

Abstract
Hypoxia-inducible factor 1 (HIF1) is a master regulator of adaptive gene expression under hypoxia. However, a role for HIF1 in the epigenetic regulation remains unknown. Genome-wide analysis of HIF1 binding sites (chromatin immunoprecipitation [ChIP] with deep sequencing) of endothelial cells clarified that HIF1 mainly binds to the intergenic regions distal from transcriptional starting sites under both normoxia and hypoxia. Next, we examined the temporal profile of gene expression under hypoxic conditions by using DNA microarrays. We clarified that early hypoxia-responsive genes are functionally associated with glycolysis, including GLUT3 (SLC2A3). Acetylated lysine 27 of histone 3 covered the HIF1 binding sites, and HIF1 functioned as an enhancer of SLC2A3 by interaction with lysine (K)-specific demethylase 3A (KDM3A). Knockdown of HIF1α and KDM3A showed that glycolytic genes are regulated by both HIF1 and KDM3A and respond to hypoxia in a manner independent of cell type specificity. We elucidated that both the chromatin conformational structure and histone modification change under hypoxic conditions and enhance the expression of SLC2A3 based on the combined results of chromatin conformation capture (3C) and ChIP assays. KDM3A is recruited to the SLC2A3 locus in an HIF1-dependent manner and demethylates H3K9me2 so as to upregulate its expression. These findings provide novel insights into the interaction between HIF1 and KDM3A and also the epigenetic regulation of HIF1.
INTRODUCTION
Oxygen deprivation leads to energy depletion and the accumulation of free radicals. Under hypoxic conditions, cells activate a number of adaptive responses to compensate for excessive metabolic demand by increasing both the oxygen supply and nonoxidative glycolytic energy production (18, 30). The transcriptional responses to hypoxia are mediated for the most part by hypoxia-inducible factor (HIF), which is a heterodimer consisting of an α-subunit (HIFα) and β-subunit (HIFβ) (14, 31). HIF recognizes the core sequence RCGTG (11, 13, 20, 29). Under normoxia, HIFα is posttranslationally degraded by prolyl hydroxylases (PHDs) (15). Under hypoxia, the α-subunit escapes degradation and forms a heterodimer with the β-subunit, exerting its hypoxic responses through binding to the RCGTG motif.
Upregulation of HIF1-regulated genes, such as by vascular endothelial growth factor (VEGF) and angiopoietin 2 (ANGPT2), promotes the initiation and maturation of angiogenesis (19). Moreover, solid tumors encounter hypoxic stress, and HIF1 plays a central role in tumor angiogenesis (6). Recently, genome-wide analyses of HIF1α, HIF1β, and HIF2α binding sites have been focused on cancer cell lines (22, 28). Therefore, HIF1-mediated gene regulation is particularly helpful when considering new strategies and therapeutic targets in ischemic diseases and cancer.
In eukaryotes, epigenetic modifications exert effects on the chromatin environment and gene expression, so that an ostensibly identical gene exhibits different patterns in a temporal and spatially dependent manner (43). Among these modifications, histone methylation and demethylation serve as regulatory markers that indicate transcriptionally active and inactive chromatin (4, 21, 26). These histone methylation marks are reversible and dynamically regulated by site-specific methyltransferases and demethylases. While the expression of various genes is regulated by histone methylation, the activities of the histone methytranseferases and demethylases are also influenced by gene expression, recruitment, and coordination with other epigenetic and posttranslational modifiers. One of the histone demethylases induced under hypoxia is lysine (K)-specific demethylase 3A (KDM3A), also known as jumonji domain-containing 1a (JMJD1a). KDM3A belongs to the JmjC domain-containing histone lysine 9 demethylases and is ubiquitously expressed (10, 42). KDM3A is one of the HIF1-mediated genes, that is, its expression is regulated by HIF1 binding under hypoxic conditions at the locus of its promoter region in cancer cell lines (3, 25). Although an analysis of KDM3A knockout mice revealed that KDM3A is essential for spermatogenesis (24) and the regulation of metabolic gene expression (36), the molecular mechanisms by which KDM3A regulates gene transcription under hypoxia have remained unclear.
Insight into the structure of chromatin is essential to an understanding of gene regulation in mammals, and cell-type-specific chromatin conformational structure and the resulting patterns of gene regulation have been reported (9). A genome-wide analysis with a chromatin interaction assay revealed that the estrogen receptor α binding sites display long-range physical interactions between genes and dispersed regulatory elements in MCF7 cells (8). However, the transcription factor-mediated chromatin interactions and the relationship between epigenetic modifiers and genome structure are at present largely unknown.
We performed chromatin immunoprecipitation with deep sequencing (ChIP-seq) to determine the genome-wide binding sites of HIF1 in human umbilical vein endothelial cells (HUVECs) under hypoxia. By utilizing a combination of ChIP-seq and microarray analyses, we found that the genes downstream of HIF1 and KDM3A are induced in the early response to hypoxic stimuli in order to regulate the genes involved in glycolytic function. Analysis of the molecular mechanism of one of these genes, SLC2A3 (solute carrier family 2A3), revealed that HIF1 and KDM3A cooperate in the regulation of gene expression by means of histone modifications and chromatin conformational changes. These data help to establish a basis for understanding the role of HIF1-induced epigenetic modifications and the regulation of the higher structure of the chromosome in the hypoxic state.
MATERIALS AND METHODS
Cell culture.
HUVECs were purchased from Lonza Japan Ltd. (Tokyo, Japan). All endothelial cells were cultured in EGM-2 MV complete medium (Lonza). Human embryonic kidney cells (HEK293; ATCC CRL-1573) and HeLa cells (ATCC CCL-2) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum (FBS). Cells were grown in a humidified atmosphere with 5% CO2 at 37°C. The hypoxic condition (1% O2 for 24 h) was brought about by means of a hypoxic cultivation incubator (Juji Field Co. Ltd., Tokyo, Japan).
ChIP.
HUVECs were cross-linked for 10 min by using 1% paraformaldehyde and sonicated into fragments. Samples were immunoprecipitated with the antibodies shown in Table S5 of the supplemental material. We used protein A-Sepharose beads (17-5138-01; GE Healthcare) and magnetic beads (Invitrogen) to immunoprecipitate the samples. The details for these methods are reported in the supplemental material.
ChIP-seq sample analysis.
Images acquired from the Illumina/Solexa sequencer were processed through the bundled Solexa image extraction pipeline, which identified polony positions, performed base-calling, and generated quality control statistics. Sequences were aligned using human genome NCBI Builder 36 (UCSC hg18) as the reference genome. All unique mapped sequences were analyzed by using QuEST 2.4 software (38). QuEST software was used according to the QuEST recommendations, with FDR analysis in the QuEST procedure, as follows: positive region size, 1,000; kernel density estimation bandwidth, 100; ChIP seeding fold enrichment, 10; ChIP extension fold enrichment, 3; ChIP-to-background fold enrichment, 3. A brief summary of the technique and minor protocol modifications are provided in the supplemental material.
DNA microarray and data analyses.
Preparation of cRNA and hybridization of the probe arrays were performed according to the manufacturer's instructions (Affymetrix, Santa Clara, CA). The raw data in the Human Genome U133 Plus 2.0 array (Affymetrix) were exported to GeneSpring11.5 and normalized by robust multiarray normalization algorithms. For identification of genes significantly altered by hypoxia, total detected entities were filtered by signal intensity value (upper cutoff, 100%; lower cutoff, 20%) and error (standard deviation, <0.1) to remove very low signal entities and to select reproducible signal values of entities among the experiments. Hierarchical clustering and kinetics analysis were performed by using the squared Euclidean uncentered algorithm with complete linkage.
3C-ChIP assay.
