

Abstract
The Wnt/β-catenin pathway promotes proliferation of neural progenitor cells (NPCs) at early stages and induces neuronal differentiation from NPCs at late stages, but the molecular mechanisms that control this stage-specific response are unclear. Pin1 is a prolyl isomerase that regulates cell signaling uniquely by controlling protein conformation after phosphorylation, but its role in neuronal differentiation is not known. Here we found that whereas Pin1 depletion suppresses neuronal differentiation, Pin1 overexpression enhances it, without any effects on gliogenesis from NPCs in vitro. Consequently, Pin1-null mice have significantly fewer upper layer neurons in the motor cortex and severely impaired motor activity during the neonatal stage. A proteomic approach identified β-catenin as a major substrate for Pin1 in NPCs, in which Pin1 stabilizes β-catenin. As a result, Pin1 knockout leads to reduced β-catenin during differentiation but not proliferation of NPCs in developing brains. Importantly, defective neuronal differentiation in Pin1 knockout NPCs is fully rescued in vitro by overexpression of β-catenin but not a β-catenin mutant that fails to act as a Pin1 substrate. These results show that Pin1 is a novel regulator of NPC differentiation by acting on β-catenin and provides a new postphosphorylation signaling mechanism to regulate developmental stage-specific functioning of β-catenin signaling in neuronal differentiation.
INTRODUCTION
Neurons are generated from neural stem/progenitor cells (NPCs) during brain development. Recently, it has been emerging that an array of extracellular factors, including Wnt, platelet-derived growth factor, vascular endothelial growth factor, and bone morphogenic proteins, trigger signaling cascades leading to activation of downstream transcription factors to regulate neuronal differentiation from NPCs (6, 16, 18, 22, 23, 33, 51, 68, 86, 91). However, little is known about how these signaling pathways are integrated to control the highly ordered events of neuronal differentiation to generate neurons in different layers or subregions of the brain during development.
A well-established key signaling pathway in neurogenesis is the Wnt/β-catenin pathway (13, 16, 18, 22, 46, 62). For example, overexpression of constitutively active β-catenin in NPCs induces neurogenesis and increases cortical size (12, 23, 90). Similarly, overexpression of Wnt3 increases neurogenesis in cortical progenitor cells (26, 43) and adult hippocampal stem/progenitor cells (34). Conversely, ablation of β-catenin inhibits neurogenesis, leading to reduced brain size (23, 90). Finally, activation of the Wnt/β-catenin pathway leads to the formation of β-catenin–TCF complexes to transactivate Wnt target genes, including many proneural transcription factors (25, 30, 48, 79). Significantly, the in vivo function of Wnt/β-catenin signaling in neuronal differentiation depends on the developmental stage during brain development. It appears to promote proliferation of early NPCs (expansion phase) but induces neuronal differentiation of NPCs in the late stage (neurogenic phase). These stage-dependent responses of NPCs might play a central role in determining the timing of differentiation and the size of the final population of each differentiated cell type (23). However, the molecular mechanisms that control this developmental stage-specific response of NPCs are not fully understood. Moreover, although the function of β-catenin is regulated by phosphorylation, nothing is known about whether β-catenin is further regulated after phosphorylation to control neuronal differentiation.
Proline-directed protein phosphorylation (pSer/Thr-Pro) is a central signaling mechanism in diverse cellular processes (8, 61, 66). Importantly, recent studies have shown that such phosphorylation events are further regulated by conformational changes after protein phosphorylation (38, 40). The pSer/Thr-Pro motifs in certain proteins exist in cis or trans conformation, whose conversion is catalyzed by the unique prolyl isomerase Pin1 (39, 42, 87). Such Pin1-catalyzed conformational regulation after phosphorylation often functions as a molecular timer, regulating many key proteins in diverse cellular processes, such as β-catenin, tau, and APP in neurons (32, 34, 35, 38, 40, 41, 64, 72, 74, 89, 92, 94). In fact, cis and trans conformation-specific functions and their regulation by Pin1 have been demonstrated directly by the development of conformation-specific antibodies (55).
Significantly, Pin1 is tightly regulated at multiple levels, and its deregulation has an important role in a growing number of pathological conditions, notably cancer and Alzheimer's disease (AD) (3, 9, 17, 19, 38, 40, 49, 78, 88). For example, we have demonstrated that Pin1 acts on the pThr231-Pro motif in tau to promote tau dephosphorylation and degradation and also on the pThr668-Pro motif in APP to reduce amyloidogenic APP processing and toxic Aβ secretion (35, 41, 45, 64, 65). Consequently, Pin1-deficient mice display both tau- and Aβ-related pathologies and neurodegeneration, in an age-dependent manner, resembling many aspects of human Alzheimer's disease (35, 64). In contrast, moderate Pin1 overexpression in postnatal neurons effectively suppresses tau-related pathology and neurodegeneration in Alzheimer's disease mouse models (34). Significantly, Pin1 is inhibited by multiple mechanisms in human Alzheimer's neurons, including downregulation, oxidation, and/or possible single nucleotide polymorphisms (35, 77, 81, 84). In contrast, the Pin1 single nucleotide polymorphism that prevents its downregulation by the brain-specific transcription factor AP4 is associated with a delayed onset of Alzheimer's disease (44). These results indicate that Pin1 plays a pivotal role in protecting against age-dependent neurodegeneration in Alzheimer's disease. In addition, Pin1 has been shown to regulate several other neuron-enriched proteins, including synphilin-1, BIMEL, gephyrin, neurofilament, and JIP3, which might contribute to neurodegeneration (7, 27, 52, 70, 71, 76). Therefore, the roles of Pin1 in various age-dependent neuropathological conditions have been studied extensively. Although Pin1 levels increase during neuronal differentiation in vitro (20), little is known about the physiological function of Pin1 during brain development.
In studying this issue, we found that increased Pin1 expression occurs in parallel with NPC differentiation in mouse embryos. Furthermore, Pin1 knockout (KO) in mice specifically inhibits neuronal differentiation in vitro and inhibits the birth of upper but not lower layer neurons in the mouse cerebral cortex, resulting in fewer upper layer neurons in the motor cortex and severely impaired motor activity during the neonatal stage. Moreover, a proteomic approach led to the identification of β-catenin as a major substrate for Pin1 in NPCs, in which Pin1 stabilizes β-catenin by controlling its protein conformation after phosphorylation to promote neuronal differentiation from NPCs. These results demonstrate for the first time that Pin1 is a novel regulator of neuronal differentiation by stabilizing β-catenin and that conformational regulation after phosphorylation is a new mechanism for controlling neuronal differentiation during certain stages of brain development.
MATERIALS AND METHODS
Animals.
Pin1 wild-type (WT) and KO mouse littermates were obtained by crossing Pin1 heterozygous male and female mice on a C57BL/6 background with each other (34).
Retroviral constructs.
The NcoI-SalI fragment from pMX-IRES-GFP was ligated with the BspHI-XhoI fragment of the puromycin resistance gene to yield pMRX-IRES-PURO. The BamHI-SnaBI fragments encoding WT and constitutively active CS2-β-cateninFlag (36) were inserted into the pMRX-IRES-PURO vector digested with NotI and blunted and digested with BamHI to yield WT and constitutively active β-catenin retroviral constructs. S246A β-catenin was generated using two-step PCR (24) with CS2-β-cateninFlag as the template, followed by insertion into the pMRX-IRES-PURO vector. The Wnt3 viral vector (33) was provided by Fred H. Gage.
NPC cultures.
Primary neural precursor cells were prepared from the cerebral hemispheres of embryonic day 15.5 (E15.5) mouse embryos. Dissected cortices transferred to Neurobasal medium (Invitrogen, Carlsbad, CA) supplemented with 2% B27 (Invitrogen), 2 mM glutamine (Invitrogen), and antibiotics were mechanically dissociated into single cells. The dissociated cells were cultured in Neurobasal medium supplemented with 2% B27, 2 mM glutamine, antibiotics, human recombinant epidermal growth factor (EGF; 20 ng/ml) (Invitrogen), and human recombinant basic fibroblast growth factor (bFGF; 20 ng/ml) (Invitrogen). For differentiation into neurons, glass coverslips were coated with poly-l-lysine (15 μg/ml) (Sigma, St. Louis, MO) and laminin (5 μg/ml) (Sigma) and were placed in 24-well plates. The dissociated cells were cultured in the presence of 1% fetal calf serum (FCS) without EGF and bFGF. For retroviral infection, cells were mixed with virus solution at 2,000 rpm for 60 min in the presence of Polybrene (20 μg/ml). For quantification of the percentage of βIII tubulin-positive neurons or GFAP-positive astrocytes, 2,000 cells were counted for each genotype. Analysis of β-catenin stability in neurosphere culture was carried out using cycloheximide (CHX; 100 μg/ml) as described previously (34) (n = 3 per group).
