in its binding partners (22). This recognition is achieved by the tankyrase ankyrin domain, comprised of multiple ankyrin repeats clustered into five conserved subdomains that are each capable of binding a motif (25, 26).
Tankyrase 1 was initially identified as a binding partner of TRF1, the double-stranded DNA telomere repeat binding protein that coats human telomeres and negatively regulates telomere length (31, 33). Tankyrase 1 binds to TRF1 via the RRCADG motif in its amino terminus (23, 31). Tankyrase 1 PARsylates TRF1 in vitro, inhibiting its ability to bind to telomeric DNA (31). Overexpression of tankyrase 1 in the nucleus evicts TRF1 from telomeres, leading to its ubiquitylation and proteasomal degradation (6) and to telomere elongation (30). RNA interference (RNAi) depletion studies show that, in addition to its role in telomere elongation, tankyrase 1 is required for resolution of sister telomere cohesion at mitosis (10). Despite its role at telomeres, only a minor fraction of endogenous tankyrase can be detected there (in prometaphase after nuclear envelope breakdown) by immunofluorescence analysis (8, 31). Tankyrase 1, which lacks a nuclear localization signal, resides mostly in the cytoplasm (29). It remains to be determined precisely when in the cell cycle tankyrase 1 localizes to telomeres.
NuMA, the nuclear matrix and mitotic apparatus protein, is another well-characterized binding partner of tankyrase 1. NuMA is required to establish and maintain spindle poles at mitosis (28). Tankyrase 1 binds to NuMA via the RTQPDG motif in its carboxy terminus (22). The endogenous proteins colocalize to spindle poles at mitosis (29); NuMA is required to recruit tankyrase 1 to the poles (5). Analysis across the cell cycle showed that NuMA associates with and is PARsylated by tankyrase 1 as cells enter mitosis (4, 5). Although the precise role of this PARsylation is not known, RNAi depletion studies indicate that tankyrase 1 is required at mitosis to establish normal spindle poles (4).
More recently, new partners of tankyrase 1 that function in signal transduction pathways, including axin (17), a key regulator in the Wnt signaling pathway, and 3BP2 (18), an adaptor protein in the SRC signaling pathway, have been identified. Tankyrase binds to axin via an RPPVPG motif (17) and to 3BP2 via an RSPPDG motif (18). These new tankyrase binding partners are distinguished by the finding that PARsylation leads directly to their ubiquitylation and degradation by the proteasome. Ubiquitylation is mediated by an E3 ligase RNF146 that recognizes PAR via its WWE domain (3, 36, 38). Hence, PARsylation by tankyrase 1 leads directly to binding of the E3 ligase and ubiquitylation and degradation of the target proteins. The finding that mutations that disrupt the RSPPDG binding site in 3BP2 underlie the inherited disorder cherubism (18) highlights the importance of this tankyrase interaction to human health.
To date, over 10 bona fide tankyrase binding partners have been identified, most by yeast two-hybrid screens (13, 16). Scrutiny of the database for RXXPDG or degenerate motifs reveals hundreds, if not thousands, of potential binding partners. Recently, a solution-based peptide library screen was used to derive an 8-amino-acid consensus RXXG/PDGXE/D TNKS-binding site (13). However, it still leaves the possibility that hundreds of proteins may be targeted by tankyrases. This complexity is compounded by the observation that tankyrase binding partners interact with only a fraction of the total tankyrase in the cell and only in a restricted window of the cell cycle (16). How these interactions are controlled and how the PARP activity of tankyrases is regulated remain to be determined.
To gain insight into this problem, we took a proteomics approach to identify major tankyrase 1 binding partners in human cells. We identified GDP-mannose 4,6-dehydratase (GMD) (21, 32) as a significant binding partner of tankyrase 1. GMD is required for the first step in the de novo synthesis of fucose (2). GDP-fucose, the donor substrate for all fucosylation reactions in the cell, is synthesized in the cytoplasm (starting with the dehydration reaction catalyzed by GMD) and transported to the endoplasmic reticulum (ER)/Golgi apparatus, where it is transferred to acceptor substrates involved in diverse biological functions, such as growth factor receptor signaling and adhesion (19). Here we identify GMD as a major partner of tankyrase 1. We show that tankyrase 1 association with GMD is prominent during interphase of the cell cycle but is reduced in mitosis when it associates with TRF1 and NuMA. Furthermore, we demonstrate that GMD inhibits tankyrase 1 PARP activity in vitro and influences tankyrase 1 stability in vivo.
MATERIALS AND METHODS
Plasmids.
MycGMD.WT contains an N-terminal myc epitope tag followed by amino acids 1 to 372 of human GMD (accession number NM_001500) cloned into the pLPCX vector (Clontech). The MycGMD.AA mutation was created by replacing the glutamic acid (D) and glycine (G) residues at positions 16 and 17 with alanine (A) residues by site-directed mutagenesis of MycGMD.WT using the oligonucletide 5′-CGGGGCTCCGGGGCCGCCGAGATGGGCAAGCCC-3′. Mutagenesis was performed using the Stratagene QuikChange site-directed mutagenesis kit according to the manufacturer's instructions. SUMO-GMD.WT contains full-length GMD cloned into the BamHI and XhoI sites of the pET28b-Sumo-6×His vector (7). SUMO-GMD.AA was generated as described above for MycGMD.AA. MycTRF1.WT (6) contains an N-terminal myc epitope tag followed by amino acids 2 to 439 in the pLPC vector (27). In the MycGMD.AA mutation (16), the D and G residues at position 17 and 18 are replaced by A residues.
Plasmid transfection.
HeLaI.2.11 cells (34) were transfected with MycGMD (wild type [WT] or AA), MycTRF1 (WT or AA), or vector (pLPC) using Lipofectamine 2000 reagent (Invitrogen) for 16 h according to the manufacturer's protocol.
Cell synchronization.
HeLaI.2.11 cells were grown in the presence of 2 mM thymidine for 16 h, washed three times with phosphate-buffered saline (PBS) and released into fresh medium for 11 h, treated again with 2 mM thymidine for 16 h, washed three times with PBS, and released into fresh medium for the tankyrase 1 chromatin immunoprecipitation (ChIP) analysis (see Fig. 3) and for the TNKS1 immunoprecipitation analysis (see Fig. 4A and B) and into medium containing 30 ng/ml nocodazole for the tankyrase 1 immunoprecipitation analysis (see Fig. 4D). Cells were then harvested by trypsinization at 2-h intervals from 0 to 16 h. Cells were synchronized in S phase by incubation in 2 mM hydroxy urea (HU) for 20 h or in mitosis by incubation in 30 ng/ml nocodazole for 20 h (see Fig. 4C).
Fluorescence-activated cell sorter (FACS) analysis.
Following trypsinization, cells were resuspended in PBS containing 2 mM EDTA, fixed with cold 70% (vol/vol) ethanol, stained with propidium iodide (50 μg/ml), and analyzed with a Becton, Dickinson FACScan and Modfit 3.0 software to determine relative DNA content.
Stable cell lines.
HTC75 cells stably expressing FlagTNKS1 (F7) were generated by stable cotransfection of pTetFLTNKS1 and the neomycin resistance plasmid pNY-HI into HTC75 cells (a hygromycin-resistant HT1080-derived clonal cell line that stably expresses the tetracycline-controlled transactivator [tTA] [33]) using calcium phosphate coprecipitation. F7, a G418-resistant clone, was expanded in the presence of doxycycline (Sigma) (100 ng/ml). For induction of FlagTNKS1, F7 cells were grown without (induced) doxycycline.