The chromatin conformation capture (3C) assay was performed as previously described (16), with certain modifications to combine ChIP with the HIF1α antibody. The details for this procedure are provided in the supplemental material. Briefly, cross-linked samples were incubated with 400 U of restriction enzyme (Csp6I) at 37°C overnight and then incubated with T4 DNA ligase (Fermentas, Glen Burnie, MD) at 16°C overnight. Samples were diluted and immunoprecipitated with the HIF1 antibody at 4°C overnight. Next, the samples were rotated with protein A-Sepharose beads for 3 h at 4°C. The beads were washed with ChIP dilution buffer, and samples were finally de-cross-linked at 65°C overnight.
Data access.
Data were analyzed according to the minimum information about a microarray experiment (MIAME) rule. Annotation of the probe numbers and targeted sequences are provided on the Affymetrix web page. These data are accessible through the National Center for Biotechnology Information, Gene Expression Omnibus (GSE35932) for microarrays, and ChIP-seqs.
Statistical analyses.
Data are reported as means ± standard errors. P values were calculated using a two-tailed unpaired Student t test. P values of <0.05 were considered significant.
RESULTS
Genome-wide occupancy of HIF1 under hypoxia in vascular endothelial cells.
HUVECs were cultured under normoxic and hypoxic conditions for 24 h. Western blotting showed an upregulation of HIF1α in HUVECs under hypoxic conditions, and the intensity of the band decreased markedly when HIF1α was knocked down by small interfering RNA (siRNA) (Fig. 1A). The knockdown efficiencies of HIF1α were 93% under normoxia and 87% under hypoxia at the mRNA level (Fig. 1B).
Fig 1.

Genome-wide analysis of HIF1 binding sites in endothelial cells. (A) Western blot analysis of HIF1 in the nuclear extract of HUVECs transfected with control or HIF1α siRNA. Antinucleoporin antibody was used as a loading control. The experiments were performed three times independently. (B) Quantitative RT-PCR analysis of HIF1 mRNA in HUVECs transfected with control or HIF1α siRNA. The experiments were performed three times independently. *, P < 0.05 compared with control siRNA. (C) Distribution of HIF1 binding sites on a genome-wide scale under hypoxia. (D) Representative HIF1 ChIP-seq results in HUVECs under normoxia and hypoxia. LOX, ENO1, VEGFA, and HK2 are well-known targets of HIF1. (E) Schematic representation of the most significantly enriched motif in the HIF1 binding sites under hypoxia. (F) Comparison of HIF1 binding and the expression value under normoxia and hypoxia. The heat map of the expression ratio (normoxia versus hypoxia) is shown on the left and is based on the induction ratio under hypoxia. Each black bar, shown in the middle, indicates HIF1 binding probes for ChIP-seq. GSEA results, shown on the right, revealed the correlation between the expression ratio (normoxia versus hypoxia) and the HIF1 binding gene profiles.
In order to identify the direct HIF1 binding sites in endothelial cells, we performed ChIP-seq. The genome precipitated by the anti-HIF1α antibody was applied to the massively parallel deep sequencer Genome Analyzer II (Illumina). Out of the total 35,558,646 (normoxia) and 38,932,254 (hypoxia) sequence reads, 56% (19,892,964 reads [normoxia]) and 51% (19,682,831 reads [hypoxia]) were uniquely aligned in the nonrepeating human genomic sequence. From the nonprecipitated DNA samples (input), we sequenced 16,194,856 reads under normoxic conditions and 28,052,794 reads under hypoxic conditions. Under normoxic conditions, 85% (13,765,628 reads) were uniquely mapped, while those uniquely mapped under hypoxic conditions comprised 82% (22,483,414 reads). We calculated the enrichment of HIF1 compared with the input and determined the significant HIF1 binding sites by using the QuEST 2.4 software (38). In total, 575 binding regions were identified as HIF1 enrichment sites under normoxia and 2,060 binding regions were identified under hypoxia (see Table S1A in the supplemental material). To investigate the correlation of HIF1 binding regions and the nearest known transcripts, we divided the regions into 10 sections based on the distance from the transcriptional starting sites (TSS) in the corresponding genes. As shown in Fig. 1C, approximately 82% (1,689 sites) were positioned within 50 kbp from the TSS under hypoxia. Of these, 12% (250 sites) were found in the proximal promoter, which is defined as the 5′ untranslated region (UTR) up to 1 kbp upstream from the TSS, and 21% (436 sites) were located in the introns. The ratio (normoxia, 8.7%; hypoxia, 12.2%) and the number of sites (normoxia, 50 sites; hypoxia, 250 sites) in the proximal promoter region were significantly increased under hypoxia than with normoxia, while that in the intergenic regions was remarkably reduced under hypoxia. The ChIP-seq data revealed that the HIF1 binding regions were not only located within the proximal promoter of each gene, but also scattered across the whole genome under hypoxia (Fig. 1C), although the proximal promoter HIF1 binding was increased under hypoxia. The representative HIF1 target genes (22) exhibited signal enrichments in the ChIP-seq data (Fig. 1D). The HIF1 ChIP-seq analysis revealed that the HIF1-recognized sequence RCGTG was the most highly enriched binding element, with an E- value of 4.5 × e−48 (Fig. 1E).
Identification of hypoxia-inducible genes in endothelial cells.
Next, we performed triplicate DNA microarrays after 24 h under hypoxia in order to focus on the gene expression profiles and their changes. We listed gene set probes that exhibited significant expression, based on results with GeneSpring 11.5 software (Agilent) (see Table S1B in the supplemental material). The highly upregulated genes included those for prostaglandin l2 synthase (PTGIS), adenylate kinase 3-like 1 (AK3L1), and angiopoietin-like 4 (ANGPTL4). Other representative HIF target genes included those for enolase 2 (ENO2), vascular endothelial growth factor A (VEGFA), glucose transporter 1 (GLUT1; solute carrier family 2A1 [SLC2A1]), adrenomedullin (ADM), and lysyl oxidase (LOX) (40).
To further investigate the correlation between HIF1 binding and subsequent gene expression, we selected the gene set probes that exhibited an average difference greater than 100 under either normoxia or hypoxia. Subsequently, 15,884 probes in total were sorted based on the ratio of the expression levels under normoxia versus hypoxia, as illustrated with a heat map (Fig. 1F, left column; see also Table S1C in the supplemental material). Upon integrating the microarray analysis and ChIP-seq data, the HIF1 binding genes were determined by deep sequencing and corresponded to each of the microarray probes on the left (Fig. 1F, right column, black bars). A list of microarray probes that were induced under hypoxia (HIF1 ChIP-seq binding sites [black bars]) are shown in Table S1D of the supplemental material. Of special note is that approximately 30% of the probes were categorized as highly expressed under hypoxia (Fig. 1F, red) and were dense with HIF1 binding in ChIP-seq under hypoxia, but not under normoxia. In contrast, one-third of the genes were downregulated under hypoxia (Fig. 1F, blue) and were sparse in ChIP-seq signals under both normoxia and hypoxia. Moreover, based on a ranking of the expression ratios, a highly significant (P < 10−6) positive gene set enrichment analysis (GSEA) score (35) was observed with HIF1 binding under hypoxia, whereas a negative score was found with HIF1 binding under normoxia. These data suggest HIF1 binding is strongly associated with the upregulation of genes under hypoxia on a genome-wide scale. These findings are consistent with the notion that HIF1 binds to hypoxia-inducible genes.
KDM3A downstream genes overlap with HIF1 downstream genes.
Among the HIF1 downstream targets, KDM3A was identified by both the microarray and ChIP-seq analyses. The upregulation of KDM3A under hypoxia was decreased 37% by the knockdown of HIF1α (Fig. 2A). HIF1 ChIP-seq showed that HIF1 binds to the KDM3A promoter region under hypoxia (Fig. 2B). We confirmed HIF1 enrichment at the promoter of KDM3A by ChIP-PCR (Fig. 2C). Because epigenetic histone modifications control gene expression (43), we focused on the histone-modifying enzymes to investigate the role of gene expression in the response to hypoxia.
Fig 2.