Western blotting, GST pulldown, and immunoprecipitation.
Total lysates were obtained from cultured NPCs as described previously (34), and Western blotting was done using anti-Pin1 (35), anti-β-catenin (BD Biosciences, San Jose, CA), antitubulin (Sigma), and anti-Ki67 antibodies. Quantification of the intensities of β-catenin signals relative to those of tubulin signals was carried out using NIH Image as described previously (75) (n = 3 per group). Cytosol and nuclear proteins were separated using a nuclear/cytosol fractionation kit (BioVision, Milpitas, CA). Glutathione S-transferase (GST) pulldown and immunoprecipitation were conducted as reported previously (74).
Immunostaining.
The primary antibodies used were goat anti-Brn1 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Brn2 (Santa Cruz), and anti-BLBP (Santa Cruz), mouse antireelin (Santa Cruz), goat antinestin (Santa Cruz), goat anti-GFAP (Santa Cruz), mouse anti-PSD95 (BD Biosciences), mouse antibromodeoxyuridine (anti-BrdU) (BD Biosciences), rat anti-BrdU (Serotec, Oxford, United Kingdom), mouse anti-β-catenin (BD Biosciences), rabbit anti-β-catenin (Cell Signaling, Danvers, MA), mouse anti-Pax6 (Abcam), guinea pig antidoublecortin (anti-DCX) (Millipore), mouse anti-βIII tubulin (Millipore), rabbit anti-βIII tubulin (TUJ1) (Covance, Princeton, NJ), mouse anti-Pin1 (35), rabbit anti-Pin1 (35), rat anti-CTIP2 (Abcam, Cambridge, MA), and rabbit anti-Tbr2 (14) (kindly provided by Robert F. Hevner) antibodies. Immunofluorescence staining and immunohistochemistry of the mouse brain and cultured cells were done essentially as described previously (35, 56, 57, 59), and samples were visualized with a confocal microscope (Zeiss LSM510). The intensities of BrdU-, DCX-, Pax6-, Tbr2-, Brn1-, and Brn2-positive signals were counted within the same area for WT and Pin1 KO mice, with WT control levels being defined as 100% (n = 2 to 7 per group). For quantitation of DCX-positive neurons, we identified cells with nuclear staining with hematoxylin to prevent counting of nonspecific signals. Neurons positive for both DCX and hematoxylin were defined as DCX-positive immature neurons and counted in the same area containing all layers in the motor cortex for WT and KO mice.
BrdU birth dating.
Timed pregnant dams received BrdU by intraperitoneal injection (100 mg/kg of body weight) and were sacrificed different time points (n = 2 to 4 per group). Brains of the pups were fixed by immersion in cold buffered 4% paraformaldehyde for 16 to 20 h at 4°C and then processed for BrdU immunostaining. Determination of the boundary between the upper and lower layers and counting of BrdU-positive cells were done essentially as described previously (31, 80). Briefly, anatomically matched sections from each mouse were selected, and fluorescence images were obtained from the motor cortex. The full cortical thickness was divided into 10 bins, with the upper 3 bins defined as upper layers (II to IV) and the remaining 7 bins defined as lower layers (V and VI).
Luciferase reporter assay.
Luciferase reporter assay was done as described previously (23). Briefly, a luciferase reporter construct containing the Ngn1 promoter (23) was transfected into control and Pin1 knockdown (KD) SH-SY5Y cells (34), and their lysates were used for measurement of luciferase activities.
Behavioral analyses.
Spontaneous motor activity of mice in a novel environment was evaluated in an open field as described previously (1, 15). Briefly, the open field was a square Plexiglas box (48 cm × 48 cm). Each mouse was placed in the center of the apparatus, and the open-field test was carried out for 5 min, essentially as described previously (58), using a videoMot2 system (TSE Systems GmbH, Bad Homburg, Germany). Total walking distance, time elapsed to circle or walk, and/or number of circles turned was measured (n = 4 to 16). A circle was counted when a pup turned through one 360° segment. The wire suspension test was carried out essentially as described previously (83) (n = 10 and 8 for WT and KO groups, respectively). The front paws of the mouse were placed on a horizontal wire, and the time taken for the animal to fall was measured.
Statistics.
Values are presented as means ± standard errors (SE). Statistical significance was assessed using two-tailed Student's t test. P values of <0.05 were considered statistically significant.
RESULTS
Pin1 is expressed in NPCs and regulates neuronal but not glial differentiation of NPCs in vitro.
We have previously shown that Pin1 is expressed in neurons but not glia in the adult brain (35, 41). To determine the role of Pin1 in brain development, we examined Pin1 expression in the mouse cerebral cortex at E15.5, postnatal day 2 (P2), and the adult stage, using immunostaining with an anti-Pin1 monoclonal antibody as described previously (35). Significantly more expression of Pin1 was found in the developmental stages (E15.5 and P2) than in the adult stage (Fig. 1A). Notably, high Pin1 levels were found in the ventricular zone (VZ) and subventricular zone (SVZ), as well as the intermediate zone (IZ) and cortical plate (CP), in the E15.5 mouse cerebral cortex (Fig. 1A and B). The VZ and SVZ are the regions where NPCs reside. To examine whether Pin1 is expressed in NPCs, we examined expression of Pin1 in nestin-positive NPCs in the E15.5 mouse cerebral cortex (Fig. 1C). Double immunofluorescence staining revealed colocalization of nestin and Pin1 in both the VZ and SVZ, suggesting high-level Pin1 expression in NPCs. To confirm these results, we isolated NPCs from E15.5 mouse brains and analyzed Pin1 expression. Both immunostaining (Fig. 1D) and Western blotting (see Fig. 5B) demonstrated that Pin1 was abundantly expressed in cultured NPCs. These data indicate that Pin1 is highly expressed in NPCs in vitro and in vivo.
Fig 1.

Pin1 promotes NPC differentiation into neurons but not astrocytes. (A and B) Higher expression of Pin1 in the developing cerebral cortex than in the adult cortex. Brain tissues were obtained from different developmental stages and cut either sagittally (A and B) or coronally (B), followed by immunostaining with mouse anti-Pin1 primary antibody. Bars, 20 μm (A) and 50 μm (B). Dotted line, pial surface; V, ventricle; Cx, cerebral cortex. (C and D) Pin1 is highly expressed in NPCs. Double immunofluorescence staining with antinestin and anti-Pin1 antibodies shows expression of Pin1 in nestin-positive NPCs in the mouse cerebral cortex at E15.5. (C) Images at lower (top) and higher (bottom) magnifications. (D) Pin1 was also expressed in cultured NPCs isolated from an E15.5 WT mouse brain. Arrowheads in panel C indicate Pin1-expressing nestin-positive cells. Bar, 10 μm. (E and F) Pin1 knockout reduces βIII tubulin-positive neurons but not GFAP-positive astrocytes differentiated from cultured NPCs. NPCs were prepared from the cerebral hemispheres of the same set of Pin1 WT and KO E15.5 mouse embryos. Dissected cortices mechanically dissociated into single cells were cultured in the presence of EGF and bFGF to induce the formation of neurospheres, and the dissociated NPCs were differentiated by being placed on glass coverslips coated with poly-l-lysine and laminin in the presence of 1% FCS without EGF and bFGF. (F) Percentages of βIII tubulin-positive neurons and GFAP-positive astrocytes were quantified. (G and H) Manipulation of Pin1 function affects neuronal differentiation of NPCs. NPCs were left untransfected (control) or transfected with EGFP, EGFP-Pin1, or EGFP-dnPin1 and then immunostained for the neuronal marker βIII tubulin on day 3 after induction of neuronal differentiation. Overexpression of EGFP-Pin1 increased the population of βIII tubulin-positive neurons compared with control NPCs and NPCs expressing EGFP alone. Conversely, overexpression of dominant-negative Pin1 (EGFP-dnPin1) inhibited neuronal differentiation. (H) The number of neurons was quantified. Arrowheads in panels E and G indicate βIII tubulin-positive neurons. *, P < 0.05.
Fig 5.