HeLa S3 cells (ATCC) stably expressing vector (pLPC) or FlagTNKS1 (pLPCFlagTNKS1) were generated by retroviral infection as described previously (15).
siRNA transfection.
F7 cells were induced (grown without doxycycline) for 9 h and then transfected with small interfering RNA (siRNA) for 48 h with Oligofectamine (Invitrogen) according to the manufacturer's protocol. The final concentration of siRNA was 100 nM. The following siRNAs (synthesized by Dharmacon Research Inc.) were used: GMD (5′-GGUCAGUAGCUAAGAUUUAUU-3′) and control (GFP Duplex I). HTC75 cells were transfected with GMD or control siRNA as described above for 48 h followed by plasmid transfection with FlagTNKS1.WT or FlagTNKS1.PD using Lipofectamine as described above for an additional 16 h.
Indirect immunofluorescence.
Cells were processed for indirect immunofluorescence as described previously (8). Briefly, cells were fixed in 2% paraformaldehyde in PBS, permeabilized in 0.5% Nonidet P-40 (NP-40)–PBS, and blocked in 1% bovine serum albumin (BSA) in PBS. Cells were incubated with rabbit anti-Flag (1.0 μg/ml) (Sigma) and detected with fluorescein isothiocyanate-conjugated donkey anti-rabbit antibodies (1:100) (Jackson Laboratories). DNA was stained with 4,6-diamino-2-phenylindole (DAPI) (0.2 μg/ml).
Image acquisition.
Images were acquired using a microscope (Axioplan 2; Carl Zeiss, Inc.) with a 20× lens (Carl Zeiss, Inc.) and a digital camera (C4742-95; Hamamatsu Photonics). Images were acquired and processed using Openlab software (Perkin Elmer).
Cell extracts.
Cells were resuspended in 4 volumes of TNE buffer (10 mM Tris [pH 7.8], 1% NP-40, 0.15 M NaCl, 1 mM EDTA, and 2.5% protease inhibitor cocktail [PIC] [Sigma]) and incubated for 1 h on ice. Suspensions were pelleted at 8,000 × g for 15 min. Twenty-five micrograms (determined by Bio-Rad protein assay) of supernatant proteins was fractionated by SDS-PAGE and analyzed by immunoblotting. For subcellular fractionation, PBS-washed cell pellets were further washed with 5 mM MgCl2-PBS and with buffer A, consisting of 10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 20% glycerol, 1 mM dithiothreitol (DTT), and PIC (Sigma). The pellet was resuspended in buffer A and homogenized on ice with a Dounce homogenizer. After centrifugation at 1,000 × g for 10 min at 4°C, the supernatant was collected as the cytoplasmic fraction. The resulting nuclear pellet was then washed twice with buffer A and resuspended in TNE buffer.
Immunoblot analysis.
Immunoblots were incubated separately with the following primary antibodies: rabbit anti-tankyrase 1 762 (1 μg/ml) (24), rabbit anti-myc (0.08 μg/ml) (Santa Cruz Biotechnologies), mouse anti-α-tubulin ascitic fluid (1:10,000) (Sigma), rabbit anti-phospho-histone H3 (ser10) (1 μg/ml) (Millipore), rabbit anti-GMD (a kind gift from Eiji Miyoshi) (1:1,000), rabbit anti-GMD (0.27 μg/ml) (Proteintech), rabbit anti-TRF1 415 (1 μg/ml) (8), mouse anti-NuMA (1 μg/ml) (Calbiochem), rabbit anti-PAR(96-10-04) (1:5,000) (Enzo Life Sciences), or rabbit anti-Flag (1.0 μg/ml) (Sigma), followed by horseradish peroxidase-conjugated donkey anti-rabbit or anti-mouse IgG (1:2,500) (Amersham) or horseradish peroxidase-conjugated goat antibiotin (1:1,000) (Cell Signaling). Bound antibody was detected by Super Signal West Pico (Thermo Scientific).
Immunoprecipitation.
Cells were lysed in 0.5 ml (per 15-cm-diameter dish) TNE buffer (10 mM Tris [pH 7.8], 1% Nonidet P-40, 0.15 M NaCl, 1 mM EDTA, and PIC) or in buffer C (20 mM HEPES-KOH [pH 7.9], 420 mM KCl, 25% glycerol, 0.1 mm EDTA, 5 mM MgCl2, 0.2% NP-40, 1 mM dithiothreitol [DTT], and PIC) on ice for 1 h and then pelleted at 8,000 × g for 10 min. Supernatants were precleared with protein G-Sepharose and rotation at 4°C for 30 min. Nonspecific protein aggregates were removed by centrifugation, and the supernatant was used for immunoprecipitation analysis or fractionated directly by SDS-PAGE. Supernatants were incubated with 0.35 μg TNKS1 762 or 1 μl of mouse anti-Myc 9B11 (Cell Signaling) for 1 h rocking at 4°C. Antigen-antibody complexes were collected on protein G beads at 4°C with rocking for 30 min. After binding, beads from TNE extracts were washed in TNE buffer and beads from buffer C extracts were washed in buffer D (20 mM HEPES, 100 mM KCl, 20% glycerol, 0.2 mM EDTA, 0.2 mM EGTA, 0.1% Triton X-100, and 0.1% NP-40). Proteins were fractionated on SDS-PAGE gels and processed for immunoblotting as described above. In the case of Flag immunoprecipitation, supernatant was incubated with anti-Flag M2 agarose (Sigma) for 4 h rocking at 4°C and, where indicated, FlagTNKS1 was eluted from the beads by incubation with TNE buffer containing 50 μg/ml Flag peptide (Sigma) for 1 h at room temperature, followed by reimmunoprecipitation with anti-TNKS1 762 as described above.
Chromatin immunoprecipitation.
HeLaI.2.11 cells in 15-cm-diameter dishes were fixed in 1% formaldehyde for 20 min with rotation at room temperature. Glycine was added to a final concentration of 0.125 M, and cells were incubated for 5 min. Cells were washed twice in ice-cold PBS and collected by scraping in 10 ml PBS containing 1 mM phenylmethylsulfonyl fluoride (PMSF), and cell pellets were lysed in 600 μl 1% SDS, 50 mM Tris-HCl, pH 8.0, 10 mM EDTA, and PIC. Lysates were sonicated to obtain chromatin fragments of <1 kb. Supernatants were collected at 5,000 rpm in a microcentrifuge for 10 min at 4°C. The protein concentration of the supernatant was determined by a DC protein assay (Bio-Rad). Twenty-five percent was saved as input. For each IP, 1 mg was diluted to 1.5 ml with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.0, 150 mM NaCl, and PIC) and samples were precleared by incubating with 20 μl protein G-Sepharose for 30 min at 4°C. Five microliters of crude serum—TNKS1 465 (31), preimmune (PI) 465, TNKS1 762 (24), or TRF1 415 (8) (465 and 762 are number designations of rabbits)—or 20 μl Myc-agarose was added, and samples were incubated overnight with rotation at 4°C. Following the addition of 40 μl protein G-Sepharose beads, samples were incubated for 30 min at 4°C and immune complexes collected at 1,000 rpm in a microcentrifuge for 1 min at 4°C. The following washes were with 1 ml of the indicated buffers for 3 min with rotation at 4°C. Beads were washed twice with ChIP dilution buffer, once with 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, once with 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, once with 0.25 M LiCl, 1% NP-40, 1% Na-deoxycholate, 1 mM EDTA, 10 mM Tris-HCl (pH 8.0), and twice with TE (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). Chromatin was eluted from the beads by the addition of 500 μl 1% SDS, 0.1 M Na2CO3. Samples were vortexed for 10 min and pelleted for 3 min at 10,000 rpm. The supernatant was collected, and after the addition of 20 μl of 5 M NaCl, cross-links were reversed for 5 h at 65°C. Following the addition of 10 μl of 1 M Tris-HCl, pH 6.5, 10 μl of 0.5 M EDTA, and 10 μg proteinase K, samples were incubated at 48°C for 1 h and phenol was extracted, and the DNA was precipitated overnight at −20°C with 1 ml ethanol. The precipitate was dissolved in 50 μl TE with 10 μg/ml RNase and incubated at 37°C for 30 min. The sample volume was adjusted to 500 μl with 6× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), denatured at 95°C for 5 min, and dot blotted onto Hybond membranes in 6× SSC (40% was loaded for the detection of telomeric [Telo] sequences and 10% for Alu sequences). Membranes were treated with 1.5 M NaCl, 0.5 N NaOH for 10 min and with 1 M NaCl, 0.5 M Tris-HCl (pH 7.0) for 10 min, and the DNA was cross-linked using a Stratalinker (Stratagene). Hybridization with a 32P-TTAGGG or Alu probe was performed in Church buffer (0.5 M sodium phosphate buffer [pH 7.2], 1% bovine serum albumin [BSA], 1 mM EDTA, 7% SDS) as described previously (9). Membranes were washed once in 40 mM sodium phosphate buffer (pH 7.2), 0.5% BSA, 1 mM EDTA, 5% SDS and once in 40 mM sodium phosphate buffer (pH 7.2), 1 mM EDTA, 1% SDS at 65°C for 10 min each and exposed to Kodak XAR film. ImageJ software was used to quantify the percentage of precipitated DNA relative to the input DNA. The Alu signal was subtracted from the Telo signal.