Target genes of KDM3A overlap with those of HIF1 and are upregulated early under hypoxia. (A) Quantitative RT-PCR of KDM3A showed upregulation under hypoxia and reduction by HIF1α knockdown. The experiments were performed three times independently. (B) HIF1 ChIP-seq data for the KDM3A locus. HIF1 binds to the promoter region of KDM3A only under hypoxia. (C) Binding of HIF1 to the KDM3A locus was validated by ChIP-PCR. The fold enrichment is shown along with the input. HBB is a negative-control primer, which was designed for the promoter regions of chromosome 2 (forward, 5205042 to 5205061; reverse, 5204825 to 5204846). *, P < 0.05 compared with normoxia; N.S., nonsignificant. (D) Quantitative RT-PCR analysis of KDM3A mRNA in HUVECs transfected with control or KDM3A siRNA. The experiments were performed three times independently. *, P < 0.05 compared with normoxia; #, P < 0.05 compared with control siRNA. (E) Western blot analysis of KDM3A in HUVECs transfected with control or KDM3A siRNA. β-Actin was used as a loading control. The experiments were performed three times independently. (F) Representative quantitative RT-PCR analysis of VEGFA and ANGPTL4 in HUVECs transfected with control or HIF1α siRNA. VEGFA and ANGPTL4 were upregulated under hypoxia when control siRNA was transfected into HUVECs, while this upregulation was reduced under hypoxia when HIF1α was knocked down. *, P < 0.05 compared with control siRNA. The experiments were performed three times independently. (G) Venn diagram depicting the overlap of HIF1 and KDM3A downstream target probes. The numbers indicate the HIF1 and KDM3A common and unique downstream target gene probe sets. The representative gene symbols are shown in the lower panel.
To survey the genes regulated by HIF1 and KDM3A in HUVECs, we transfected HUVECs with siRNA to knock down HIF1α or KDM3A. The knockdown efficiency of KDM3A by siRNA was 90% under normoxia and 75% under hypoxia (Fig. 2D). We confirmed that KDM3A was upregulated under hypoxia both at the mRNA (5.9-fold change [Fig. 2D]) and protein (Fig. 2E) level. To comprehensively identify the HIF1- and KDM3A-regulated genes, we employed microarrays performed in parallel using HUVECs transfected with control siRNA, HIF1α siRNA, or KDM3A siRNA. To find the genes upregulated under hypoxia, we set the criteria as such: the base 2 log fold change from normoxia to hypoxia was higher than 1.6 in the control siRNA. In order to identify the genes regulated by HIF1α among them, we focused on the genes for which the base 2 log fold change from control siRNA to HIF1α siRNA was lower than 1.2 under hypoxia (480 probes) (see Table S2A in the supplemental material). HIF1α knockdown under hypoxia for 24 h resulted in reduction of the representative HIF1 target genes (VEGFA and ANGPTL4). The findings of this microarray analysis were subsequently validated by quantitative real-time PCR (Fig. 2F). We similarly identified KDM3A downstream genes according to the criterion. Among them, we focused on the genes for which the base 2 log fold change from control siRNA to KDM3A siRNA was lower than 1.2 under hypoxia (174 probes). We identified 123 probes (see Table S2A) that were regulated by both HIF1 and KDM3A, for which the representative gene symbols are shown in Fig. 2G.
Early hypoxia-responsive genes are associated with glycolysis and are downstream targets of HIF1 and KDM3A.
In order to analyze how the HIF1 downstream target genes are upregulated by hypoxic stimuli, we determined the temporal profile of gene expression under hypoxia. We performed DNA microarray analysis after 1, 2, 4, 8, 12, and 24 h of hypoxic incubation. The findings of the microarray analysis were subsequently validated by reverse transcription-PCR (RT-PCR) (Fig. 3A). We divided the genes into two groups based on the criterion of a greater than 2-fold induction at each time point compared with normoxia (Fig. 3B). Group I was defined as early hypoxia-responsive genes, i.e., those which were upregulated more than 2.5 logs at after 1, 2, 4, and 8 h. Group II was defined as late hypoxia-responsive genes, which were upregulated more than 2.5 logs after 12 h and 24 h (see Table S2B in the supplemental material). In order to examine the relationship between hypoxic response and gene function, we performed functional clustering using the Database for Annotation, Visualization, and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/). Group I included the claudin 3 gene (CLDN3), the DNA damage-inducible transcript 4 gene (DDIT4), and ANGPTL4, which are associated with the hypoxia response and oxygen level (Fig. 3C). Group I also included SLC2A3, SLC2A1, the phosphoglucomutase 1 gene (PGM1), the hexokinase 2 gene (HK2), and ENO2, which are associated with glycolysis. KDM3A was also included in group I. On the other hand, group II included genes for interleukin 8 (IL-8), vascular cell adhesion molecule 1 (VCAM1), intercellular adhesion molecule 1 (ICAM1), E-selectin (SELE), and serpin peptidase inhibitor, clade E, member 1 (SERPINE1), which are typically expressed on endothelial cells. These functional clustering patterns made it evident that the early hypoxia-responsive genes in group I are associated with the response to oxygen levels and glycolysis, whereas the late responsive genes in group II are associated with angiogenesis and blood vessel development (see Table S2C).
Fig 3.

HIF1 regulates fundamental responses, such as glycolysis, independently of cell specificity in the early phase of hypoxia. (A) Quantitative RT-PCR showed a time-dependent increase in VEGFA and SLC2A1 under hypoxia. *, P < 0.05 compared with normoxia. The experiments were performed three times independently. (B) The left panel is a heat map that shows the probe lists for the hypoxia-responsive genes. The purple bars in the right lane show probes commonly identified as the HIF1 and KDM3A downstream targets. The pink bars in the right lane represent HIF1-specific target gene probes, while the blue bars represent KDM3A-specific gene probes. The pie graphs on the right show a comparison of HIF1 and KDM3A downstream genes in groups I and II. The numbers in the pie charts represent the percentages of each target gene. The genes commonly regulated by HIF1 and KDM3A were more frequently included in group I (25%) than in group II (2%) (P < 0.0001). (C) Functional annotations for each group and the representative gene symbols for each category are shown in the left and middle panels. The enrichment scores of each group from DAVID are shown in the bar graphs on the right. The background colors of each gene symbol correspond to those of the HIF1 and KDM3A target probes in panel B. (D) Venn diagram depicting the overlap of the HIF1 binding genes between MCF7 cells and HUVECs, both of which are upregulated under hypoxia. (E) Enrichment scores for the HIF1 binding sites in HUVECs and MCF7 cells derived using DAVID. The HIF1 binding genes that are common in HUVECs and MCF7 cells are functionally associated with glycolysis. (F) Percentage distribution of HIF1 binding genes of group I and group II in HUVECs and MCF7 cells. The HIF1 binding genes in common in HUVECs and MCF7 cells (16%) were significantly more frequently included in group I that in group II (2%) (P < 0.0001).
In order to examine whether the two groups were regulated by both HIF1 and KDM3A, we compared them with the HIF1 and KDM3A downstream target genes. As shown in Fig. 3B, the early hypoxia-responsive genes in group I included significantly more common HIF1 and KDM3A downstream targets than the late hypoxia-responsive genes in group II.
Early hypoxia-responsive genes are present in HUVECs and MCF7 cells.
In order to clarify the cell-specific HIF1 binding sites, we utilized the recently reported HIF1 ChIP-seq data in a breast cancer cell line, MCF7 (28), and compared the HIF1 association patterns in the whole genome between HUVECs and MCF7 cells. As a result of analysis of HIF1 binding sites in MCF7 cells by using QuEST 2.4, of the total HIF1α binding genes (366) in MCF7 cells, 188 genes were upregulated under hypoxia when the criterion for upregulation for the microarray data was a fold change of >0.3 log. As a result, 59 genes were found to overlap between MCF7 and HUVECs under the hypoxic condition (Fig. 3D). The binding genes of the common and cell-type-specific HIF1 binding genes are shown in Table S3 of the supplemental material.