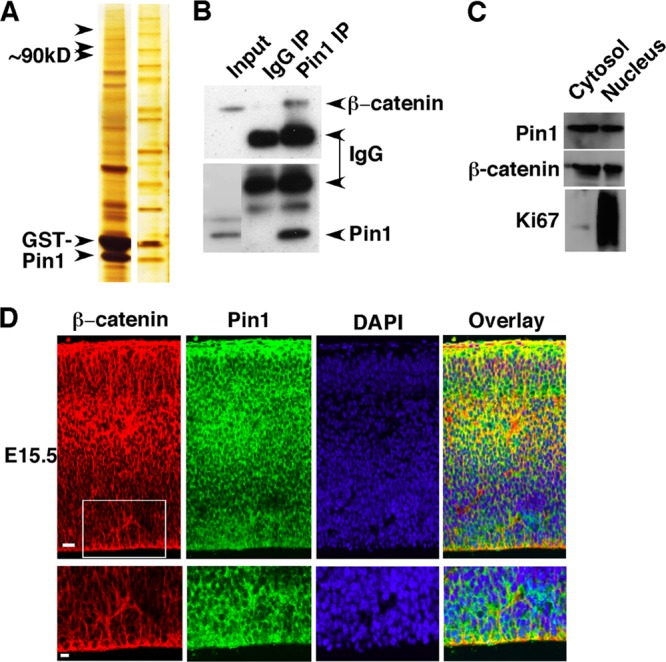
Proteomic identification of β-catenin as a major Pin1 substrate in NPCs. (A) Proteomic identification of β-catenin as a Pin1-binding protein in NPCs. NPCs were cultured under serum-free conditions in the presence of EGF or bFGF2 for 6 days to give rise to neurospheres and then subjected to GST-Pin1 pulldown experiments, followed by SDS-PAGE and silver staining. Ninety-kilodalton bands were identified as containing β-catenin by electrospray ionization-mass spectrometry analysis. (B) Endogenous Pin1 and β-catenin form stable complexes in NPCs. Cell lysates from NPCs were subjected to immunoprecipitation with anti-Pin1 antibody or nonspecific IgG, followed by immunoblotting with β-catenin antibody. (C) Pin1 and β-catenin are detected in both cytosol and nuclei of NPCs. Western blot analysis was performed on cytosol and nuclear fractions of cultured NPCs, using antibodies against Pin1, β-catenin, and the nuclear marker Ki67. (D) Colocalization of Pin1 and β-catenin in E15.5 mouse cerebral cortex. Double immunofluorescence staining of the sagittal sections of the mouse brain was performed with anti-β-catenin and anti-Pin1 antibodies. Bars, 20 μm (top) and 10 μm (bottom).
Given the high level of Pin1 expression in NPCs, Pin1 might affect their neuronal differentiation. To examine this possibility, we isolated primary NPCs from Pin1 WT and Pin1 KO littermates at E15.5 and compared their neuronal differentiation abilities in vitro. Compared with WT controls, Pin1 KO NPCs had a significantly lower percentage of differentiated neurons, as indicated by the marker βIII tubulin, but not astrocytes, as indicated by the marker GFAP (Fig. 1E and F), suggesting that Pin1 knockout might specifically impair neuronal but not glial differentiation of NPCs. To further support these findings, we infected NPCs with a retrovirus expressing Pin1 or its dominant-negative mutant. EGFP-Pin1 resulted in a significantly higher percentage of βIII tubulin-positive neurons than those in non-infected or enhanced green fluorescent protein (EGFP) vector-infected NPCs, whereas dominant-negative Pin1 strongly inhibited neuronal differentiation (Fig. 1G and H). The impaired differentiation into neurons was also observed in vivo, because there were fewer βIII tubulin-positive neurons in Pin1 KO mice (data not shown). These results indicate that Pin1 promotes neuronal but not glial differentiation of NPCs.
Pin1 expression is increased in parallel with NPC differentiation, and its knockout inhibits NPC differentiation during mouse embryonic development.
Given that Pin1 knockout significantly inhibits neuronal differentiation of NPCs in vitro, we wondered whether Pin1 plays any role in neuronal differentiation in vivo. To address this question, we examined Pin1 expression and the effects of Pin1 knockout on neuronal differentiation and on each neuronal differentiation step from NPCs to immature neurons in the mouse cerebral cortex, using well-established markers for progressive neuronal differentiation. There are two types of neurogenic progenitors, i.e., radial glia (RGC) and intermediate progenitors (IPC), during embryonic development. Some RGC function as progenitors that make major contributions to cortical neuronal differentiation, either directly through mitoses to produce neurons or indirectly through the production of proliferating IPC (51). IPC divide symmetrically to produce immature neurons. These immature neurons migrate into the IZ and CP and eventually become mature neurons. Neuronal differentiation in the cerebral cortex of the mouse occurs under strict temporal control during E11.5 to E17.5, in an “inside-out” fashion (4, 10, 67); that is, neurons born earlier are destined mainly for lower (deeper) cortical layers (layers V and VI), whereas those born later are destined for upper (superficial) cortical layers (layers II to IV) (6, 86). At E15.5, when most upper layer neurons are born, Pin1 expression in NPCs was recognized, whereas it was almost below the detection limit at E12.5 (Fig. 2A), when most lower layer neurons are born. Thus, Pin1 might affect NPC differentiation into upper layer neurons at E15.5.
Fig 2.

Pin1 expression is increased in parallel with NPC differentiation, and its knockout inhibits NPC differentiation to immature neurons during embryonic development. (A) Immunofluorescence staining of cortical sections from E12.5 and E15.5 mouse brains with anti-Pin1 antibody. The Pin1 level was much higher at E15.5 than at E12.5. (B to F) Emergence of DCX-positive immature neurons, but not NPCs, is impaired in Pin1 KO mice. Immunostaining of E15.5 mouse cerebral cortex was performed with a Pax6 (B), Tbr2 (C), or DCX (D and E) antibody. The same detection gains were applied for Pin1 WT and KO mice to take confocal images. For immunohistochemistry (D and E), hematoxylin was used for counterstaining. Magnified images of DCX staining are also shown in panel E. (F) The relative numbers of Pax6-, Tbr2-, and DCX-positive cells were quantified. DCX-positive immature neurons, but not RGCs and IPCs, were decreased in the KO mice. Bars, 20 μm (A) and 50 μm (B). Dotted line, pial surface; SVZ, subventricular zone; VZ, ventricular zone; V, ventricle. *, P < 0.05.
To examine this possibility, we immunostained Pin1 WT and KO embryos at E15.5 for various neuronal differentiation markers, including DCX, a marker of migrating immature neurons; Pax6, a marker of RGC; and Tbr2, a marker of IPC (14). Although the expression of neither Pax6 (Fig. 2B) nor Tbr2 (Fig. 2C) was affected in Pin1 KO mice, the number of DCX-positive cells was reduced dramatically in Pin1 KO embryos at E15.5 compared with WT controls (Fig. 2D and E). The number of DCX-positive immature neurons in Pin1 KO embryos was only 35% of that in WT embryos (Fig. 2F). These results indicate that Pin1 knockout does not affect the production of NPCs but specifically inhibits their differentiation into migrating immature neurons at E15.5, which is consistent with the pattern of Pin1 expression.
Pin1 knockout inhibits birth of upper layer neurons in the mouse cerebral cortex.
Neural progenitor cells divide to produce immature neurons. Enhanced NPC proliferation might also result in a defect of differentiation. However, this seems unlikely, because the expression of neither Pax6 (Fig. 2B) nor Tbr2 (Fig. 2C) was affected. Thus, if neuronal differentiation from NPCs was impaired in Pin1 KO mice, their mitotic activity would likely be reduced. To evaluate the mitotic activity of NPCs, we used acute incorporation of BrdU, a marker of DNA replication. BrdU was injected into pregnant females at E15.5, and embryonic cortices were analyzed for BrdU incorporation 1 h later. Pin1 KO mice had significantly fewer BrdU-positive cells. The difference was more apparent in the most basal area of the periventricular region, corresponding with the SVZ territory, with a reduction of 35% (65.1% ± 9.1% of the WT level; P < 0.05) (Fig. 3A and B). Indeed, when we measured the percentage of BrdU-positive cells in the SVZ among the BrdU-positive cells in the entire cortex, it was lower for the KO mice (data not shown). These results indicate that fewer SVZ cells divide to differentiate into neurons in Pin1 KO mice.
Fig 3.

