

Abstract
The low-density lipoprotein receptor-related protein 1 (LRP-1) is a large endocytic receptor mediating the clearance of various molecules from the extracellular matrix. In the field of cancer, LRP-1-mediated endocytosis was first associated with antitumor properties. However, recent results suggested that LRP-1 may coordinate the adhesion-deadhesion balance in malignant cells to support tumor progression. Here, we observed that LRP-1 silencing or RAP (receptor-associated protein) treatment led to accumulation of CD44 at the tumor cell surface. Moreover, we evidenced a tight interaction between CD44 and LRP-1, not exclusively localized in lipid rafts. Overexpression of LRP-1-derived minireceptors indicated that the fourth ligand-binding cluster of LRP-1 is required to bind CD44. Labeling of CD44 with EEA1 and LAMP-1 showed that internalized CD44 is routed through early endosomes toward lysosomes in a LRP-1-dependent pathway. LRP-1-mediated internalization of CD44 was highly reduced under hyperosmotic conditions but poorly affected by membrane cholesterol depletion, revealing that it proceeds mostly via clathrin-coated pits. Finally, we demonstrated that CD44 silencing abolishes RAP-induced tumor cell attachment, revealing that cell surface accumulation of CD44 under LRP-1 blockade is mainly responsible for the stimulation of tumor cell adhesion. Altogether, our data shed light on the LRP-1-mediated internalization of CD44 that appeared critical to define the adhesive properties of tumor cells.
INTRODUCTION
The low-density lipoprotein receptor-related protein 1 (LRP-1) is a large multifunctional endocytic receptor, composed of a large extracellular ligand-binding subunit (515-kDa α-chain) noncovalently linked to a transmembrane subunit encompassing a short cytoplasmic tail (85-kDa β-chain) (21). The extracellular domain contains four ligand-binding clusters that mediate the binding of various ligands associated with the extracellular matrix (ECM), including lipoproteins, ECM macromolecules, proteinases, and proteinase-inhibitor complexes. LRP-1-mediated endocytosis of soluble ligands is usually followed by intracellular lysosomal routing and catabolism. Thus, LRP-1 is now recognized as a main regulator of extracellular proteolytic cascades involved in tumor progression (14, 15, 50). Beyond internalization and lysosome delivery, ligands binding to LRP-1 may also induce phosphorylation of residues within its intracellular domain. This is crucial not only for initiation and regulation of endocytosis but also for binding of intracellular molecular adaptors involved in signal transduction (19, 30, 47).
LRP-1 is able to interact with other membrane-anchored proteins, including the amyloid protein precursor, the urokinase-type plasminogen activator receptor (uPAR), the platelet-derived growth factor receptor β (PDGFR-β), and some integrins, to regulate their trafficking and the associated intracellular signals (3, 6, 10, 37, 46, 51). This leads to a variety of cellular responses, such as modulating blood-brain-barrier integrity (60), contributing to vasculature protection (3–5), or regulating cellular migration under various physiopathological conditions (7, 8, 12, 29). In fibrosarcoma cells, neutralizing LRP-1 led to increased extracellular signal-regulated kinase (ERK) activity, which stimulates cell migration and invasion (57). The ability of LRP-1 to bind the cell surface uPA/uPAR system is frequently evoked to explain the control of ERK phosphorylation (32, 57, 62). Blockade of LRP-1-mediated internalization of uPAR could indeed strengthen the assembly of an active uPAR-integrin signaling complex, affecting the subsequent intracellular signals. The ability of LRP-1 to allow β1-integrin recruitment and the following stimulation of integrin-linked kinases may control the actin cytoskeleton dynamics during retraction of migrating cells (24). Cao and colleagues have proposed an exciting molecular model in macrophages in which LRP-1 facilitates cell detachment at the trailing edge by mediating the internalization of integrin-containing adhesion complexes (7). Cooperation at the cell surface between LRP-1 and β2-integrin was also reported to mediate the adhesion of leukocytes (46, 51). The main distribution of LRP-1 at the leading edge and at the rear of the cell could therefore orchestrate cell polarization and support directional migration of various cell types (7, 8). LRP-1 was proposed to sustain the contractile activity of fibroblasts by activating the myosin light-chain kinase by an ERK-dependent mechanism (53) and was involved in focal adhesion disassembly in response to thrombospondin through FAK-dependent signaling and RhoA inactivation (43). Besides, we have previously demonstrated that LRP-1 is required for both FAK and paxillin targeting into focal contacts to support migration of thyroid carcinoma cells (12). Furthermore, our recent data highlighted that LRP-1 contributes to maintaining malignant cells in an adhesive state favorable to tumor invasion by controlling both ERK- and c-Jun N-terminal protein kinase (JNK)-dependent pathways (28).