To determine whether these cell-type-specific hypoxia-responsive genes could provide insight into functional relevance, we performed functional annotation clustering using DAVID. We found that the HIF1 binding genes in both MCF7 cells and HUVECs were associated with various functional annotation groups, which were significantly enriched for glycolysis and the response to hypoxia (Fig. 3E). On the other hand, HUVEC-specific HIF1 binding genes were correlated with angiogenesis, blood vessel, and vascular development, while MCF7-specific HIF1 binding genes were associated with metabolic process and regulation of signal transduction. These findings suggest that HIF1 binding genes are commonly associated with glycolysis, which is a fundamental response to hypoxia beyond cell specificity, while cell-specific HIF1 binding genes are uniquely associated with certain cell-specific responses to hypoxia, such as angiogenesis and apoptosis.
Furthermore, we compared the HIF1 binding of early and late hypoxia-responsive genes in HUVECs and MCF7 cells. The early hypoxia-responsive genes in group I were significantly associated with HIF1 binding in both HUVECs and MCF7 cells (Fig. 3F) compared with the late responsive genes in group II (group I, 16%; group II, 2%).
Taken together, these results suggest that HIF1 regulates certain fundamental responses, such as glycolysis, independently of cell specificity in the early phase of hypoxia. In addition, the early hypoxia-responsive genes in group I were regulated by HIF1 both in HUVECs and MCF7 cells, while the functions and HIF1 binding of the late responsive genes in group II were cell type specific.
SLC2A3 enhances glucose uptake under hypoxic conditions.
Focusing on the functional role of the SLC2A3 regulation carried out by both HIF1 and KDM3A in endothelial cells, we evaluated the functional relevance of SLC2A3, which is included in group I (Fig. 3C). SLC2A3 encodes glucose transporter isoform 3, one of the solute carrier family (34). Under hypoxic conditions, cells need to increase glucose uptake in order to perform anaerobic glycolysis so as to produce energy. Knockdown of SLC2A3 by siRNA (93% reduction) (Fig. 4A) decreased the amount of glucose uptake into cells (a 43% reduction compared with control siRNA under hypoxia) (Fig. 4B). In addition, when SLC2A3 was knocked down by siRNA, the glucose consumption rate in HUVECs was also decreased (42% reduction) (Fig. 4C). Furthermore, in order to examine the effects of SLC2A3 on angiogenesis in endothelial cells, we performed a tube formation assay. As shown in Fig. 4D, we demonstrated that knockdown of SLC2A3 disturbed tube formation in HUVECs (27% reduction compared with control siRNA), while tube formation was enhanced by hypoxia (2-fold increase compared with normoxia). The inhibition of tube formation by knockdown of SLC2A3 was observed under both normoxic (35% reduction) and hypoxic (27% reduction) conditions.
Fig 4.
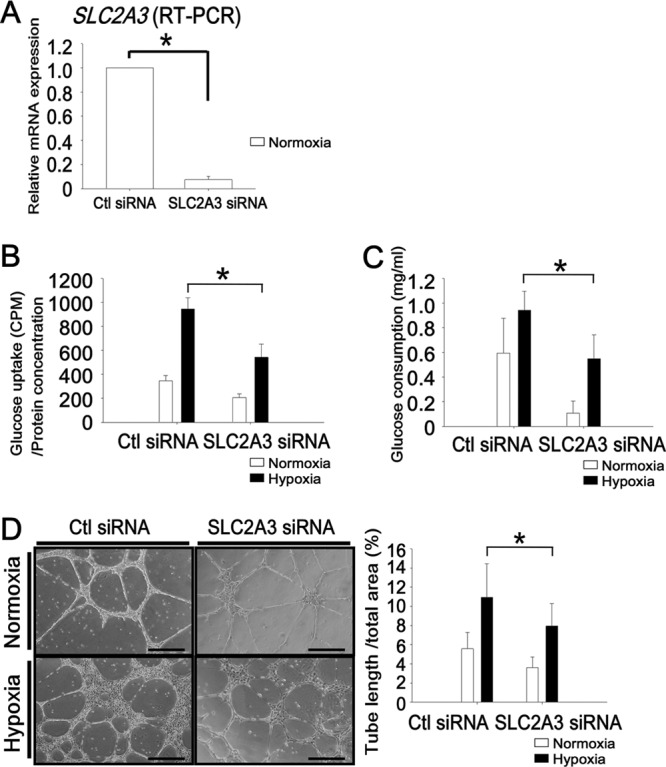
SLC2A3 is essential for glucose transport into the cell and tube formation in HUVECs. (A) Expression of SLC2A3 was significantly reduced at the mRNA level compared with control siRNA. The experiments were performed three times independently. (B) Glucose uptake, measured using a radioactive isotope. The experiments were performed three times independently. *, P < 0.05 compared with control siRNA under hypoxia. (C) Glucose consumption rates in HUVECs. The experiments were performed three times independently. *, P < 0.05 compared with control siRNA under hypoxia. (D) Representative tube formation assay using Matrigel (left) and quantification of the total tube length (in mm) versus the total area (right). Bar, 100 μm. The experiments were performed three times independently. *, P < 0.05 compared with control siRNA under hypoxia.
HIF binding to the TSS and kbp −35 region upstream of the SLC2A3 locus is critical for expression.
As the result of the microarray analysis (Fig. 2G), SLC2A3 was found to be one of the HIF1 and KDM3A downstream target genes. We performed quantitative RT-PCR and confirmed that SLC2A3 mRNA expression was upregulated under hypoxia (6.6-fold induction compared with normoxia) when control siRNA was transfected into HUVECs, while SLC2A3 mRNA expression decreased (HIF1α siRNA, 70% reduction; KDM3A siRNA, 31% reduction [compared with control siRNA]) when HIF1α and KDM3A siRNA were transfected (Fig. 5A). This result as well as the functional role of SLC2A3 in HUVECs stimulated us to investigate the mechanism of the regulation of gene expression by HIF1 in detail. The HIF1 ChIP-seq results revealed that there were associations detected at kbp −35 under both normoxia and hypoxia and at kbp −24 under hypoxia (Fig. 5B). To determine whether these regions were in active chromatin states, we investigated the distribution of the monomethylated lysine 4 of histone 3 (H3K4me1), acetylated lysine 27 of histone 3 (H3K27ac), trimethylated lysine 4 of histone 3 (H3K4me3), acetylated histone 3 (H3ac), and acetylated histone 4 (H4ac) (Fig. 5C) using their specific antibodies (17a). As shown in Fig. 5C, the SLC2A3 promoter indicated a profound enrichment with enhancer marks (H3K4me1 and H3K27ac) and active marks (H3K4me3, H3ac, and H4ac) in association with the recruitment of HIF1 (Fig. 5D). Importantly, the H3K4me1 and H3K27ac signals increased under hypoxia in the enhancer regions in the locus (kbp −35), while the HBB locus, which is completely silent under both normoxia and hypoxia in endothelial cells, did not.
Fig 5.

HIF binding to the TSS and distal enhancer 1 at the SLC2A3 loci under normoxia and hypoxia. (A) Quantitative RT-PCR analysis of SLC2A3 mRNA in HUVECs transfected with control siRNA or HIF1 and KDM3A siRNA. The experiments were performed three times independently. *, P < 0.05 compared with control siRNA under hypoxia. (B) HIF ChIP-seq data at the SLC2A3 loci, derived using an integrated genome browser. Genome-wide analysis results of histone modifications (H3K4me1, H3K27ac, and H3K4me3) are displayed for both normoxia and hypoxia conditions. (C) ChIP-PCR of histone modification at the SLC2A3 locus. The experiments were performed three times independently. *, P < 0.05 compared with normoxia. (D) ChIP-PCR of HIF1 on TSS and enhancer 1 (kbp −35) revealed an enrichment of HIF1 compared with HBB. HIF1 was significantly enriched under hypoxia compared with normoxia. The experiments were performed three times independently. *, P < 0.05 compared with normoxia. (E) Schematic representation of the SLC2A3 promoter-luc, enhancer-luc, and RCGTG motifs. There is one RCGTG motif in the SLC2A3 promoter, five RCGTG motifs in enhancer 1 (kbp −35), and 1 RCGTG motif in enhancer 2 (kbp −24). (F) Reporter assay of the SLC2A3 promoter in combination with enhancer 1 in HUVECs. The experiments were performed three times independently. *, P < 0.001 compared with the activity from the core promoter-luc under normoxia; N.S., nonsignificant. (G) Reporter assay results for the SLC2A3 promoter in HEK293 cells overexpressing HIF1. The experiments were performed three times independently. *, P < 0.01 compared with activity from the core promoter-luc of the control vector.