PARP assays.
For tankyrase 1 PARP assays, samples containing recombinant tankyrase 1 (0.1 to 0.2 μg) (31) and recombinant GMD (2 μg) or TRF1 (2 μg) (31) were incubated for 30 min at 25°C in 10 μl PARP reaction buffer (20 mM Tris [pH 8.0], 4 mM MgCl2, 0.2 mM dithiothreitol) containing 25 μM biotinylated NAD+ (Trevigen) or 1 μM 32P-NAD+ (Perkin Elmer). Recombinant GMD was expressed and purified from Escherichia coli BL21 cells according to standard protocols. Digestion and removal of the SUMO protein tag were performed with SUMO protease 1 according to the manufacturers' instructions (Life Sensors), except where the SUMO protein tag was not removed for GMD.WT or GMD.AA (see Fig. 6B). Reactions were terminated by the addition of 2× sample buffer and fractionated by SDS/PAGE. Gels were either dried and autoradiographed or transferred electrophoretically to nitrocellulose and probe with antibiotin horseradish peroxidase (1:1,000) (Cell Signaling). For PARP-1, PARP assay samples containing 1.25 units PARP-1 (Trevigen) and 0.2 μg activated DNA (Trevigen) were incubated and processed as described above.
RESULTS
GMD is a novel binding partner of tankyrase 1.
We used an inducible (tetracycline-controlled) HTC75 stable cell line (F7) expressing a Flag epitope-tagged tankyrase 1 allele (FlagTNKS1) to identify tankyrase 1 binding partners. Cells were grown with and without induction and subject to immunoprecipitation with anti-Flag beads under low-stringency conditions. As shown in Fig. 1A, FlagTNKS1 was immunoprecipitated specifically from induced cells. In addition, we detected a single polypeptide migrating ahead of the 45-kDa marker that uniquely coimmunoprecipitated with FlagTNKS1 from induced cells. The identity of the polypeptide, determined by mass spectrometry of tryptic polypeptides derived from the excised gel slice, was GDP-mannose 4,6-dehydratase (GMD).
Fig 1.
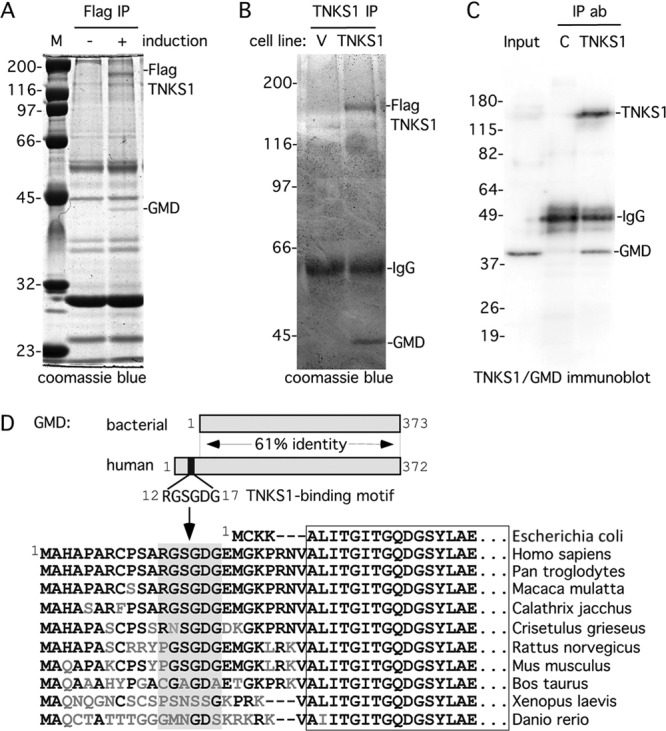
GMD is a novel binding partner of tankyrase 1. (A and B) GMD coimmunoprecipitated with FlagTNKS1 using a (A) one- or (B) two-step protocol. (A) Coomassie-stained gel of proteins immunoprecipitated from an HTC75 cell line (F7) expressing inducible FlagTNKS1 that was grown with (+) or without (−) induction and subject to immunoprecipitation with anti-Flag beads. (B) Coomassie-stained gel of proteins isolated from two rounds of immunoprecipitation from HeLa S3 cell lines stably expressing a vector (V) or FlagTNKS1. Cell lysates were immunoprecipitated with anti-Flag beads, bound proteins were eluted with Flag peptide, and the eluates were subject to immunoprecipitation with anti-TNKS1 762 antibody. (A and B) The band indicated as GMD was excised and its identity was determined by mass spectrometry. (C) Endogenous GMD coimmunoprecipitated with endogenous tankyrase 1. HeLaI.2.11 cell lysates were immunoprecipitated with control or anti-TNKS1 762 antibodies and analyzed by immunoblotting with anti-TNKS1 762 or anti-GMD antibodies. (D) Schematic diagram comparing bacterial and human GMD. Human GMD has an amino-terminal extension that contains the TNKS-binding motif RDSGDG. Below, alignment of the amino termini of GMD from Escherichia coli (NCBI protein accession number ADV17654), Homo sapiens (protein accession number AAH00117), Pan troglodytes (reference sequence number XP_518203), Macaca mulatta (protein accession number AFE65973), Callithrix jacchus (reference sequence number XP_002746325), Crisetulus grieseus (reference sequence number NP_001233625), Rattus norvegicus (protein accession number AAI04709), Mus musculus (protein accession number AAH93502), Bos taurus (protein accession number AAI03031), Xenopus laevis (protein accession number AAI57412), and Danio rerio (protein accession number BAF73663). Identical amino acids are in black.