Pin1 knockout inhibits birth of upper but not lower layer neurons in the mouse cerebral cortex. (A and B) Acute BrdU incorporation is decreased in NPCs of Pin1 KO mice at E15.5. Immunofluorescence staining of E15.5 mouse cerebral cortex was performed with anti-BrdU antibody. One hour of BrdU pulse labeling was performed. BrdU was injected intraperitoneally into pregnant mice, and embryos were collected 1 h later. Brains of the pups were fixed and processed for BrdU immunostaining. (B) Percentage of BrdU-positive cells in the SVZ. (C and D) Neuronal differentiation of upper layer neurons, but not lower layer neurons, is decreased in Pin1 KO mice at E13.5 and E15.5. The distribution of BrdU birth date-labeled cells was examined in the E18.5 cortex after BrdU injection at E13.5 and in the P2 cortex after BrdU injection at E15.5. Brains of the pups were fixed and processed for BrdU immunostaining. (D) BrdU-positive cells were quantified separately in the upper layers and lower layers. Determination of the boundary between the upper and lower layers and counting of BrdU-positive cells were done essentially as described previously (31, 80). Significant differences were found only in the upper layers. (E and F) The number of upper layer neurons in the motor cortex in Pin1 KO mice is reduced at E18 and P2. Coronal sections of motor cortex were immunostained with anti-Brn1 antibody, followed by quantification of Brn1-positive cells in the upper layers. *, P < 0.05; **, P < 0.01. Bars, 50 μm. Dotted line, pial surface; CP, cortical plate; IZ, intermediate zone; L, lower layers; SVZ, subventricular zone; U, upper layer; V, ventricle; VZ, ventricular zone.
The lower mitotic activity described above might lead to a reduced efficiency of production of neurons. To determine the impact of Pin1 knockout on the birth of the neurons destined for each cortical layer in vivo, we performed migrational analysis using BrdU birth dating. As described previously (31, 80), since progenitor cells that divide to generate neurons can incorporate BrdU, those neurons that are born at the time when BrdU is injected will be BrdU positive, thus indicating the birth date of those neurons. When BrdU was injected at E13.5, when most lower layer neurons and some upper layer neurons are born (51), the majority of BrdU-positive cells were observed in lower layers at E18.5 (Fig. 3C), as expected. Importantly, there was no significant difference in BrdU-positive cell numbers in lower layers between Pin1 WT and KO mice (105% ± 6.4% of WT level; P > 0.05) (Fig. 3C and D). However, BrdU-positive cells in upper layers were significantly reduced (by 39%) in Pin1 KO mice compared with Pin1 WT controls (61.3% ± 9.6% of WT level; P < 0.05) (Fig. 3C and D). This difference became even more obvious when BrdU was injected at E15.5, the peak time when upper layer (II/III) neurons are born (51). The number of BrdU-positive cells in upper layers was decreased by 50% in Pin1 KO mice compared with WT controls (49.2% ± 5.5% of WT level; P = 0.0001), although there were no differences in lower layers (74.9% ± 12.6% of WT level; P > 0.05) (Fig. 3C and D). These results indicate that Pin1 knockout does not affect the birth of lower layer neurons but specifically inhibits the birth of upper layer neurons. These data are consistent with the increased Pin1 expression in NPCs at E15.5 and further support the notion that Pin1 knockout inhibits NPC differentiation into immature neurons destined for upper layers.
The finding that fewer upper layer neurons were generated in Pin1 KO mice suggests that these mutant mice might have fewer upper layer neurons at late embryonic stages and in the early postnatal period. To examine this possibility, we immunostained the cerebral cortex with anti-Brn1 and anti-Brn2 antibodies that specifically recognize upper layer neurons and part of the lower layer neurons during development (50, 51, 80). We also stained the samples with anti-CTIP2 antibody, a marker of lower layer subcerebral projection neurons (5). As a control, anti-GFAP antibody, a marker of glia, was used. Although similar results were obtained for different regions of the cerebral cortex, we chose to focus on the motor cortex for easier quantification. There was no apparent difference in the distribution of CTIP- or GFAP-positive cells between Pin1 WT and KO mice at E18.5 or P2 (data not shown), indicating that Pin1 knockout does not affect NPC differentiation into either astrocytes or lower layer neurons. However, the number of upper layer neurons, as indicated by Brn1 immunostaining, was reduced in Pin1 KO mice at E18 (64.9% ± 10.1% of WT level; P = 0.02) and P2 (65.2% ± 7.5% of WT level; P = 0.004) (Fig. 3E and F). These findings were further confirmed by Brn2 immunostaining at neonatal stages, with the positive neurons in the upper layers significantly reduced in Pin1 KO mice (41.0% ± 2.9% of WT level; P < 0.001). Thus, Pin1 knockout in mice not only impairs the birth of upper layer neurons but also leads to fewer upper layer neurons in the motor cortex at late embryonic stages and in the neonatal period.
Pin1 knockout leads to severely impaired neonatal motor activity in mice.
Given the fewer upper layer neurons in the motor cortex in Pin1 KO mice in the neonatal period, we wondered whether there were any functional consequences related to the upper layers in the motor cortex. The main excitatory pathway in the mouse motor cortex (layers II/III to V) is fractionated into distinct pathways, targeting corticospinal and corticostriatal neurons, which are involved in motor control (2). We compared spontaneous motor activities of Pin1 WT and KO littermates at different postnatal days in a novel environment in an open field, as described previously (1, 15), because the number of upper layer neurons in the motor cortex is reduced in Pin1 mutant mice (Fig. 3E and F). At P5, when mice cannot walk and their movement involves circling in the open field, the latency to circling and the number of circles turned were measured. Although the latency to circling was not significantly different in Pin1 KO mice (P > 0.05), the number of circles turned was dramatically reduced at P5 (P = 0.04) (Fig. 4A). After P9, when mice can walk, the latency to walking and walking distance were measured. Pin1 KO mice had longer latencies (P < 0.05) and walked shorter distances (P < 0.05) than control mice at both P9 and P11 (Fig. 4B to D). To exclude the possibility that weaker muscle tone contributed to the reduced motor activity in Pin1 KO mice, we performed a wire suspension test with P11 mice and recorded the time taken before animals fell. No difference was found between the two genotypes (P > 0.05) (Fig. 4E). Therefore, the reduced motor activity in Pin1 KO mice was not due to weaker muscle tone in Pin1 KO mice. The lower general motor activity suggests that synaptic organization might be immature in Pin1 KO mice at P9. Since glutamate is a crucial transmitter that regulates motor function in the motor cortex (82), we performed immunofluorescence staining of coronal sections of the motor cortex with anti-PSD95 antibody, a marker of postsynapses of glutamatergic neurons. Compared with WT controls, PSD95 signals in the upper layers were weaker in Pin1 KO mice at P9 (Fig. 4F), suggesting that synapses that underlie general motor activity are immature in P9 Pin1 KO mice.
Fig 4.
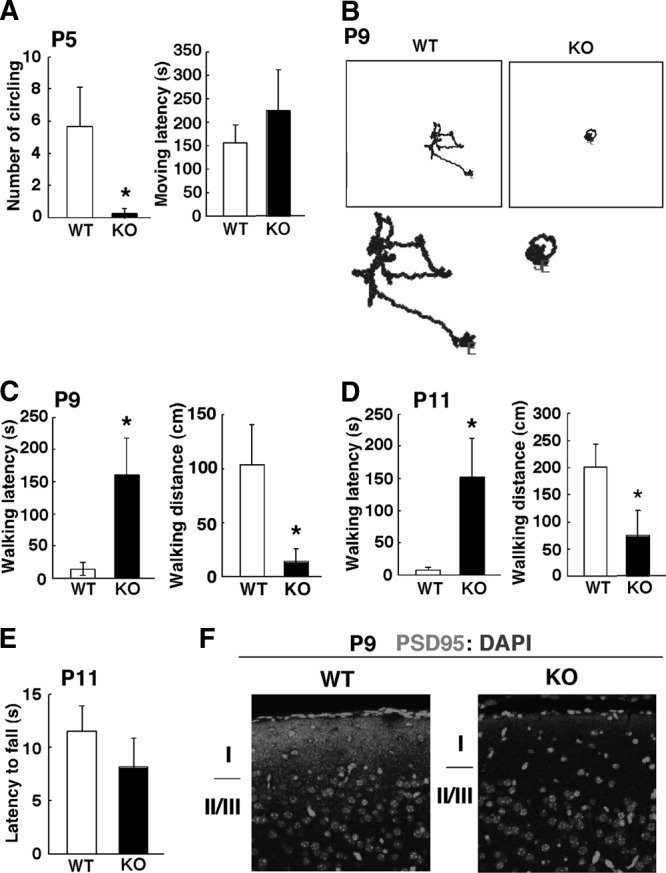
Impaired spontaneous motor activity in Pin1 KO neonatal mice. (A to D) Decreased general motor activity in Pin1 KO mice at P5, P9, and P11. An open-field test was performed at P5 (A), P9 (B and C), and P11 (D) to measure spontaneous motor activity of mice in a novel environment in an open field. The open field was a square box (48 cm × 48 cm). Each mouse was placed in the center of the apparatus, and the open-field test was carried out for 5 min, essentially as described previously (58). Since P5 mice cannot walk and their movement involves circling in the open field, the latency to circling and number of circles turned were measured for P5 mice. A circle was counted when a pup turned through one 360° segment. For P9 and P11 mice, the latency to walking and walking distance were measured. (B) Representative examples of the tracks of Pin1 WT and KO mice at P9. Lower panels show a higher magnification. *, P < 0.05. S, start point; E, endpoint of movement. (E) There was no difference in the wire suspension test between Pin1 WT and KO mice at P11. The wire suspension test was carried out to examine muscle tone. The front paws of each mouse were placed on a horizontal wire, and the time taken for the animal to fall was measured. (F) PSD95 expression is reduced in Pin1 KO mice at P9. Coronal sections of a P9 mouse cerebral cortex were immunostained with anti-PSD95 antibody. I, layer I; II/III, layers II and III.