HIF1 ChIP-seq data suggested that HIF1 was recruited to the proximal promoter and two enhancer regions far from the TSS in the SLC2A3 locus. Reporter assays were employed to determine whether the HIF1 binding RCGTG motif is required for SLC2A3 gene expression. We isolated fragments from the human SLC2A3 promoter located from −651 to +240 relative to the TSS and enhancer 1 (kbp −35) located from −35162 to −33862 relative to the TSS (Fig. 5E). These were cloned into the reporter vector pGL3. The SLC2A3 promoter and enhancer 1 include RCGTG motifs (promoter, 1 motif [bp +128]; enhancer 1, 5 motifs) (Fig. 5E). The SLC2A3 promoter-luc and enhancer 1-luc constructs were transiently transfected into HUVECs. The enhancer 1-luc construct was upregulated 13-fold more than the core promoter-luc in HUVECs under hypoxia (Fig. 5F). A comparison of a series of mutated (CGT→AAA) constructs demonstrated that mutation of motif 4 of enhancer 1 [E1 (mut 4)-luc] resulted in a 69% reduction of luciferase activity. Mutation of all five sites [E1 (mut all)-luc] reduced promoter activity by 72%. In addition, E1-luc of SLC2A3 was 2-fold upregulated by HIF1 overexpression compared with the control vector in HEK293 cells, while E1 (mut 4)-luc abrogated the upregulation of luciferase activity by HIF1 overexpression (Fig. 5G). It was thus demonstrated that the RCGTG motif 4 in enhancer 1 (kbp −35) is essential for the induction of SLC2A3 by HIF1 under hypoxia.
HIF1 functions as an enhancer by changing the level of histone modification under hypoxia.
Epigenetic modifications are associated with gene expression, and the modifications associated with H3K4me1 and H3K27ac are considered to be the enhancers of actively transcribed genes (27, 44). To examine whether histone enhancers mark changes under hypoxia, we compared the HIF1 binding sites and H3K27ac under normoxia and hypoxia on a genome-wide scale. The number of total reads and uniquely mapped sequences of the ChIP-seq for histone marks are shown in Table S4 in the supplemental material. H3K27ac sites overlapped at 12,498 sites between the state of normoxia (14,679 sites) and hypoxia (16,366 sites) on a genome-wide scale (Fig. 6A). Of the total number of HIF1 binding sites (2,060 sites), H3K27ac covered 1,332 sites (65%). We found that the HIF1 binding sites colocalized with the enhancer mark H3K27ac and that H3K27ac increased under hypoxia at the HIF1 binding region loci (Fig. 6B).
Fig 6.

HIF1 binding to distal enhancers plays roles in the chromatin conformational change under hypoxia. (A) Genome-wide analysis of H3K27ac binding sites under normoxia (14,679 sites) and hypoxia (16,366 sites). A total of 12,498 sites overlapped under normoxic and hypoxic conditions. (B) Comparison of H3K27ac binding among the HIF1 binding sites, based on the ChIP-seq results under normoxia and hypoxia. H3K27ac increased at the HIF1 binding sites only under hypoxia. The numbers above or below each bar in the graph are the HIF1 binding sites. (C) ChIP-PCR for distal enhancer 2 (kbp −24) showed enrichment of HIF1 and H3K27ac compared with HBB. The experiments were performed three times independently. *, P < 0.05 compared with normoxia. (D) Reporter assay of the SLC2A3 promoter in combination with enhancer 1 and enhancer 2 under hypoxia. Enhancer 2-luc in combination with enhancer 1 was transiently transfected into HUVECs and assayed for luciferase activity. The combination with enhancer 2-luc upregulated luciferase activity both with and without mutation 4 (mut 4) in enhancer 1. The experiments were performed three times independently. *, P < 0.001 compared with the activity from the core promoter-luc under normoxia; #, P < 0.05. (E) Reporter assay results with the SLC2A3 promoter in combination with enhancer 1 and enhancer 2 in HEK293 cells overexpressing HIF1. The experiments were performed three times independently. *, P < 0.001 compared with activity from the core promoter-luc under normoxia; #1, P < 0.05 compared with activity from the E1-luc-vector under hypoxia; #2, P < 0.05 compared with activity from the E1 (mut 4)-luc vector under hypoxia; #3, P < 0.05 compared with activity from the E2-E1-luc vector under hypoxia. (F) Long-range interactions between the SLC2A3 promoter and enhancer, determined using 3C-ChIP. The vertical black bars represent each Csp6I fragment at the corresponding region. The numbers depict the distance (in kbp) from the TSS. 3C products were amplified by specific primers of the SLC2A3 loci. Bacterial artificial chromosomes were used as positive controls for the specific primers in the 3C-ChIP. The site at kbp +91 served as a negative control. The experiments were performed three times independently.
The results of the HIF1 ChIP-seq analysis provided data of hypoxic enhancer 2 (kbp −24 from the TSS) at the SLC2A3 loci (Fig. 5B). HIF1 and H3K27ac were found to be significantly enriched at the site of enhancer 2 by ChIP-PCR (Fig. 6C). Next, we examined whether HIF1 binding to distal enhancer 2 had transcriptional activity under hypoxia. We isolated fragments from −24862 to −23362 relative to the TSS (Fig. 5E) and cloned them into pGL3. One RCGTG motif (−24668) was included in enhancer 2. We made reporter constructs in which enhancer 2 and enhancer 1 were closely linked (E2-E1-luc) (Fig. 6D). Distal enhancer 2 (E2-E1-luc) exhibited a 2-fold increase in luciferase activity under hypoxia compared with enhancer 1 (E1-luc). When both of the RCGTG motifs in enhancer 1 and enhancer 2 were mutated [E2 (mut)-E1 (mut 4)-luc], luciferase activity was significantly reduced (66% reduction) under hypoxia compared with the E2-E1-luc vector. To ensure that HIF1 had direct transcriptional activity within E2, we overexpressed HIF1 in HEK293 cells. The combination vector (E2-E1-luc) resulted in a 1.2-fold increase in the luciferase activity compared with E1-luc, while mutation of both RCGTG motifs in enhancer 1 and enhancer 2 [E2 (mut)-E1 (mut 4)-luc] led to a 61% reduction of luciferase activity under the condition of overexpression of HIF1 (Fig. 6E). These results suggest that distal HIF1 binding to enhancer 2 is required for full-blown transcriptional activity under hypoxia.
HIF1-mediated higher-order chromatin conformational change induces SLC2A3 expression.
The experimental data described above showed that distal enhancer 2 was functionally active under hypoxic conditions, suggesting that enhancer 2 has an especially close proximity to the promoter of SLC2A3 under hypoxic conditions. Therefore, we tried to examine whether hypoxia changes chromatin conformation three-dimensionally at the hypoxia-responsive gene loci. We performed chromatin conformation capture combined with ChIP (3C-ChIP) assays (33). To compare the HIF1-dependent chromatin structure between normoxic and hypoxic conditions by 3C-ChIP assay, we picked 5 different sites (kbp −36, −35, and −24, the TSS, and kbp +91) near the SLC2A3 locus. As shown in Fig. 6F, the 3C-ChIP product allowed PCR amplification not only under normoxia but also under hypoxia for enhancer 1 and TSS. Thus, the interaction of enhancer 1 and TSS was established under both normoxia and hypoxia, while the enhancer 1-enhancer 2 interaction was observed only under hypoxia. Because 3C-ChIP products were immunoprecipitated by HIF1α antibody, this result demonstrated that the chromatin conformation at the SLC2A3 loci was changed by the complex, including HIF1 under hypoxia.