Since the immunoprecipitation analysis shown in Fig. 1A yielded many nonspecific (independent of induction) polypeptides, we sought a more stringent approach for identifying tankyrase 1 binding partners. Stable HeLa S3 cell lines expressing a vector control or FlagTNKS1 were subject to immunoprecipitation with anti-Flag beads (affinity resin). FlagTNKS1 (and any bound factors) was then eluted from the beads with Flag peptide, immunoprecipitated with anti-tankyrase 1 antibody, and fractionated by SDS-PAGE. As shown in Fig. 1B, the background was greatly reduced, as there were very few proteins in either lane. Once again, we detected a single polypeptide migrating ahead of the 45-kDa marker that uniquely coimmunoprecipitated with FlagTNKS1, determined by mass spectrometry to be GMD. To determine if endogenous tankyrase 1 was associated with GMD, tankyrase 1 was immunoprecipitated from HeLaI.2.11 cells and analyzed by immunoblotting with anti-GMD antibodies. As shown in Fig. 1C, endogenous GMD specifically coimmunoprecipitated with endogenous tankyrase 1. Together these data indicate that GMD is a robust binding partner of tankyrase 1.
Inspection of the primary sequence of human GMD revealed an RGSGDG motif in its amino terminus (Fig. 1D). GMD is highly conserved, with 61% identity between the bacterial and human proteins. One distinction of GMD from nonbacterial sources is a small amino-terminal extension. It is this extension that houses the RGSGDG motif (Fig. 1D). The motif is conserved in monkeys and some mammals but not in frog or zebrafish (Fig. 1D).
To determine if the RGSGDG motif in GMD was required for binding to tankyrase 1, we generated a double point mutation converting DG to AA (GMD.AA) (Fig. 2A). A similar mutation in TRF1 (TRF1.AA) (Fig. 2A) was shown previously to abrogate its ability to bind tankyrase 1 (16). Wild-type (WT) or mutant (AA) MycGMD and MycTRF1 plasmids were transfected into HeLaI.2.11 cells. Cell extracts were immunoprecipitated with Myc beads (affinity resin) and subjected to immunoblot analysis. As shown in Fig. 2B, GMD.WT, but not GMD.AA, efficiently coimmunoprecipitated endogenous tankyrase 1. Similarly, TRF1.WT, but not TRF1.AA, efficiently coimmunoprecipitated endogenous tankyrase 1.
Fig 2.

The RGSGDG motif in GMD is required for binding to tankyrase 1. (A) Schematic diagram showing the TNKS binding motif in GMD and TRF1 with the double point mutations indicated. (B) The TNKS binding motif is required for binding of GMD and TRF1. Lysates from HeLaI.2.11 cells transfected with vector (V) or MycGMD (WT or AA) or MycTRF1 (WT or AA) were immunoprecipitated with anti-myc beads and analyzed by immunoblotting with anti-TNKS1 762 or anti-Myc antibodies. *, breakdown product of MycTRF1. (C) Tankyrase 1 binding partners TRF1 and GMD fractionate to distinct subcellular compartments. HeLaI.2.11 cells were fractionated into nuclear and cytosol extracts and analyzed by immunoblotting with anti-TNKS 762, anti-TRF1 415, anti-GMD, and anti-α-tubulin antibodies.
While TRF1 and GMD both interact with tankyrase 1 (Fig. 2B), the proteins localize to distinct subcellular compartments, with TRF1 in the nucleus and GMD in the cytoplasm. We confirmed this distinct localization by fractionating cells into nuclear and cytosolic fractions and analyzing by immunoblotting. As shown in Fig. 2C, tankyrase 1 fractionated to both compartments with the majority in the cytosol, whereas GMD fractionated exclusively to the cytosol and TRF1 fractionated exclusively to the nucleus.
Distinct association of tankyrase 1 with GMD versus TRF1 and NuMA across the cell cycle.
We next asked if tankyrase 1 interaction with GMD and TRF1 occurred in the same or different windows of the cell cycle. While we can readily analyze association of endogenous tankyrase 1 and GMD by coimmunoprecipitation (shown in Fig. 1C), analysis of the association between endogenous tankyrase 1 and TRF1 has proven difficult, likely due to the low abundance of the complex (8). We have shown by immunofluorescence analysis that tankyrase 1 localizes with TRF1 at telomeres in prometaphase spreads, but we have not detected tankyrase 1 at telomeres at all stages of the cell cycle. We thus sought to use a more sensitive assay, chromatin immunoprecipitation (ChIP), to detect association of tankyrase 1 with TRF1/telomeres across the cell cycle. In this assay, protein-associated telomeric DNA is detected by quantitative hybridization of a telomeric TTAGGG repeat probe to DNA dot blots. Alu repeats are used as a negative control.
To establish conditions for telomeric ChIP of endogenous tankyrase 1, we took advantage of a previous observation where we showed by immunofluorescence analysis that overexpression of TRF1.WT but not TRF1.AA recruited endogenous tankyrase 1 to telomeres (16). Hence, HeLaI.2.11 cells were mock transfected or transfected with MycTRF1.WT or MycTRF1.AA. ChIP analysis with anti-TRF1 antibody showed that, as expected, TRF1 was enriched at telomeres in all three cases (Fig. 3A and B). ChIP analysis with anti-Myc antibody showed MycTRF1 enrichment at telomeres in cells transfected with MycTRF1.WT or Myc.TRF1.AA TRF1 but not in control cells (Fig. 3A and B). The observation that TRF1.WT and TRF1.AA were similarly enriched at telomeres confirms previous immunofluorescence analysis demonstrating that TRF1 association with telomeres is independent of its TNKS-binding site. When ChIP was performed with anti-TNKS1 antibody, tankyrase 1 was enriched at telomeres, but only in cells expressing TRF1.WT, not in those expressing TRF1.AA or vector control (Fig. 3A and B). Together, these results demonstrate that we can use the ChIP assay to detect endogenous tankyrase 1 at telomeres.
Fig 3.

Tankyrase 1 is recruited to telomeres by TRF1 and localizes there in G2/M. (A and B) TRF1 recruits tankyrase 1 to telomeres. Telomeric DNA ChIP analysis of HeLaI.2.11 cells mock transfected (C) or transfected with MycTRF1 (WT or AA) using the indicated beads or antibodies: beads (protein G-Sepharose), Myc beads (Myc-agarose), TRF1 415 (raised against baculovirus-derived full-length TRF1), TNKS1 465 (raised against E. coli-derived tankyrase 1 amino acids 973 to 1149), PI 465 (preimmune serum). Dot blots with the immunoprecipitated DNA were analyzed by Southern blotting with 32P-labeled telomeric or Alu repeat probes. Autoradiographs were cropped from the same experiment. (B) Graphical representation of the percentage of immunoprecipitated telomeric DNA relative to total input DNA derived from three independent experiments; error bars indicate standard deviations. (C to E) Tankyrase localizes to telomeres in G2/M. (C) HeLaI.2.11 cells were synchronized in G1/S by a double thymidine block, released, collected at 2-h intervals, and analyzed by FACS analysis (y axis, cell numbers, 0 to 200; x axis, relative DNA content based on propidium iodide staining, 0 to 600) and by immunoblotting with antibodies against α-tubulin or phospho-histone H3 (Ser10) to mark entry into G2/M. (D) Telomeric DNA ChIP analysis of staged cell cycle extracts using the following antibodies: TRF1 415, TNKS1 465, PI 465, and TNKS1 762 (raised against E. coli-derived tankyrase 1 amino acids 973 to 1149). Dot blots with the immunoprecipitated DNA were analyzed by Southern blotting with 32P-labeled telomeric or Alu repeat probes. Autoradiographs were cropped from the same experiment. (E) Graphical representation of the percentage of immunoprecipitated telomeric DNA relative to total input DNA derived from three independent experiments; error bars indicate standard deviations using the indicated antibodies: anti-TNKS1 465 or 762 antibodies, x axis on the left; anti-TRF1 415 antibody, x axis on the right.