Proteomic approach identifies β-catenin as a major Pin1 substrate in NPCs.
The above results demonstrate the major role for Pin1 in the control of neuronal differentiation from NPCs both in vitro and in vivo. A key question is the molecular mechanism underlying the action of Pin1 on NPCs. To answer this question, we used a proteomic approach to identify Pin1 substrates in NPCs, using a GST-Pin1 affinity purification procedure, under high-salt and -detergent conditions (42, 87), which has been used to identify almost all known Pin1 substrates (40). Cell lysates from cultured NPCs were subjected to GST pulldown analysis with GST-fused Pin1, and captured proteins were separated by SDS-PAGE, followed by silver staining. We found bands at around 90 kDa, a molecular mass similar to that of β-catenin (Fig. 5A). Electrospray ionization-mass spectrometry (ESI-MS) analysis revealed significant matches of these 90-kDa bands to sequences derived from β-catenin. Coimmunoprecipitation analysis using cell lysates demonstrated that endogenous Pin1 associated with β-catenin in NPCs (Fig. 5B). The interaction of the two molecules is possible, as both were detected in the cytosol and nuclei of cultured NPCs (Fig. 5C). Moreover, Pin1 and β-catenin were also colocalized in both the VZ and SVZ in the E15.5 mouse cerebral cortex, where NPCs reside, as shown by double immunofluorescence staining (Fig. 5D). These results indicate that Pin1 and β-catenin interact in NPCs.
Pin1 stabilizes β-catenin in NPCs.
It has been shown that in cancer cells, Pin1 acts on the Ser246-Pro motif in β-catenin to prevent its degradation, leading to enhanced Wnt/β-catenin signaling to promote oncogenesis (74). To examine the effects of Pin1 on β-catenin levels in NPCs, NPCs were derived from Pin1 WT and KO E15.5 embryos and subjected to Western blotting (Fig. 6A) and immunostaining (Fig. 6B) with anti-β-catenin antibody. Both assays showed that β-catenin levels were significantly reduced in cultured Pin1 KO NPCs compared with those in Pin1 WT controls. Given the reduced amount of β-catenin, we assessed the effects of Pin1 knockout on protein stability of β-catenin in NPCs by using cycloheximide as described previously (74). In this CHX chase assay, the rate at which β-catenin steady-state levels decline reflects the half-life of the protein. In Pin1 KO NPCs, β-catenin was much less stable, with a half-life of ∼4.5 h, than that in Pin1 WT controls, with a half-life of ∼9 h (Fig. 6C and D). These results suggest that Pin1 might control the β-catenin level by increasing its protein stability in NPCs.
Fig 6.

Pin1 controls protein stability of β-catenin in NPCs in vitro and in vivo. (A and B) Pin1 knockout reduces β-catenin levels in cultured NPCs. NPCs were derived from Pin1 WT and KO E15.5 embryos and subjected to Western blotting (A) and immunostaining (B) with anti-β-catenin antibody. (A) Intensities of β-catenin signals were quantified relative to those of tubulin. Values are presented as means ± SE for 3 independent experiments. *, P < 0.05. (C and D) Pin1 knockout reduces protein stability of β-catenin in cultured NPCs. Pin1 WT and KO NPCs were cultured in the presence of CHX for 0, 3, 6, and 9 h, followed by immunoblotting using anti-β-catenin and antitubulin antibodies. (D) Intensities of β-catenin signals relative to those of tubulin were quantified for three independent experiments. Values are presented as means ± SE for three independent experiments. (E and F) Overexpression or inhibition of Pin1 increases or reduces β-catenin level in NPCs, respectively. NPCs were infected retrovirally with empty vector (EV) or Pin1 (E) or with GFP, GFP-Pin1, or GFP-dnPin1 (F) and selected with puromycin for 48 h. Cells were then subjected to immunoblotting with anti-β-catenin antibody, with Coomassie brilliant blue staining as a loading control (E), or to immunostaining with anti-Pin1 or anti-β-catenin antibody (F). GFP-Pin1 and GFP-dnPin1 increased and decreased the β-catenin level, respectively. (G and H) Pin1 knockout in mice reduces β-catenin levels in nestin-positive neuronal progenitor cells (G) and BLBP-positive radial glia (H) in the VZ and SVZ in the cerebral cortex at E15.5. Sagittal mouse brain sections were subjected to double immunofluorescence staining with antinestin or anti-BLBP and anti-β-catenin antibodies. Bar, 20 μm. V, ventricle.
To further confirm this possibility, we examined the impact of manipulating Pin1 function on β-catenin levels in NPCs in vitro and in vivo. When NPCs were transfected with Pin1 or empty vector and subjected to immunoblotting, Pin1 overexpression led to increased β-catenin (Fig. 6E). Similarly, NPCs infected retrovirally with GFP-Pin1 showed higher β-catenin levels than those of GFP-expressing controls, as determined by immunostaining (Fig. 6F). In contrast, β-catenin levels were reduced in NPCs infected with GFP-tagged dominant-negative Pin1 (Fig. 6F). Moreover, compared with Pin1 WT controls, β-catenin levels in Pin1 KO E15.5 mouse embryos were reduced dramatically in nestin-positive NPCs (Fig. 6G) and BLBP-positive RGC (Fig. 6H) in both the VZ and the SVZ. These results together demonstrate that Pin1 is a major regulator of β-catenin protein stability in NPCs, both in vitro and in vivo.
Pin1 controls neuronal differentiation by stabilizing β-catenin.
Because the Wnt/β-catenin pathway has a well-established role in neuronal differentiation during development (13, 16, 18, 22, 46, 62), the above results suggest that decreased neuronal differentiation in Pin1 KO NPCs might be due to reduced β-catenin levels. If so, we would expect that restoration of Wnt/β-catenin signaling would rescue the defects in neuronal differentiation of Pin1 KO NPCs and that the ability of Pin1/β-catenin to promote neuronal differentiation depends on the ability of β-catenin to act as a Pin1 substrate and to activate downstream transcription.
To examine whether restoration of Wnt/β-catenin signaling would rescue the defects in neuronal differentiation of Pin1 KO NPCs, we infected Pin1 KO NPCs with a retrovirus expressing wild-type β-catenin, constitutively active S33Y β-catenin, or Wnt3. In contrast to the vector control, all three expression constructs promoted neuronal differentiation in Pin1 KO NPCs, to levels that were indistinguishable from those in Pin1 WT cells (Fig. 7A and B). These results indicate that upregulating Wnt/β-catenin is sufficient to overcome the defects in neuronal differentiation in Pin1 KO NPCs. Binding of Pin1 to β-catenin is postulated to alter the protein conformation of β-catenin. To examine the importance of the ability of β-catenin to act as a Pin1 substrate in neuronal differentiation and to study if a β-catenin mutant to which Pin1 cannot bind would work or not, we introduced a β-catenin S246A point mutant that fails to act as a Pin1 substrate due to a mutation at the Pin1 binding site (74) into Pin1 KO NPCs. Unlike WT β-catenin or its constitutively active mutant, S246A β-catenin completely failed to promote neuronal differentiation (Fig. 7A and B). These results demonstrate that S246A β-catenin cannot rescue the defects in neuronal differentiation in Pin1 KO NPCs, indicating that the ability of Pin1 to act on β-catenin is essential for neuronal differentiation from NPCs. Finally, to examine the importance of β-catenin transcriptional activity for Pin1 to promote NPC neuronal differentiation, we used truncated TCF4 (DN-TCF4), which has been shown to act as a dominant-negative mutant, to block β-catenin activity (21, 29). When cotransfected into NPCs with Pin1, the dominant-negative TCF4 mutant completely abolished the ability of Pin1 to enhance neuronal differentiation of NPCs, as estimated by use of another neuronal marker, MAP2 (Fig. 7C and D). These results suggest that the enhancing effect of Pin1 is β-catenin/TCF4 pathway dependent. Furthermore, the observations that β-catenin binds to the neurogenin 1 (Ngn1) promoter and that a mutation in a putative TCF binding site in the Ngn1 promoter lowers the promoter activity in NPCs (23) led us to assess Ngn1 promoter activity in Pin1 KD SH-SY5Y cells. The luciferase activity in the Pin1 KD cells was significantly lower than that in the control cells (Fig. 7E). These results collectively demonstrate that Pin1 controls neuronal differentiation of NPCs by stabilizing β-catenin.