KDM3A regulates SLC2A3 gene expression by interacting with HIF1.
Since SLC2A3 is one of the HIF1 and KDM3A downstream target genes (Fig. 5A), we examined KDM3A enrichment at SLC2A3 loci by ChIP-PCR with a KDM3A antibody. KDM3A was recruited to the TSS, enhancer 1, and enhancer 2 under hypoxia, and KDM3A recruitment to these loci was reduced when HIF1α was knocked down by siRNA (Fig. 7A). These results suggest that KDM3A is essential to regulate SLC2A3 gene expression under hypoxia and that the recruitment of KDM3A to these sites is dependent on HIF1 occupancy.
Fig 7.

KDM3A interacts with HIF1 and is recruited to the SLC2A3 loci. (A) ChIP-PCR of KDM3A at the TSS, enhancer 1, and enhancer 2 showed enrichment of KDM3A compared with the negative-control region (kbp +91). The experiments were performed three times independently. *, P < 0.05 compared with control siRNA under normoxia; #, P < 0.05 compared with control siRNA under hypoxia; N.S., nonsignificant. (B) ChIP-PCR results for H3K9me2 at the SLC2A3 loci. The experiments were performed three times independently. *, P < 0.05 compared with normoxia. (C) ChIP-PCR results for H3K9me2 at the SLC2A3 loci when KDM3A was knocked down by siRNA. The experiments were performed three times independently. N.S., nonsignificant. (D) ChIP-PCR results of H3K9me2 at the SLC2A3 loci when HIF1α was knocked down by siRNA. The experiments were performed three times independently. N.S., nonsignificant. (E) HUVECs were immunoprecipitated with an HIF1α antibody or rabbit IgG and subsequently immunoblotted (IB) with a KDM3A antibody. The data are representative of three independent experiments. (F) Whole-cell lysates of HEK293 cells cultured under normoxia or hypoxia were untreated (input) or immunoprecipitated with a HIF1α antibody and subsequently immunoblotted with a KDM3A antibody. The experiments were performed three times independently. (G) The combination of either pFlag or pcDNA3 or that of pFlag-HIF1 or pcDNA-KDM3A was cotransfected into HeLa cells under normoxia. Extracted whole-cell lysates from the cells were untreated (input) or precipitated with HIF1α, KDM3A antibody, or control IgG and subsequently immunoblotted with the KDM3A antibody.
To clarify the function of KDM3A at the SLC2A3 loci, we performed ChIP-PCR with a dimethyl histone 3 lysine 9 (H3K9me2) antibody, because KDM3A was shown to demethylate H3K9 (for the most part H3K9me2) (42). Under hypoxia, H3K9me2 was reduced at the loci of the TSS and the distal enhancer 1 and enhancer 2 regions (Fig. 7B). The reduction of H3K9me2 under hypoxia did not occur when KDM3A was knocked down by siRNA (Fig. 7C), demonstrating that KDM3A demethylates the repressive histone H3K9me2 and thus activates SLC2A3 expression. In addition, the decrease of H3K9me2 under hypoxia was also absent when HIF1α was knocked down, suggesting that KDM3A demethylates H3K9me2 at SLC2A3 loci only when HIF1 binds to these sites (Fig. 7D). These data are consistent with the findings described above that indicated HIF1 is essential for KDM3A recruitment.
In order to confirm that HIF1 and KDM3A do in fact interact with each other under hypoxia, we performed coimmunoprecipitation using HUVECs. We observed immunoprecipitation of endogenous KDM3A by an HIF1 antibody under hypoxia (Fig. 7E). We also confirmed the interaction between KDM3A and HIF1 under hypoxia in HEK293 cells (Fig. 7F). In support of these observations, KDM3A was immunoprecipitated by the HIF1 antibody when both HIF1 and KDM3A were overexpressed in HeLa cells under normoxia (Fig. 7G). These results suggested that HIF1 makes a complex that contains KDM3A under hypoxic conditions, enabling the regulation of robust SLC2A3 expression by a coordinated recruitment of the complex to the enhancer regions of SLC2A3.
HIF1 binds to the RCGTG motif and retains the chromatin conformation under normoxia.
We found that HIF1 also binds 575 regions under normoxia, and our Western blotting detected HIF1 in the nucleus not only under hypoxia but also under normoxia (Fig. 1A). For example, HIF1 ChIP-seq showed that HIF1 binds to the promoter regions of NR2F1 (nuclear receptor subfamily 2, group F, member 1) and FLI1 (Friend leukemia virus integration 1) under normoxia (Fig. 8A). We confirmed the specificity of our observation by ChIP-PCR analysis, which demonstrated a significant reduction of the HIF1 binding levels to these promoters under normoxia when HIF1α was knocked down by siRNA (NR2F1, 86% reduction; FLI1, 85% reduction) (Fig. 8A). These regions contained consensus RCGTG motifs (Fig. 8B), and microarray analysis showed that both genes were expressed in HUVECs under normoxia (the average difference for the NR2F1 probe [209505_at] was 210.4, and for the FLI1 probe [210786_s_at] it was 823.7).
Fig 8.

HIF1 binds to the TSS and enhancer 1 (kbp −35) under normoxia and maintains the chromatin conformation. (A) Representative HIF1 binding sites and ChIP-PCR validations under normoxia (NR2F1 and FLI1). The promoter regions of NR2F1 and FLI1 were validated by ChIP-PCR to confirm HIF1 binding under normoxia. When HIF1α was knocked down by siRNA, the enrichment of HIF1 at the TSS of NR2F1 and FLI1 was significantly reduced. *, P < 0.05 compared with control siRNA. The experiments were performed three times independently. (B) HIF1 binding RCGTG motif in the NR2F1 and FLI1 promoters. (C) ChIP-PCR validation of the HIF1 binding sites under normoxia at the SLC2A3 loci. TSS and enhancer 1 (kbp −35) were validated by ChIP-PCR to confirm HIF1 binding under normoxia. When HIF1α was knocked down by siRNA, the enrichment of HIF1 at the TSS and enhancer 1 (kbp −35) was significantly reduced under normoxia. *, P < 0.05. The experiments were performed three times independently. (D) Reporter assay of the SLC2A3 enhancer 1 (kbp −35) in combination with the pGL3-promoter vector under normoxia. Enhancer 1 (kbp −35)-luc and enhancer 1 (kbp −35) with a mutation in motif 4 (mut 4) were transiently transfected into HUVECs and assayed for luciferase activity. *, P < 0.001 compared with the activity from pGL3 promoter-luc under normoxia. The experiments were performed three times independently. (E) Long-range interactions between the SLC2A3 promoter and enhancer as measured with 3C-ChIP. The vertical black bars represent each Csp6I fragment at target sites. The numbers depict the distance (in kbp) from the TSS. The 3C-ChIP assay was conducted in HUVECs under normoxia. The interaction of enhancer 1 (kbp −35) and the TSS under normoxia disappeared when HIF1α was knocked down by siRNA. The site at kbp +91 served as a negative control. The experiments were performed three times independently.
To clarify the function of HIF1 binding under normoxia, we focused on HIF1 binding at the SLC2A3 loci. Microarray analysis showed that SLC2A3 was expressed in HUVECs at a relatively low level under normoxia (the average difference of SLC2A3 probe [202497_x_at] ratios was 129.9), and HIF1 ChIP-seq revealed the normoxic binding of HIF1 to the TSS and kbp −35 regions (Fig. 5B). This enrichment of ChIP-PCR was significantly reduced by treatment with HIF1α siRNA (Fig. 8C). To examine whether RCGTG motif 4 in the kbp −35 region (E1-mut 4) had transcriptional activity not only under hypoxia (Fig. 5F) but also under normoxia, we performed a luciferase reporter assay. Mutation of the RCGTG motif 4 in the enhancer 1 led to a 76% reduction of transcriptional activity by HIF1 under normoxia (Fig. 8D). As shown above, 3C-ChIP under normoxia revealed that the kbp −35 region is in close proximity to the TSS under both normoxia and hypoxia (Fig. 6F). We additionally confirmed that this 3C-ChIP product was abolished by treatment with HIF1α siRNA under normoxia (Fig. 8E). This result indicated that HIF1 is necessary to keep the close proximity of chromatin conformation at the kbp −35 regions of SLC2A3 loci. Taking all of the findings together, under normoxia, the kbp −35 region functions as an enhancer to induce the expression of SLC2A3 at a relatively low level and retains chromatin conformation also under normoxia.