To analyze association of tankyrase 1 with telomeres (in untransfected cells) across the cell cycle, HeLaI.2.11 cells were synchronized by a double thymidine block, released, and collected every 2 h. Cells were analyzed at each time point by FACS, immunoblotting, and ChIP. As shown in Fig. 3C, FACS analysis and immunoblotting with antibody against histone H3 phosphorylated on serine 10 (a mitosis-specific mark) indicated that cells were in G2/M between 8 and 12 h. ChIP analysis with anti-TRF1 antibody indicated that TRF1 was enriched at telomeres throughout the cell cycle (Fig. 3D) (35). ChIP analysis using antibodies against TNKS1 from two different rabbits (rabbit 465 or rabbit 762) showed that tankyrase 1 was enriched at telomeres at the 10-h time point (Fig. 3D and E). Comparison of the amount of tankyrase 1 at telomeres at 2 versus 10 h indicates a 10-fold enrichment of tankyrase 1. TRF1 was also increased at mitosis but to a lesser (2-fold) extent. Together, these data indicate that tankyrase 1 localizes to telomeres at mitosis.
To determine when in the cell cycle tankyrase 1 interacted with GMD, we synchronized cells as described above and assayed the fractions by immunoprecipitation with anti-TNKS1 antibody. Analysis of whole-cell extracts across the cell cycle showed that tankyrase 1 was expressed at similar levels across the cell cycle, with a shift to a more slowly migrating phosphorylated form at mitosis (10 to 12 h) (Fig. 4A) (5). Immunoblot analysis with anti-GMD antibody shows that GMD is expressed at constant levels across the cell cycle (Fig. 4A). Immunoprecipitation with anti-TNKS1 antibody followed by immunoblotting with anti-GMD antibody shows that GMD associates with tankyrase 1 throughout interphase but is reduced in G2/M (10 to 12 h) (Fig. 4B). Conversely, another tankyrase 1 binding partner, NuMA, does not associate with tankyrase 1 throughout the cell cycle but, rather, only in G2/M (10 to 12 h) (Fig. 4B) (5).
Fig 4.

Association of GMD with tankyrase 1 is lost at mitosis. (A and B) Immunoprecipitation analysis across the cell cycle. HeLaI.2.11 cells were synchronized in G1/S by a double thymidine block, released, and collected at 2-h intervals, and whole-cell extracts (Input) generated in TNE buffer were analyzed by immunoblotting with antibodies to TNKS 762, GMD, NuMA, and α-tubulin. *, more slowly migrating form of tankyrase 1 that marks entry into mitosis at the 10- and 12-h time points. (B) Staged extracts generated in TNE buffer were immunoprecipitated with anti-TNKS1 762 and analyzed by immunoblotting with antibodies to TNKS 762, GMD, and NuMA. (C) Immunoprecipitation analysis in cells arrested in S phase versus mitosis. HelaI.2.11 cells were untreated (−) or incubated with hydroxy urea (HU) or nocodazole (Noc) for 20 h. Cell extracts generated in buffer C were analyzed directly (Input) or were immunoprecipitated with control (C) or anti-TNKS1 762 antibody and analyzed by immunoblotting with antibodies to TNKS1 762, GMD, or NuMA. A shorter exposure of the GMD blot is indicated. (D) HeLaI.2.11 cells were synchronized in G1/S by a double thymidine block, released into nocodazole, and collected at 2-h intervals. Staged extracts generated in buffer C were immunoprecipitated with anti-TNKS1 762 and analyzed by immunoblotting with antibodies to TNKS 762 and GMD. GMD levels relative to tankyrase 1 and normalized to the zero time point are indicated below the blot.
For a more robust demonstration of the differential cell cycle association of GMD and NuMA with tankyrase 1, we arrested cells in S phase with hydroxy urea (HU) and in mitosis with nocodazole (Noc) and immunoprecipitated the cell extracts (generated under more stringent buffer conditions; see Materials and Methods) with anti-TNKS1. As shown in Fig. 4C, tankyrase 1 coimmunoprecipitated GMD in S phase but not in mitosis. Conversely, tankyrase 1 coimmunoprecipitated NuMA in mitosis but not in S phase. Finally, we analyzed association of GMD and tankyrase 1 across the cell cycle by synchronizing cells with a double thymidine block, followed by release into nocodazole and collection every 2 h. As shown in Fig. 4D, GMD association with tankyrase 1 is reduced at the 10- and 12-h time points (G2/M). Together, these data demonstrate that tankyrase 1 has distinct cell cycle associations with its binding partners.
GMD is not an acceptor of PARsylation by tankyrase 1.
Together, the data described above indicate that GMD interacts with tankyrase 1 in the cytoplasm during interphase, but upon entry into mitosis the association is reduced and tankyrase interacts with NuMA at spindle poles and with TRF1 at telomeres. We showed previously that TRF1 and NuMA were acceptors of PARsylation by tankyrase 1. We thus asked if GMD was an acceptor of PARsylation by tankyrase 1 by performing an in vitro PARP assay with purified proteins. Recombinant human GMD purified from E. coli was incubated with recombinant tankyrase 1 purified from baculovirus and 32P-NAD+ substrate. Reaction products were fractionated by SDS-PAGE, stained with Coomassie blue to visualize the proteins (Fig. 5A, left panel), and autoradiographed to visualize ADP-ribosylated proteins (Fig. 5A, right panel). Tankyrase 1 underwent automodification, as measured by the appearance of 32P-labeled protein (Fig. 5A, right panel, lane 1). However, addition of GMD to the reaction did not result in its ADP ribosylation (Fig. 5A, right panel, lanes 3 and 4). The addition of excess unlabeled substrate (NAD+) resulted in a more slowly migrating smear of 32P-labeled tankyrase 1, indicating PARsylation, but again GMD was not modified (Fig. 5A, right panel, lane 5). In contrast, addition of a similar amount of the known acceptor TRF1 led to its PARsylation (compare Fig. 5A, right panel, lanes 5 and 6). Even upon a much longer exposure, specific labeling of GMD was not detected (Fig. 5A, longer exposure). Together, these data indicate that GMD is not an acceptor of PARsylation by tankyrase 1.
Fig 5.

GMD is not an acceptor of PARsylation by tankyrase 1. (A) Purified recombinant GMD is not PARsylated by tankyrase 1 in vitro. Recombinant tankyrase 1 was incubated with the substrate 32P-NAD+ in the absence (−) or presence of recombinant GMD or TRF1. Unlabeled NAD+ was added where indicated. The products were fractionated by SDS-PAGE and visualized by Coomassie blue stain (left panel) and autoradiography (right panel; longer exposure, lower right panel). *, breakdown product of MycTRF1. (B) GMD isolated as a tankyrase 1 complex from human cells is not PARsylated by tankyrase 1 in vitro. Extracts from HeLaI.2.11 cells untransfected (−) or transfected with vector (V), MycGMD (WT or AA), or MycTRF1 (WT or AA) were immunoprecipitated with Myc beads. The beads were incubated with recombinant tankyrase 1 and NAD+ substrate, fractionated by SDS-PAGE, and analyzed by immunoblotting with antibodies to Myc (top panel), TNKS1 762 (middle panel), and PAR (lower panel).