Fig 7.

Pin1 controls neuronal differentiation through β-catenin. (A and B) Restoration of β-catenin signaling fully rescues neuronal differentiation defects in Pin1 KO NPCs. (A) Pin1 WT and KO NPCs were infected with retroviruses expressing control vector, Wnt3, WT β-catenin, constitutively active β-catenin, or S246A mutant β-catenin, followed by neuronal differentiation assay. (B) Two thousand cells were counted for each genotype, and percentages of βIII tubulin-positive neurons were quantified. Pin1 knockout reduced neuronal differentiation, which was fully rescued by expression of WT β-catenin, constitutively active β-catenin, or Wnt3 but not by S246A mutant β-catenin, which cannot act as a Pin1 substrate (74). (C and D) Inhibition of β-catenin transcription activity suppresses the ability of Pin1 to promote neuronal differentiation from NPCs. (C) NPCs stably infected with EGFP or EGFP-Pin1 were dissociated and transfected with control vector or dominant-negative TCF4 (29) by use of Nucleofector, followed by immunostaining for MAP2. (D) The number of MAP2-positive differentiated neurons was quantified. Data from three independent experiments were averaged. Arrowheads indicate differentiated neurons. (E) Lower Ngn1 promoter activity in Pin1 knockdown SH-SY5Y cells. A luciferase reporter construct containing the Ngn1 promoter was transfected into control and Pin1 KD SH-SY5Y cells, and luciferase activity was measured. *, P < 0.05.
DISCUSSION
While Pin1 plays a critical role in age-dependent neurodegeneration, its role in normal brain development is not known. We have shown that Pin1 is highly expressed in NPCs at the late developmental stage and has a major impact on their neuronal differentiation. Without affecting gliogenesis, Pin1 knockout in mice does not affect production of NPCs but specifically inhibits their differentiation into upper but not lower layer neurons in the cerebral cortex, resulting in a significant decrease in upper layer neurons in the motor cortex and severely impaired motor activity during the neonatal stage. Moreover, our proteomic approach to search for the substrate of Pin1 in NPCs led to the finding that Pin1 stabilizes β-catenin in NPCs at late but not early stages during mouse brain development. Finally, stimulation of the Wnt/β-catenin pathway rescues the neuronal differentiation defect of Pin1 KO NPCs, in a Pin1 binding site-dependent manner. Conversely, blockade of signaling by dominant-negative TCF4 abolishes the increased neuronal differentiation seen with Pin1 overexpression. These results not only identify for the first time a major function for Pin1 in regulating neuronal differentiation of NPCs but also uncover a previously unappreciated mechanism by which Wnt/β-catenin signaling is further regulated after phosphorylation to regulate neuronal differentiation.
The characterization of the unique peptidyl-prolyl cis-trans isomerase Pin1, which specifically regulates the conformation of certain Pro-directed phosphorylation sites in a subset of proteins, has led to the discovery of a new postphosphorylation regulatory mechanism (39, 69, 73, 87). Such Pin1-dependent conformational regulation has been shown to regulate many key signaling molecules in many cellular events (32, 34, 35, 38, 40, 41, 65, 72, 74, 89, 92, 94). As for Pin1 function in neurons, it has been shown that Pin1 is expressed in most neurons in the brain but is present at an especially low level in the neurons that are most vulnerable to neurodegeneration in AD (35). Indeed, Pin1 has been shown to play an important role in age-dependent neurodegeneration in AD and other related disorders. Although Pin1 expression is increased when neuron-like SY5Y cells are induced to undergo neuronal differentiation by use of either retinoic acid or nerve growth factor (20), little is known about the role of Pin1 in neuronal differentiation and in brain development. In this study, we have shown for the first time that Pin1 protein levels in the brain are much higher during developmental stages than those in the adult, and we uncovered a novel physiological function of Pin1 in cortical neuronal differentiation. Thus, Pin1 plays a critical role during early neuronal development, in addition to its protective role against age-dependent neurodegeneration.
Cortical NPCs sequentially undergo expansion, neurogenic, and gliogenic phases during development. The fate decision of NPCs between proliferation and differentiation determines the number of differentiated cells and the size of each region of the brain. β-Catenin has been shown to play a major role in regulating NPC proliferation and differentiation in mouse embryos, depending on the developmental stage (13, 16, 18, 22, 46, 62). For example, during the expansion phase, transgenic overexpression of stabilized β-catenin in NPCs, under the control of the nestin enhancer, which is known to become active at around E8.5, has been shown to expand the NPC population, resulting in enlarged brains (12). Using the D6 promoter, which is active from around E11, ectopic expression of a β-catenin–LEF1 fusion protein delays the onset of expression of the neurogenic factors Pax6, Ngn2, and Tbr2 and subsequent neurogenesis, whereas conditional ablation of β-catenin accelerates expression of the same neurogenic genes (47). Likewise, sustained β-catenin activity in conditionally transgenic animals by use of nestin-Cre driver lines delays maturation of RGC into intermediate progenitors, specifically for the production of upper layer cortical neurons (85).
On the other hand, at later time points during the neurogenic phase, Wnt signaling instructively promotes neuronal differentiation of cortical NPCs prepared from the E13.5 mouse neocortex (23). Clonal analysis and serial observations of living clones have revealed that signaling via the canonical Wnt pathway, through activation of β-catenin and TCF, increases the neuronal population at the expense of multipotential NPCs, even under conditions not affecting survival or proliferation of neuronal progenitors. Overexpression of Wnt7a or a stabilized form of β-catenin in mouse cortical NPC cultures from E13.5 induces neuronal differentiation. In contrast, inhibition of Wnt signaling by axin or DKK1 leads to suppression of neuronal differentiation of cortical NPCs in vitro and in the developing mouse neocortex (23). These results together indicate developmental stage-dependent functions of Wnt/β-catenin signaling in NPCs (22).
Consistent with this idea, we found impaired neuronal differentiation in Pin1 KO mice at E15.5, with no apparent indication of primary migration defects, as indicated by normal reelin expression in Pin1 KO mice (data not shown), although Pin1 might also contribute to promigratory neuronal functions. Furthermore, at E12.5, there was little Pin1 expression and Pin1 knockout had no effects on β-catenin levels in NPCs (data not shown). However, at E15.5, Pin1 levels were significantly elevated and Pin1 knockout significantly reduced β-catenin levels in NPCs. These results suggest that Pin1 may act mainly during late developmental stages (neurogenic phase) to promote neuronal differentiation, rather than during the earlier NPC expansion phase. In line with our results, a recent paper (54) suggests that activation of the Wnt/β-catenin pathway by overexpression of Wnt3a promotes differentiation of intermediate progenitors into neurons. However, electroporation of Wnt3a also led to RGC expansion at E16.5 (54). Thus, balance among multiple ligands for Wnt/β-catenin signaling, as well as the developmental stage, might also influence the consequence of the balance between proliferation and differentiation of NPCs.
We showed that Pin1 controls NPC differentiation by binding and stabilizing β-catenin. These results are consistent with the previous finding that Pin1 increases β-catenin levels by inhibiting its degradation, with Pin1 expression tightly correlating with β-catenin levels in many human cancer tissues and cells (11, 28, 60, 63, 74, 93). Tau and APP are also substrates for Pin1, and the Aβ peptide increases the number of newborn hippocampal neurons, with no changes in the rate of cell death or proliferation, suggesting that the Aβ peptide could act on hippocampal neuronal progenitors and promote their differentiation into neurons (37). Because Pin1 reduces neurotoxic Aβ peptides (64), it does not seem likely that the impaired neuronal differentiation in Pin1 KO mice is due to APP. Instead, a similar mechanism was reported for embryonic stem cells, in which Pin1 binds and stabilizes Nanog, a transcription factor crucial for the self-renewal of embryonic stem cells (53). Thus, Pin1 may regulate protein stability of multiple substrates in different stem cells.
Significantly, Pin1 KO mice showed severely impaired motor activity during the neonatal period. Abnormal motor activities during the neonatal period are rarely reported. An example is Dyrk1A transgenic mice, which show a longer latency to initiation of walking at P7 and P10 (1). Since differentiation from NPCs to immature neurons is impaired in Pin1 KO mice, axonal and dendritic development is also likely to be inhibited, such that synaptic organization is immature during the neonatal stage. Indeed, PSD95 staining showed that synaptic organization in Pin1 KO mice was immature at P9. Pin1 KO mice would be useful for studying the mechanisms of neonatal defects in motor activities.