DISCUSSION
Genome-wide approaches to determine both transcription factor binding and functional regions have been reported for endothelial cells (16, 37). In this paper, we analyzed the HIF1 occupancy in HUVECs under hypoxic stimuli by ChIP-seq and demonstrated that HIF1 binds not only to the TSS, but also to the intergenic regions (Fig. 1C), with active histone marks (Fig. 5B). We found that more than 60% of the HIF1 binding sites were covered by the enhancer histone mark H3K27ac and that H3K27ac was increased under hypoxia at the loci of HIF1 binding regions (Fig. 6B). Although previous studies have reported HIF1 binding in intergenic regions (40), the biological significance of these intergenic binding regions has remained unclear. Here we showed that HIF1 binds to the kbp −35 region (normoxia and hypoxia) and the kbp −24 region (hypoxia) within the SLC2A3 loci, regions which have HIF1-dependent transcriptional activities (Fig. 5 and 6). These results suggest that a comprehensive analysis may be able to determine the hypoxia response regions corresponding to changes in histone modification on a genome-wide scale.
As HIF1 binding regions in intergenic regions were functionally active, we speculated that they would contribute to the chromatin conformational changes that occur in response to hypoxia. Recent technological advances have allowed analysis of the chromatin conformation in living cells by using the 3C assay (5, 41). In this paper, a 3C-ChIP assay showed that the TSS and enhancer 1 (kbp −35) region of SLC2A3 are closely similar in terms of chromatin structure under normoxia, and this structure changed with the proximity of the enhancer 2 (kbp −24) region to the TSS-enhancer 1 conformation under hypoxic conditions (Fig. 6F). Some reports have suggested that regulatory elements can act over large genomic distances (8). This is the first work that has shown that not only hypoxia-induced change in HIF1 binding but also changes to the chromatin conformation accompany histone modifications in a HIF-dependent manner.
We demonstrated that one of the histone demethylases, KDM3A, was recruited to the SLC2A3 gene loci (Fig. 7A). Previous studies reported the development of obesity and hyperlipidemia in KDM3A knockout mice, suggesting that KDM3A may regulate metabolic gene expression (12). Our observation that KDM3A is one of the HIF1 target genes is consistent with recent reports for cancer cell lines (3, 25, 39). Here, we demonstrated for the first time that KDM3A interacts with HIF1 and is recruited to the distal enhancers of SLC2A3 loci under hypoxic conditions, subsequently changing the histone modifications and chromatin conformation (Fig. 7). We propose a new regulatory mechanism of KDM3A under hypoxia, which induces a robust expression in hypoxia-responsive genes along with HIF1. Interestingly, KDM3A is induced under hypoxia in both HUVECs and MCF7 cells. Therefore, we speculate that a KDM3A-HIF1 interaction plays an essential role in the response to hypoxia in various cell types, because most of the genes downstream of KDM3A and HIF1 belong to the “response to hypoxia” and “glycolysis” categories (Fig. 3C). We also demonstrated that the early hypoxia-responsive genes in group I (Fig. 3B) are commonly regulated by both HIF1 and KDM3A. These findings suggest that hypoxia-responsive histone modifications induced by histone-modifying enzymes, such as KDM3A, and subsequent genome-wide chromatin modifications are necessary for cell survival in the early period during hypoxia.
Furthermore, we found that HIF1 also binds 575 regions under normoxia and that HIF1 exists not only under hypoxia but also under normoxia in the nucleus (Fig. 1A). For example, HIF1 binds to the promoter regions of NR2F1 and FLI1 under normoxia (Fig. 8A). These regions contain consensus RCGTG motifs (Fig. 8B), and ChIP-PCR revealed binding signals that were reduced when HIF1α was knocked down by siRNA under normoxia. These findings verified that the HIF1 ChIP-seq signals under normoxia are specific.
While previous studies have mostly focused on the role of HIF1 under hypoxic conditions, recent reports have suggested a biological role for HIF1 under normoxia in endothelial cells (7) and cancer cells (2). Real-time imaging studies have shown that HIF1α undergoes biexponential clearance under normoxic conditions, as it is readily separated into rapid (mean t1/2, 4 to 6 min) and slow (mean t1/2, ∼200 min) kinetic phase components (23), and a small amount of HIF1α exists in the cell during a certain period of time. Although SLC2A3 is robustly expressed under hypoxia, it is also transcribed under normoxia, and HIF1 ChIP-seq revealed the normoxic binding of HIF1 to the TSS and kbp −35 regions. This enrichment of ChIP-PCR was profoundly reduced by treatment with HIF1α siRNA (Fig. 8C). Luciferase reporter analysis demonstrated that RCGTG motif 4 in the kbp −35 region has transcriptional activity under normoxia (Fig. 8D), and 3C-ChIP under normoxia revealed that the kbp −35 region is in close proximity to the TSS under both normoxia and hypoxia (Fig. 6F). This 3C-ChIP product was abolished by treatment with HIF1α siRNA (Fig. 8E). Taking all of the findings together, under normoxia, the kbp −35 region functions as an enhancer to induce the expression of SLC2A3 at a relatively low level.
In this report, we analyzed the comprehensive ChIP-seq data for HUVECs and MCF7 cells. While the commonly bound genes were classified as early response genes, which were functional in glycolysis and the response to hypoxia, cell-type-specific binding genes had cell-type-specific functions, such as angiogenesis, in endothelial cells (Fig. 3E). These results suggested that HIF1 binds to genes with fundamental functions in the early phase of hypoxia, independently of cell type. In contrast, late response genes were related to cell-type-specific functions (Fig. 3F). The differences in the HIF1-bound genes between HUVECs and MCF7 cells may be attributable to epigenetic differences between the cell types. In this paper, we found that the regions to which HIF1 is recruited under hypoxia have active histone marks, even under normoxia (Fig. 5B), and so the late response genes may be epigenetically prepared for hypoxic stimuli in a cell-type-specific manner.
In order to investigate the regulatory mechanisms of gene expression by HIF1 in detail, we focused on SLC2A3, because it is the target gene of both HIF1 and KDM3A. The function of SLC2A3 belongs to the metabolic category (32), metabolism being a common response to hypoxia in the various cell types. Glucose is transported across the lipid bilayer of the cell membrane by glucose transporters, including SLC2A3. SLC2A3 is expressed in a variety of cell types (1, 17), and here the knockdown of SLC2A3 siRNA was shown to suppress glucose uptake, glucose consumption, and matrix tube formation in HUVECs. A novel finding is that the HIF1-SLC2A3 axis is necessary for endothelial function, which suggests that one of the angiogenic mechanisms induced by HIF1 is related to glucose metabolism.
In summary, the data suggest that there is a novel mechanism of epigenetic regulation of SLC2A3 loci under both normoxia and hypoxia in HUVECs (Fig. 9). Under normoxia, HIF1 binds to the TSS, and enhancer 1 (kbp −35) is conformationally close to the TSS. Under hypoxia, enhancer 2 (kbp −24) comes into proximity with both the TSS and enhancer 1 via hypoxia-responsive HIF1 binding. Upon chromatin conformational changes, KDM3A is recruited to SLC2A3 in an HIF1-dependent manner, by which KDM3A demethylates H3K9me2 and reduces the repressive histone marks. These changes are associated with an increase in the enhancer H3K27ac and result in the robust upregulation of SLC2A3.
Fig 9.