The findings described above were unexpected, since most tankyrase 1 binding partners described to date have been shown to be acceptors of PARsylation. One possibility was that the E. coli-produced recombinant GMD protein was not competent for PARsylation. To address this issue, we asked if tankyrase 1-bound GMD from human cells was an acceptor of PARsylation. HeLaI.2.11 cells were transfected with MycGMD.WT or MycGMD.AA and, as a control, MycTRF1.WT or MycTRF1.AA. Cell extracts were immunoprecipitated with anti-Myc beads. Recombinant tankyrase 1 was added to the beads and incubated with unlabeled NAD+ substrate (Fig. 5B, Protocol). The products were fractionated by SDS-PAGE, and PARsylation was detected by immunoblotting with anti-PAR antibodies. As shown in Fig. 5B, similar amounts of MycGMD and MycTRF1 proteins were immunoprecipitated and the same amount of tankyrase 1 was added to each (middle panel). However, immunoblot analysis with anti-PAR antibody showed that GMD.WT was not PARsylated, whereas a similar amount of TRF1.WT was (Fig. 5B, bottom panel, compare lanes 3 and 5). These data show that human GMD that associates with tankyrase 1 in vivo is not an acceptor of PARsylation.
GMD inhibits tankyrase 1 PARP activity in vitro.
Upon examination of the PARsylation reaction shown in Fig. 5A, we noticed that addition of 5-fold more GMD reduced the automodification of tankyrase 1 (compare lanes 3 and 4), suggesting that GMD might inhibit tankyrase 1. To address this question, we performed PARsylation reactions with tankyrase 1, increasing amounts of GMD, and biotinylated NAD+ substrate. The reaction products were fractionated by SDS-PAGE, transferred to nitrocellulose, stained with amido black to visualize the proteins (Fig. 6A, top panel), and probed with antibiotin to visualize PARsylated proteins (Fig. 6A, bottom panel). Addition of the smallest amount of GMD (0.5 μg) inhibited auto-PARsylation of tankyrase 1, and this inhibition increased with increasing amounts of GMD (Fig. 6A, lanes 1 to 5). Inclusion of TRF1 in the reaction showed that PARsylation of TRF1 was also reduced by the addition of GMD (lane 7) (although not to as great a degree as tankyrase 1 auto-PARsylation), indicating that GMD inhibits tankyrase 1 automodification as well as tankyrase 1 PARsylation of other acceptors.
Fig 6.
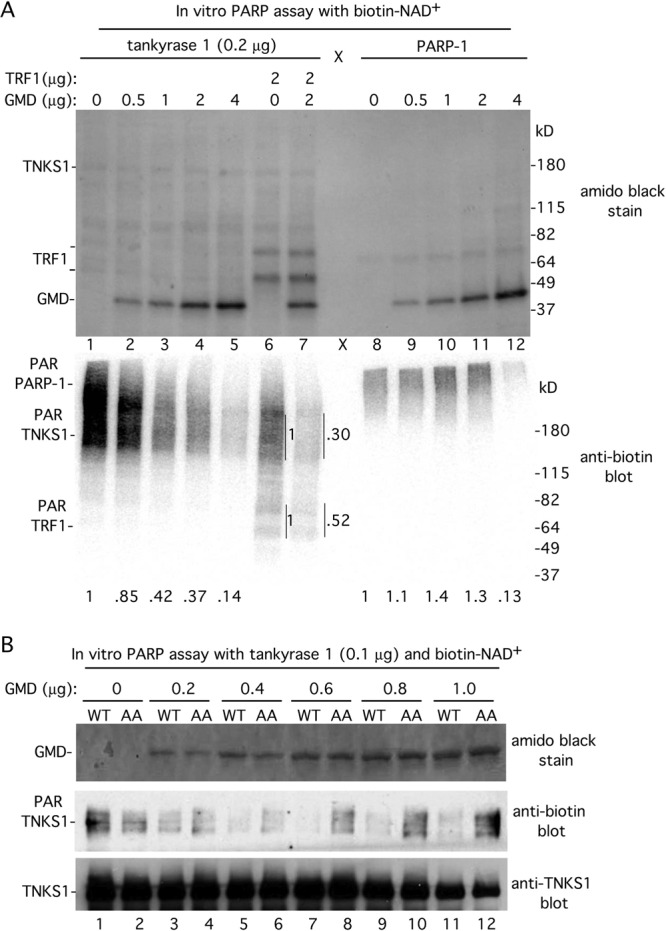
GMD inhibits tankyrase 1 PARP activity. (A) GMD inhibits tankyrase 1 but not PARP-1 in vitro. Recombinant tankyrase 1 or PARP-1 was incubated with the substrate biotin-NAD+ in the absence (−) or presence of recombinant GMD and/or TRF1. Proteins were fractionated by SDS-PAGE, transferred to nitrocellulose, and visualized by staining with amido black (top panel) and by immunoblotting with antibiotin antibody (bottom panel). PAR-TNKS1 levels normalized to lane 1 and PAR-PARP-1 levels normalized to lane 8 are indicated below the blot. PAR-TNKS1 and PAR-TRF1 levels in lane 7 are normalized to lane 6 and indicated on the side of the blot. (B) GMD inhibition of tankyrase 1 depends on its TNKS1 binding site. Recombinant tankyrase 1 was incubated with the substrate biotin-NAD+ in the absence or presence of increasing amounts of recombinant GMD (WT or AA). Proteins were fractionated by SDS-PAGE, transferred to nitrocellulose, and visualized by staining with amido black (top panel) and by immunoblotting with anti-biotin (middle panel) or anti-TNKS1 762 antibodies.
To determine if GMD inhibition was specific for tankyrase 1, we tested its ability to inhibit a different PARP, PARP-1. PARsylation reactions were performed with recombinant PARP-1 and increasing amounts of GMD. As shown in Fig. 6A, in contrast to tankyrase 1, PARP-1 automodification was not inhibited by 0.5, 1, or 2 μg of GMD (compare lanes 8 to 11 with 1 to 4). We did observe inhibition of PARP-1 upon addition of the largest amount of GMD (4 μg) (lane 12). However, as this amount of GMD is a large excess, the inhibition may be nonspecific.
The data described above suggested that GMD was a specific inhibitor of tankyrase 1 rather than a general inhibitor of PARPs. Thus, a prediction is that the inhibition should be dependent on the ability of GMD to bind to tankyrase 1. To address this possibility, we generated and purified mutant recombinant GMD (GMD.AA) from E. coli that cannot bind tankyrase 1. PARsylation reactions were performed with tankyrase 1, biotinylated NAD+ substrate, and increasing amounts of GMD.WT or GMD.AA proteins. As shown in Fig. 6B, lane 3, addition of the smallest amount of GMD.WT (0.2 μg) inhibited tankyrase 1 auto-PARsylation. In contrast, addition of 0.2 μg GMD.AA (Fig. 6B, lane 4) had no effect on PARsylation of tankyrase 1. Even at the largest amount of GMD (1 μg) where GMD.WT fully inhibited tankyrase 1 PARsylation, GMD.AA had no effect (Fig. 6B, lanes 11 and 12). These data indicate that the ability of GMD to inhibit tankyrase 1 PARP activity depends on its tankyrase 1 binding domain.
GMD influences tankyrase 1 stability in vivo.