In summary, we have shown that Pin1 expression is increased in NPCs during certain developmental stages and that Pin1 knockout in mice inhibits the birth of upper but not lower layer neurons in the cerebral cortex, without affecting gliogenesis. Moreover, our proteomic approach identified β-catenin as a major substrate for Pin1 in NPCs, in which Pin1 stabilizes β-catenin by inducing conformational changes after phosphorylation. Consequently, Pin1 knockout leads to reduced β-catenin in NPCs at late but not early stages during brain development. Functionally, overexpression of Wnt or β-catenin, but not a β-catenin mutant that cannot act as a Pin1 substrate, fully rescues the deficits in neuronal differentiation of Pin1 knockout NPCs in vitro. These results not only identify for the first time a major function for Pin1 in regulating neuronal differentiation of NPCs but also uncover a previously unappreciated mechanism by which Wnt/β-catenin signaling is further regulated after phosphorylation to orchestrate the developmental stage-specific response of NPCs during neuronal differentiation.
ACKNOWLEDGMENTS
We thank Xi He for constructive advice and Robert F. Hevner, Fred H. Gage, Tatsuya Saito, Yukiko Gotoh, and Xe He for providing many critical reagents.
D.A.S. was supported by the Glenn Foundation for Medical Research and is a Senior Scholar of the Ellison Medical Foundation. This study was supported by NIH grants AG017870, AG039405, and CA167677 to K.P.L.
Footnotes
Published ahead of print 29 May 2012
REFERENCES
- 1. Altafaj X, et al. 2001. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum. Mol. Genet. 10:1915–1923 [DOI] [PubMed] [Google Scholar]
- 2. Anderson CT, Sheets PL, Kiritani T, Shepherd GM. 2010. Sublayer-specific microcircuits of corticospinal and corticostriatal neurons in motor cortex. Nat. Neurosci. 13:739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson P. 2005. Pin1: a proline isomerase that makes you wheeze? Nat. Immunol. 6:1211–1212 [DOI] [PubMed] [Google Scholar]
- 4. Angevine JB, Jr, Sidman RL. 1961. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature 192:766–768 [DOI] [PubMed] [Google Scholar]
- 5. Arlotta P, et al. 2005. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45:207–221 [DOI] [PubMed] [Google Scholar]
- 6. Arnold SJ, et al. 2008. The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 22:2479–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Becker EB, Bonni A. 2006. Pin1 mediates neural-specific activation of the mitochondrial apoptotic machinery. Neuron 49:655–662 [DOI] [PubMed] [Google Scholar]
- 8. Blume-Jensen P, Hunter T. 2001. Oncogenic kinase signalling. Nature 411:355–365 [DOI] [PubMed] [Google Scholar]
- 9. Butterfield DA, et al. 2006. Pin1 in Alzheimer's disease. J. Neurochem. 98:1697–1706 [DOI] [PubMed] [Google Scholar]
- 10. Caviness VS, Jr, Takahashi T. 1995. Proliferative events in the cerebral ventricular zone. Brain Dev. 17:159–163 [DOI] [PubMed] [Google Scholar]
- 11. Chen SY, et al. 2006. Activation of beta-catenin signaling in prostate cancer by peptidyl-prolyl isomerase Pin1-mediated abrogation of the androgen receptor–beta-catenin interaction. Mol. Cell. Biol. 26:929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chenn A, Walsh CA. 2002. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297:365–369 [DOI] [PubMed] [Google Scholar]
- 13. Clevers H. 2006. Wnt/beta-catenin signaling in development and disease. Cell 127:469–480 [DOI] [PubMed] [Google Scholar]
- 14. Englund C, et al. 2005. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci. 25:247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fotaki V, Martinez De Lagran M, Estivill X, Arbones M, Dierssen M. 2004. Haploinsufficiency of Dyrk1A in mice leads to specific alterations in the development and regulation of motor activity. Behav. Neurosci. 118:815–821 [DOI] [PubMed] [Google Scholar]
- 16. Gage FH. 2010. Molecular and cellular mechanisms contributing to the regulation, proliferation and differentiation of neural stem cells in the adult dentate gyrus. Keio J. Med. 59:79–83 [DOI] [PubMed] [Google Scholar]
- 17. Goutagny N, Severa M, Fitzgerald KA. 2006. Pin-ning down immune responses to RNA viruses. Nat. Immunol. 7:555–557 [DOI] [PubMed] [Google Scholar]
- 18. Grigoryan T, Wend P, Klaus A, Birchmeier W. 2008. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 22:2308–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hajnoczky G, Hoek JB. 2007. Cell signaling. Mitochondrial longevity pathways. Science 315:607–609 [DOI] [PubMed] [Google Scholar]
- 20. Hamdane M, et al. 2006. Pin1 allows for differential Tau dephosphorylation in neuronal cells. Mol. Cell. Neurosci. 32:155–160 [DOI] [PubMed] [Google Scholar]
- 21. He TC, et al. 1998. Identification of c-MYC as a target of the APC pathway. Science 281:1509–1512 [DOI] [PubMed] [Google Scholar]
- 22. Hirabayashi Y, Gotoh Y. 2005. Stage-dependent fate determination of neural precursor cells in mouse forebrain. Neurosci. Res. 51:331–336 [DOI] [PubMed] [Google Scholar]
- 23. Hirabayashi Y, et al. 2004. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 131:2791–2801 [DOI] [PubMed] [Google Scholar]
- 24. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59 [DOI] [PubMed] [Google Scholar]
- 25. Israsena N, Hu M, Fu W, Kan L, Kessler JA. 2004. The presence of FGF2 signaling determines whether beta-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev. Biol. 268:220–231 [DOI] [PubMed] [Google Scholar]
- 26. Kalani MY, et al. 2008. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. U. S. A. 105:16970–16975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kesavapany S, et al. 2007. Inhibition of Pin1 reduces glutamate-induced perikaryal accumulation of phosphorylated neurofilament-H in neurons. Mol. Biol. Cell 18:3645–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim CJ, et al. 2005. Pin1 overexpression in colorectal cancer and its correlation with aberrant beta-catenin expression. World J. Gastroenterol. 11:5006–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Korinek V, et al. 1997. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 275:1784–1787 [DOI] [PubMed] [Google Scholar]
- 30. Kuwabara T, et al. 2009. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 12:1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lai T, et al. 2008. SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron 57:232–247 [DOI] [PubMed] [Google Scholar]
- 32. Lee TH, et al. 2009. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat. Cell Biol. 11:97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lie DC, et al. 2005. Wnt signalling regulates adult hippocampal neurogenesis. Nature 437:1370–1375 [DOI] [PubMed] [Google Scholar]
- 34. Lim J, et al. 2008. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J. Clin. Invest. 118:1877–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liou Y-C, et al. 2003. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424:556–561 [DOI] [PubMed] [Google Scholar]
- 36. Liu C, et al. 1999. Beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc. Natl. Acad. Sci. U. S. A. 96:6273–6278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lopez-Toledano MA, Shelanski ML. 2004. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J. Neurosci. 24:5439–5444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu KP, Finn G, Lee TH, Nicholson LK. 2007. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 3:619–629 [DOI] [PubMed] [Google Scholar]
- 39. Lu KP, Hanes SD, Hunter T. 1996. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380:544–547 [DOI] [PubMed] [Google Scholar]
- 40. Lu KP, Zhou XZ. 2007. The prolyl isomerase Pin1: a pivotal new twist in phosphorylation signalling and human disease. Nat. Rev. Mol. Cell. Biol. 8:904–916 [DOI] [PubMed] [Google Scholar]
- 41. Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. 1999. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399:784–788 [DOI] [PubMed] [Google Scholar]
- 42. Lu PJ, Zhou XZ, Shen M, Lu KP. 1999. A function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science 283:1325–1328 [DOI] [PubMed] [Google Scholar]
- 43. Lyu J, Yamamoto V, Lu W. 2008. Cleavage of the Wnt receptor Ryk regulates neuronal differentiation during cortical neurogenesis. Dev. Cell 15:773–780 [DOI] [PubMed] [Google Scholar]
- 44. Ma SL, et al. 2012. A PIN1 polymorphism that prevents its suppression by AP4 associates with delayed onset of Alzheimer's disease. Neurobiol. Aging 33:804–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ma SL, Pastorino L, Zhou XZ, Lu KP. 2012. Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3β (GSK3β) activity: novel mechanism for Pin1 to protect against Alzheimer disease. J. Biol. Chem. 287:6969–6973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. MacDonald BT, Tamai K, He X. 2009. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17:9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Machon O, et al. 2007. A dynamic gradient of Wnt signaling controls initiation of neurogenesis in the mammalian cortex and cellular specification in the hippocampus. Dev. Biol. 311:223–237 [DOI] [PubMed] [Google Scholar]