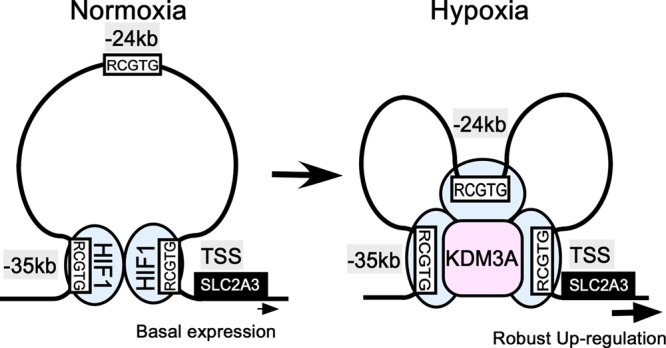
Schematic of the regulation of SLC2A3 expression under hypoxia. Under normoxia, the enhancer 1 (kbp −35) region is structurally close to the TSS bound by HIF1. Under hypoxia, HIF1 binding to enhancer 2 (kbp −24) increases, and KDM3A is recruited via interaction with HIF1, followed by subsequent chromatin structural changes. Finally, the expression of SLC2A3 is robustly upregulated under hypoxia.
Supplementary Material
ACKNOWLEDGMENTS
I.M. is a Research Fellow of the Japan Society for the Promotion of Science. This research was supported by a grant from the Japan Society for the Promotion of Science (JSPS) through the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program), initiated by the Council for Science and Technology Policy (CSTP) (T.K.). This work was also supported by Grants-in-Aid for Scientific Research (B) 22310117 (Y.W.) and 21390260 and 24390213 (M.N.) and Grant-in-Aid for Exploratory Research 23659050 (Y.W.) from the Japan Society for the Promotion of Science and also Grant-in-Aid for Scientific Research on Innovative Areas 23125503 (Y.W.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
We are grateful to Hiroshi Kimura (Osaka University) for kindly gifted histone antibodies and Genta Nagae, Takayuki Isagawa, Aya Nonaka, Satoshi Goda, and Kaori Shina (The University of Tokyo) for technical assistance on ChIP-seq and microarrays. Pacific Edit reviewed the manuscript prior to submission.
I.M., M.N., Y.K., T.I., A.I., M.K., T.K., and Y.W. performed and designed the experiments; I.M., M.N., Y.K., T.S., T.K., S.Y., T.F., T.S., and T.K. analyzed the data; I.M., Y.K., Y.W., M.N., and T.K. wrote the paper.
Footnotes
Published ahead of print 29 May 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Baumann MU, Zamudio S, Illsley NP. 2007. Hypoxic upregulation of glucose transporters in BeWo choriocarcinoma cells is mediated by hypoxia-inducible factor-1. Am. J. Physiol. Cell Physiol. 293:C477–C485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berra E, et al. 2003. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 22:4082–4090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. 2008. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 283:36542–36552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Calvani M, Trisciuoglio D, Bergamaschi C, Shoemaker RH, Melillo G. 2008. Differential involvement of vascular endothelial growth factor in the survival of hypoxic colon cancer cells. Cancer Res. 68:285–291 [DOI] [PubMed] [Google Scholar]
- 5. Dekker J, Rippe K, Dekker M, Kleckner N. 2002. Capturing chromosome conformation. Science 295:1306–1311 [DOI] [PubMed] [Google Scholar]
- 6. Du R, et al. 2008. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13:206–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fijalkowska I, et al. 2010. Hypoxia inducible-factor1α regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am. J. Pathol. 176:1130–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fullwood MJ, et al. 2009. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462:58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heintzman ND, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459:108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoog C, Schalling M, Grunder-Brundell E, Daneholt B. 1991. Analysis of a murine male germ cell-specific transcript that encodes a putative zinc finger protein. Mol. Reprod. Dev. 30:173–181 [DOI] [PubMed] [Google Scholar]
- 11. Huang LE, Gu J, Schau M, Bunn HF. 1998. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. U. S. A. 95:7987–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Inagaki T, et al. 2009. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 14:991–1001 [DOI] [PubMed] [Google Scholar]
- 13. Ivan M, et al. 2001. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468 [DOI] [PubMed] [Google Scholar]
- 14. Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. 1996. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 271:17771–17778 [DOI] [PubMed] [Google Scholar]
- 15. Kaelin WG, Jr, Ratcliffe PJ. 2008. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30:393–402 [DOI] [PubMed] [Google Scholar]
- 16. Kanki Y, et al. 2011. Epigenetically coordinated GATA2 binding is necessary for endothelium-specific endomucin expression. EMBO J. 30:2582–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kayano T, et al. 1988. Evidence for a family of human glucose transporter-like proteins. Sequence and gene localization of a protein expressed in fetal skeletal muscle and other tissues. J. Biol. Chem. 263:15245–15248 [PubMed] [Google Scholar]
- 17a. Kimura H, Hayashi-Takanaka Y, Goto Y, Takizawa N, Nozaki N. 2008. The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct. Funct. 33:61–73 [DOI] [PubMed] [Google Scholar]
- 18. Majmundar AJ, Wong WJ, Simon MC. 2010. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40:294–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manalo DJ, et al. 2005. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105:659–669 [DOI] [PubMed] [Google Scholar]
- 20. Mimura I, Nangaku M. 2010. The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 6:667–678 [DOI] [PubMed] [Google Scholar]
- 21. Mimura I, Tanaka T, Wada Y, Kodama T, Nangaku M. 2011. Pathophysiological response to hypoxia. From the molecular mechanisms of malady to drug discovery: epigenetic regulation of the hypoxic response via hypoxia-inducible factor and histone-modifying enzymes. J. Pharmacol. Sci. 115:453–458 [DOI] [PubMed] [Google Scholar]
- 22. Mole DR, et al. 2009. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 284:16767–16775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moroz E, et al. 2009. Real-time imaging of HIF-1α stabilization and degradation. PLoS One 4:e5077 doi:10.1371/journal.pone.0005077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Okada Y, Scott G, Ray MK, Mishina Y, Zhang Y. 2007. Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature 450:119–123 [DOI] [PubMed] [Google Scholar]
- 25. Pollard PJ, et al. 2008. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1α. Biochem. J. 416:387–394 [DOI] [PubMed] [Google Scholar]
- 26. Pouyssegur J, Dayan F, Mazure NM. 2006. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441:437–443 [DOI] [PubMed] [Google Scholar]
- 27. Rada-Iglesias A, et al. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470:279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schodel J, et al. 2011. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117:e207–e217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schofield CJ, Ratcliffe PJ. 2004. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5:343–354 [DOI] [PubMed] [Google Scholar]
- 30. Semenza GL. 2011. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 365:537–547 [DOI] [PubMed] [Google Scholar]
- 31. Semenza GL. 2003. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3:721–732 [DOI] [PubMed] [Google Scholar]
- 32. Semenza GL, Roth PH, Fang HM, Wang GL. 1994. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269:23757–23763 [PubMed] [Google Scholar]
- 33. Simonis M, Kooren J, de Laat W. 2007. An evaluation of 3C-based methods to capture DNA interactions. Nat. Methods 4:895–901 [DOI] [PubMed] [Google Scholar]
- 34. Simpson IA, et al. 2008. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 295:E242–E253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Subramanian A, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102:15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tateishi K, Okada Y, Kallin EM, Zhang Y. 2009. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458:757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tozawa H, et al. 2011. Genome-wide approaches reveal functional interleukin-4-inducible STAT6 binding to the vascular cell adhesion molecule 1 promoter. Mol. Cell. Biol. 31:2196–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valouev A, et al. 2008. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods 5:829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wellmann S, et al. 2008. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem. Biophys. Res. Commun. 372:892–897 [DOI] [PubMed] [Google Scholar]
- 40. Xia X, et al. 2009. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. U. S. A. 106:4260–4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu J, et al. 2010. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 24:783–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamane K, et al. 2006. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125:483–495 [DOI] [PubMed] [Google Scholar]
- 43. Yang J, et al. 2009. Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann. N. Y. Acad. Sci. 1177:185–197 [DOI] [PubMed] [Google Scholar]
- 44. Zentner GE, Tesar PJ, Scacheri PC. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 21:1273–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.