Tankyrase 1-mediated PARsylation has been shown to influence protein degradation of a number of proteins. In particular, auto-PARsylation of tankyrase 1 led to its own ubiquitylation and proteasomal degradation in the cytoplasm (37). To determine if GMD influenced tankyrase 1 in vivo, we depleted GMD using siRNA and determined the effect on tankyrase 1 expression. Induced F7 cells (the HTC75 cell line stably expressing inducible FlagTNKS1) (Fig. 1A) were transiently transfected with control or GMD siRNA for 48 h and harvested for immunofluorescence and immunoblot analysis. Immunofluorescence analysis of cells transfected with control siRNA showed that FlagTNKS1 was expressed in the cytoplasm in the majority of cells (Fig. 7A, upper left panel). In contrast, in GMD siRNA-transfected cells, FlagTNKS1 expression levels were reduced in individual cells and there was an overall reduction in the number of FlagTNKS1-expressing cells (Fig. 7A, upper right panel, and quantification in Fig. 7B). This reduction was confirmed by immunoblot analysis; we observed a 50% reduction in FlagTNKS1 in GMD-depleted cells (Fig. 7C, lane 2). This reduction was fully rescued by treatment with the proteasome inhibitor MG132 (Fig. 7C, lane 3), indicating that GMD depletion led to degradation of tankyrase 1 by the proteasome. To determine if the reduction required tankyrase 1 PARP activity, HTC75 cells were transfected first, with control or GMD siRNA for 48 h, followed by transfection with a plasmid expressing FlagTNKS1 wild type (WT) or a PARP-dead allele (PD) (8) for an additional 16 h. As shown in Fig. 7D, GMD depletion led to a reduction in wild-type FlagTNKS1 (Fig. 7D, lane 2) but not the PARP-dead allele (Fig. 7D, lane 4). Together, these data are consistent with the notion that GMD inhibits tankyrase 1 auto-PARsylation, preventing its degradation in the cytoplasm.
Fig 7.
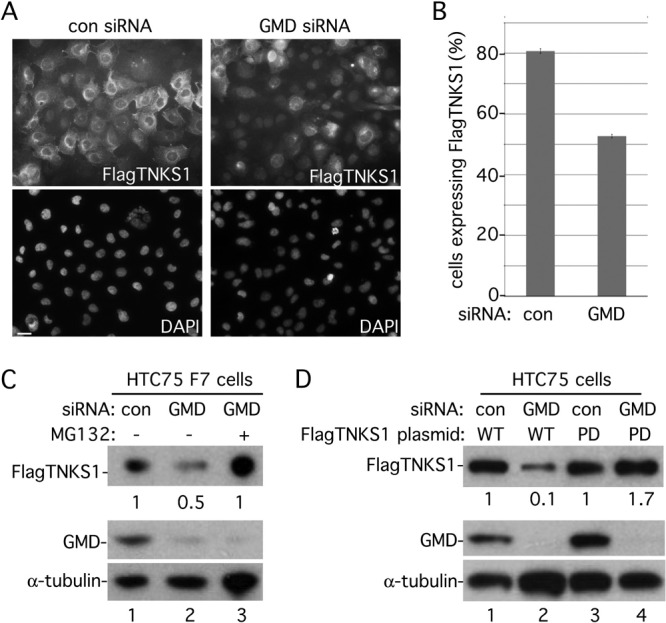
GMD influences tankyrase 1 stability in vivo. (A to C) Induced F7 cells were transfected with control (con) or GMD siRNA for 48 h and analyzed by immunofluorescence (A and B) or immunoblotting (C). (A) Cells were formaldehyde fixed and stained with anti-Flag antibody (top panels) or DAPI (bottom panels). Bar, 100 μm. (B) Graphical representation of the frequency of cells expressing FlagTNKS1 in control versus GMD siRNA cells. Data represent the means ± standard deviations of the results of three independent experiments (n = 300 cells or more each). (C) Depletion of GMD leads to degradation of tankyrase 1 by the proteasome. Cell extracts were analyzed by immunoblotting with anti-Flag, anti-GMD, or anti-α-tubulin antibodies. 12.5 μM MG132 was added 5 h prior to harvest. FlagTNKS1 protein levels relative to α-tubulin and normalized to the siRNA control are indicated below the blot. (D) Loss of FlagTNKS1 depends on its catalytic PARP domain. HTC75 cells were transfected first with control or GMD siRNA for 48 h, followed by transfection with a plasmid expressing FlagTNKS1 wild type (WT) or a PARP-dead allele (PD) for an additional 16 h. Cell extracts were analyzed by immunoblotting with anti-Flag, anti-GMD, or anti-α-tubulin antibodies. FlagTNKS1 protein levels relative to α-tubulin and normalized to the siRNA control are indicated below the blot.
DISCUSSION
We have identified GMD, the enzyme required for the first step of fucose synthesis in the cytoplasm, as a major partner of tankyrase 1. Like previous tankyrase 1 binding partners, GMD has a strong consensus TNKS binding motif, RGSGDG. However, unlike most other TNKS binding partners, GMD does not serve as an acceptor of PARsylation. In fact, GMD inhibits the catalytic PARP activity of tankyrase 1. GMD inhibited tankyrase 1 automodification as well as tankyrase 1-mediated PARsylation of its acceptor TRF1. The inhibition was specific for tankyrase 1: GMD did not inhibit PARP-1, and inhibition required an intact TNKS1 binding motif. Finally, we show that depletion of GMD led to degradation of tankyrase 1 dependent on its catalytic PARP activity, consistent with the notion that GMD inhibits tankyrase 1 PARP activity in vivo. We speculate that the GMD-tankyrase 1 complex may serve as a ready pool of tankyrase 1 that is kept in an inactive form that can be tapped by other binding partners.
A previous study identified another TNKS binding partner, myeloid cell leukemia 1 protein (Mcl-1), that, like GMD, is not an acceptor of PARsylation by tankyrase 1 (1). Mcl-1 was found to suppress PARsylation of TRF1 by tankyrase 1 in vitro. Mcl-1 also suppressed auto-PARsylation of tankyrase 1 but to a lesser extent. Mcl-1 contains a RPPPIG TNKS-1 binding motif, but it was not determined if the inhibition depended on this motif. Whether Mcl-1 and GMD influence tankyrase 1 activity by the same or different mechanisms remains to be determined.
We found that association between GMD and tankyrase 1 was prominent in interphase. Coimmunoprecipitation analysis across the cell cycle showed that GMD was complexed to tankyrase 1 in the G1, S, and G2 phases but was reduced in mitosis. In contrast, the association between NuMA or TRF1 and tankyrase 1 was enriched in mitosis. Tankyrase 1 does not contain a nuclear localization signal, and the bulk of the protein localizes to the cytoplasm. Hence, nuclear proteins such as NuMA and TRF1 may have to await mitosis and nuclear envelope breakdown to gain full access to tankyrase 1. It remains to be determined how tankyrase 1 is freed from its association with GMD. NuMA and TRF1 may compete with GMD for binding to tankyrase 1. Alternatively, the phosphorylation of tankyrase 1 that occurs upon entry into mitosis (5) could release GMD.
GMD is required for fucosylation, a modification that acts in a number of signal transduction pathways that include Notch and epidermal growth factor (EGF) (19) and, more recently, the TRAIL (TNF-related apoptosis-inducing ligand) pathway (11). TRAIL-mediated killing of tumor cells holds promise as a tumor therapy. Studies show that some human colon cancer cells lack GMD and have a defect in fucosylation, suggesting that loss of GMD could be a common mechanism to evade TRAIL-mediated killing (14, 20). Our studies suggest that in the absence of GMD, tankyrase 1 is turned over more rapidly. Hence, loss of GMD could impact multiple distinct pathways that influence cancer.
Since its initial identification as a partner of the telomere repeat protein TRF1, tankyrase 1 and its closely related homolog tankyrase 2 have been found to associate with a variety of proteins involved in a broad range of biological functions. PARsylation by tankyrase 1 can lead to eviction from DNA in the case of TRF1 (30) or to proteasomal degradation in the case of axin and 3BP2 (17, 18). Studies have shown that pharmacological inhibition of tankyrases by the specific small molecule inhibitor XAV939 results in dramatic consequences (via stabilization of axins) ranging from targeted killing of tumor cells (17) to enhanced myelin regeneration in nerve cells (12). Here we have identified a partner of tankyrase 1 that inhibits its PARP activity. Elucidation of the mechanism of tankyrase 1 inhibition by GMD may provide insights for new tankyrase inhibition strategies in the future.