- 48. Machon O, van den Bout CJ, Backman M, Kemler R, Krauss S. 2003. Role of beta-catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience 122:129–143 [DOI] [PubMed] [Google Scholar]
- 49. Maudsley S, Mattson MP. 2006. Protein twists and turns in Alzheimer disease. Nat. Med. 12:392–393 [DOI] [PubMed] [Google Scholar]
- 50. McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG. 2002. Transcriptional regulation of cortical neuron migration by POU domain factors. Science 295:1528–1532 [DOI] [PubMed] [Google Scholar]
- 51. Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. 2007. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8:427–437 [DOI] [PubMed] [Google Scholar]
- 52. Moretto Zita M, et al. 2007. Post-phosphorylation prolyl isomerisation of gephyrin represents a mechanism to modulate glycine receptors function. EMBO J. 26:1761–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moretto-Zita M, et al. 2010. Phosphorylation stabilizes Nanog by promoting its interaction with Pin1. Proc. Natl. Acad. Sci. U. S. A. 107:13312–13317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ. 2011. Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J. Neurosci. 31:1676–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nakamura K, et al. 2012. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell 149:232–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nakamura K, et al. 2007. CD3 and immunoglobulin G Fc receptor regulate cerebellar functions. Mol. Cell. Biol. 27:5128–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nakamura K, et al. 2006. Requirement of tryptophan hydroxylase during development for maturation of sensorimotor gating. J. Mol. Biol. 363:345–354 [DOI] [PubMed] [Google Scholar]
- 58. Nakamura K, et al. 2001. Enhancement of hippocampal LTP, reference memory and sensorimotor gating in mutant mice lacking a telencephalon-specific cell adhesion molecule. Eur. J. Neurosci. 13:179–189 [DOI] [PubMed] [Google Scholar]
- 59. Nakamura K, Sato T, Ohashi A, Tsurui H, Hasegawa H. 2008. Role of a serotonin precursor in development of gut microvilli. Am. J. Pathol. 172:333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakashima M, et al. 2004. Cyclin D1 overexpression in thyroid tumours from a radio-contaminated area and its correlation with Pin1 and aberrant beta-catenin expression. J. Pathol. 202:446–455 [DOI] [PubMed] [Google Scholar]
- 61. Nigg EA. 2001. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell. Biol. 2:21–32 [DOI] [PubMed] [Google Scholar]
- 62. Nusse R, et al. 2008. Wnt signaling and stem cell control. Cold Spring Harb. Symp. Quant. Biol. 73:59–66 [DOI] [PubMed] [Google Scholar]
- 63. Pang R, et al. 2004. PIN1 overexpression and beta-catenin gene mutations are distinct oncogenic events in human hepatocellular carcinoma. Oncogene 23:4182–4186 [DOI] [PubMed] [Google Scholar]
- 64. Pastorino L, et al. 2006. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 440:528–534 [DOI] [PubMed] [Google Scholar]
- 65. Pastorino L, et al. 2012. Alzheimer's disease-related loss of Pin1 function influences the intracellular localization and the processing of AβPP. J. Alzheimers Dis. 30:277–297 [DOI] [PubMed] [Google Scholar]
- 66. Pawson T, Scott JD. 2005. Protein phosphorylation in signaling—50 years and counting. Trends Biochem. Sci. 30:286–290 [DOI] [PubMed] [Google Scholar]
- 67. Rakic P. 1974. Neurons in rhesus monkey visual cortex: systematic relation between time of origin and eventual disposition. Science 183:425–427 [DOI] [PubMed] [Google Scholar]
- 68. Rakic P, Ayoub AE, Breunig JJ, Dominguez MH. 2009. Decision by division: making cortical maps. Trends Neurosci. 32:291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ranganathan R, Lu KP, Hunter T, Noel JP. 1997. Structural and functional analysis of the mitotic peptidyl-prolyl isomerase Pin1 suggests that substrate recognition is phosphorylation dependent. Cell 89:875–886 [DOI] [PubMed] [Google Scholar]
- 70. Rudrabhatla P, Albers W, Pant HC. 2009. Peptidyl-prolyl isomerase 1 regulates protein phosphatase 2A-mediated topographic phosphorylation of neurofilament proteins. J. Neurosci. 29:14869–14880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rudrabhatla P, et al. 2008. Pin1-dependent prolyl isomerization modulates the stress-induced phosphorylation of high molecular weight neurofilament protein. J. Biol. Chem. 283:26737–26747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rustighi A, et al. 2009. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat. Cell Biol. 11:133–142 [DOI] [PubMed] [Google Scholar]
- 73. Ryo A, et al. 2002. Pin1 is an E2F target gene essential for the Neu/Ras-induced transformation of mammary epithelial cells. Mol. Cell. Biol. 22:5281–5295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ryo A, Nakamura N, Wulf G, Liou YC, Lu KP. 2001. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 3:793–801 [DOI] [PubMed] [Google Scholar]
- 75. Ryo A, et al. 2003. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 12:1413–1426 [DOI] [PubMed] [Google Scholar]
- 76. Ryo A, et al. 2006. Prolyl-isomerase Pin1 accumulates in Lewy bodies of Parkinson disease and facilitates formation of alpha-synuclein inclusions. J. Biol. Chem. 281:4117–4125 [DOI] [PubMed] [Google Scholar]
- 77. Segat L, et al. 2007. Pin1 promoter polymorphisms are associated with Alzheimer's disease. Neurobiol. Aging 28:69–74 [DOI] [PubMed] [Google Scholar]
- 78. Shaw PE. 2007. Peptidyl-prolyl cis/trans isomerases and transcription: is there a twist in the tail? EMBO Rep. 8:40–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Solberg N, Machon O, Krauss S. 2008. Effect of canonical Wnt inhibition in the neurogenic cortex, hippocampus, and premigratory dentate gyrus progenitor pool. Dev. Dyn. 237:1799–1811 [DOI] [PubMed] [Google Scholar]
- 80. Sugitani Y, et al. 2002. Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev. 16:1760–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sultana R, et al. 2006. Oxidative modification and down-regulation of Pin1 in Alzheimer's disease hippocampus: a redox proteomics analysis. Neurobiol. Aging 27:918–925 [DOI] [PubMed] [Google Scholar]
- 82. Svenningsson P, et al. 2007. Involvement of AMPA receptor phosphorylation in antidepressant actions with special reference to tianeptine. Eur. J. Neurosci. 26:3509–3517 [DOI] [PubMed] [Google Scholar]
- 83. Thullier F, Lalonde R, Cousin X, Lestienne F. 1997. Neurobehavioral evaluation of lurcher mutant mice during ontogeny. Brain Res. Dev. Brain Res. 100:22–28 [DOI] [PubMed] [Google Scholar]
- 84. Wang S, et al. 2007. The significance of Pin1 in the development of Alzheimer's disease. J. Alzheimers Dis. 11:13–23 [DOI] [PubMed] [Google Scholar]
- 85. Wrobel CN, Mutch CA, Swaminathan S, Taketo MM, Chenn A. 2007. Persistent expression of stabilized beta-catenin delays maturation of radial glial cells into intermediate progenitors. Dev. Biol. 309:285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wu SX, et al. 2005. Pyramidal neurons of upper cortical layers generated by NEX-positive progenitor cells in the subventricular zone. Proc. Natl. Acad. Sci. U. S. A. 102:17172–17177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yaffe MB, et al. 1997. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278:1957–1960 [DOI] [PubMed] [Google Scholar]
- 88. Yeh ES, Means AR. 2007. PIN1, the cell cycle and cancer. Nat. Rev. Cancer 7:381–388 [DOI] [PubMed] [Google Scholar]
- 89. Zacchi P, et al. 2002. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 419:853–857 [DOI] [PubMed] [Google Scholar]
- 90. Zechner D, et al. 2003. Beta-catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev. Biol. 258:406–418 [DOI] [PubMed] [Google Scholar]
- 91. Zhang CL, Zou Y, He W, Gage FH, Evans RM. 2008. A role for adult TLX-positive neural stem cells in learning and behaviour. Nature 451:1004–1007 [DOI] [PubMed] [Google Scholar]
- 92. Zheng H, et al. 2002. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419:849–853 [DOI] [PubMed] [Google Scholar]
- 93. Zhou CX, Gao Y. 2006. Aberrant expression of beta-catenin, Pin1 and cylin D1 in salivary adenoid cystic carcinoma: relation to tumor proliferation and metastasis. Oncol. Rep. 16:505–511 [PubMed] [Google Scholar]
- 94. Zhou XZ, et al. 2000. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol. Cell 6:873–883 [DOI] [PubMed] [Google Scholar]