ACKNOWLEDGMENTS
We thank Tom Meier for comments on the manuscript and helpful discussion. We are grateful to Eiji Miyoshi for anti-GMD antibody. We thank the NYU Protein Analysis Facility in the Skirball Institute for the mass spectrometric analysis. We thank Susan Hsiao for generation of the HeLa S3 FlagTNKS1 and Vector cell lines. S.S. is grateful to Joachim Lingner and members of the Lingner lab for discussion and support during her stay in the Lingner laboratory.
This work was supported by NIH grant R01 CA095099.
Footnotes
Published ahead of print 29 May 2012
REFERENCES
- 1. Bae J, Donigian JR, Hsueh AJ. 2003. Tankyrase 1 interacts with Mcl-1 proteins and inhibits their regulation of apoptosis. J. Biol. Chem. 278:5195–5204 [DOI] [PubMed] [Google Scholar]
- 2. Becker DJ, Lowe JB. 2003. Fucose: biosynthesis and biological function in mammals. Glycobiology 13:41R–53R [DOI] [PubMed] [Google Scholar]
- 3. Callow MG, et al. 2011. Ubiquitin ligase RNF146 regulates tankyrase and axin to promote Wnt signaling. PLoS One 6:e22595 doi:10.1371/journal.pone.0022595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang P, Coughlin M, Mitchison TJ. 2005. Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat. Cell Biol. 7:1133–1139 [DOI] [PubMed] [Google Scholar]
- 5. Chang W, Dynek JN, Smith S. 2005. NuMA is a major acceptor of poly(ADP-ribosyl)ation by tankyrase 1 in mitosis. Biochem. J. 391:177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang W, Dynek JN, Smith S. 2003. TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev. 17:1328–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y, et al. 2008. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science 319:1092–1096 [DOI] [PubMed] [Google Scholar]
- 8. Cook BD, Dynek JN, Chang W, Shostak G, Smith S. 2002. Role for the related poly(ADP-ribose) polymerases tankyrase 1 and 2 at human telomeres. Mol. Cell. Biol. 22:332–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Lange T. 1992. Human telomeres are attached to the nuclear matrix. EMBO J. 11:717–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dynek JN, Smith S. 2004. Resolution of sister telomere association is required for progression through mitosis. Science 304:97–100 [DOI] [PubMed] [Google Scholar]
- 11. Falschlehner C, Emmerich CH, Gerlach B, Walczak H. 2007. TRAIL signalling: decisions between life and death. Int. J. Biochem. Cell Biol. 39:1462–1475 [DOI] [PubMed] [Google Scholar]
- 12. Fancy SP, et al. 2011. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat. Neurosci. 14:1009–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guettler S, et al. 2011. Structural basis and sequence rules for substrate recognition by tankyrase explain the basis for cherubism disease. Cell 147:1340–1354 [DOI] [PubMed] [Google Scholar]
- 14. Haltiwanger RS. 2009. Fucose is on the TRAIL of colon cancer. Gastroenterology 137:36–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Houghtaling BR, Cuttonaro L, Chang W, Smith S. 2004. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr. Biol. 14:1621–1631 [DOI] [PubMed] [Google Scholar]
- 16. Hsiao SJ, Smith S. 2008. Tankyrase function at telomeres, spindle poles, and beyond. Biochimie 90:83–92 [DOI] [PubMed] [Google Scholar]
- 17. Huang SM, et al. 2009. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461:614–620 [DOI] [PubMed] [Google Scholar]
- 18. Levaot N, et al. 2011. Loss of tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 147:1324–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyoshi E, Moriwaki K, Nakagawa T. 2008. Biological function of fucosylation in cancer biology. J. Biochem. 143:725–729 [DOI] [PubMed] [Google Scholar]
- 20. Moriwaki K, et al. 2009. Deficiency of GMDS leads to escape from NK cell-mediated tumor surveillance through modulation of TRAIL signaling. Gastroenterology 137:188–198 [DOI] [PubMed] [Google Scholar]
- 21. Ohyama C, et al. 1998. Molecular cloning and expression of GDP-d-mannose-4,6-dehydratase, a key enzyme for fucose metabolism defective in Lec13 cells. J. Biol. Chem. 273:14582–14587 [DOI] [PubMed] [Google Scholar]
- 22. Sbodio JI, Chi NW. 2002. Identification of a tankyrase-binding motif shared by IRAP, TAB182, and human TRF1 but not mouse TRF1. NuMA contains this RXXPDG motif and is a novel tankyrase partner. J. Biol. Chem. 277:31887–31892 [DOI] [PubMed] [Google Scholar]
- 23. Sbodio JI, Lodish HF, Chi NW. 2002. Tankyrase-2 oligomerizes with tankyrase-1 and binds to both TRF1 (telomere-repeat-binding factor 1) and IRAP (insulin-responsive aminopeptidase). Biochem. J. 361:451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scherthan H, et al. 2000. Mammalian meiotic telomeres: protein composition and redistribution in relation to nuclear pores. Mol. Biol. Cell 11:4189–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seimiya H, Muramatsu Y, Smith S, Tsuruo T. 2004. Functional subdomain in the ankyrin domain of tankyrase 1 required for poly(ADP-ribosyl)ation of TRF1 and telomere elongation. Mol. Cell. Biol. 24:1944–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seimiya H, Smith S. 2002. The telomeric poly(ADP-ribose) polymerase, tankyrase 1, contains multiple binding sites for telomeric repeat binding factor 1 (TRF1) and a novel acceptor, 182-kDa tankyrase-binding protein (TAB182). J. Biol. Chem. 277:14116–14126 [DOI] [PubMed] [Google Scholar]
- 27. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593–602 [DOI] [PubMed] [Google Scholar]
- 28. Silk AD, Holland AJ, Cleveland DW. 2009. Requirements for NuMA in maintenance and establishment of mammalian spindle poles. J. Cell Biol. 184:677–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith S, de Lange T. 1999. Cell cycle dependent localization of the telomeric PARP, tankyrase, to nuclear pore complexes and centrosomes. J. Cell Sci. 112:3649–3656 [DOI] [PubMed] [Google Scholar]
- 30. Smith S, de Lange T. 2000. Tankyrase promotes telomere elongation in human cells. Curr. Biol. 10:1299–1302 [DOI] [PubMed] [Google Scholar]
- 31. Smith S, Giriat I, Schmitt A, de Lange T. 1998. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 282:1484–1487 [DOI] [PubMed] [Google Scholar]
- 32. Sullivan FX, et al. 1998. Molecular cloning of human GDP-mannose 4,6-dehydratase and reconstitution of GDP-fucose biosynthesis in vitro. J. Biol. Chem. 273:8193–8202 [DOI] [PubMed] [Google Scholar]
- 33. van Steensel B, de Lange T. 1997. Control of telomere length by the human telomeric protein TRF1. Nature 385:740–743 [DOI] [PubMed] [Google Scholar]
- 34. van Steensel B, Smogorzewska A, de Lange T. 1998. TRF2 protects human telomeres from end-to-end fusions. Cell 92:401–413 [DOI] [PubMed] [Google Scholar]
- 35. Verdun RE, Crabbe L, Haggblom C, Karlseder J. 2005. Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol. Cell 20:551–561 [DOI] [PubMed] [Google Scholar]
- 36. Wang Z, et al. 2012. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 26:235–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yeh TY, et al. 2006. Tankyrase recruitment to the lateral membrane in polarized epithelial cells: regulation by cell-cell contact and protein poly(ADP-ribosyl)ation. Biochem. J. 399:415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y, et al. 2011. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 13:623–629 [DOI] [PubMed] [Google Scholar]