LRP-1 henceforth emerges as an endocytic receptor regulating cellular matrix attachment sites and coordinating the adhesion-deadhesion balance. However, the small number of transmembrane proteins identified as being associated with LRP-1 in a tumor context is insufficient to understand how LRP-1 controls cell-matrix interactions.
Considering the ability of the hyaluronan receptor CD44 to modulate cell adhesion, migration, and tumor progression (13, 34, 41), we investigated in this study whether this adhesion receptor may constitute a new cell surface partner for LRP-1 to control adhesion dynamics of cancer cells. Our data demonstrate for the first time that LRP-1 binds to CD44 at the plasma membrane and reveal that the LRP-1-mediated uptake of CD44 contributes to regulating carcinoma cell attachment to the ECM.
MATERIALS AND METHODS
Antibodies and chemicals.
Anti-LRP-1 α-chain (mouse, clone 8G1), anti-LRP-1 β-chain (mouse, clone 5A6), monoclonal anti-CD44 (rat, A020), monoclonal anti-receptor-associated protein (anti-RAP) (mouse, clone 7F1), and nonreactive IgGs used as a negative control for immunoprecipitation (HP6030) were obtained from Merck Biosciences (distributed by VWR International, Strasbourg, France). Antibodies raised against EEA-1 (ab2900), lysosome-associated membrane protein 1 (LAMP-1) (ab24170), CD44 (F10-44-2), or the transferrin receptor (ab8598) were purchased from Abcam (Paris, France) and used for immunoblotting. Anti-PTRF (rabbit, A301-269A) was from Bethyl Laboratories (distributed by Euromedex, Souffelweyersheim, France), and anti-caveolin-1 (rabbit, 3238) was obtained from Cell Signaling Technology (distributed by Ozyme, Montigny-Le-Bretonneux, France). Horseradish peroxidase (HRP)-conjugated anti-rabbit (7074) and anti-mouse (NA931V) antibodies were from Cell Signaling Technology and Amersham Biosciences (GE Healthcare Europe, Velizy-Villacoublay, France), respectively. Anti-rat (sc2006) and anti-β-actin (sc-1616) antibodies were from Santa Cruz Biotechnology (distributed by Tebu-Bio, Le Perray en Yvelines, France). Alexa Fluor 488, Alexa Fluor 568-phalloidin, and Prolong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (P36935) were from Molecular Probes (distributed by Invitrogen, Cergy Pontoise, France). Human α2 M and fluorescein isothiocyanate (FITC)-labeled human α2 M were purchased from BioMac (Leipzig, Germany). EZ-Link sulfo-NHS-LC-biotin, d-biotin, and monomeric avidin-agarose beads were obtained from Thermo Fisher Scientific (Illkirch, France). Methyl-β-cyclodextrin (MβCD), dynasore (D7693), anti hemagglutinin (anti-HA) tag antibody (mouse, clone HA-7) and other chemicals were from Sigma-Aldrich (Saint Quentin Fallavier, France).
Plasmid constructs and silencing sequences.
All the coding regions for human LRP-1 (hLRP-1) were amplified by PCR from human brain cDNA. First, the hLRP-1 signal peptide and an HA tag were amplified using the following primer pairs (boldface letters indicate restriction sites): SP-LRP-1-F, ATATATCTCGAGATGCTGACCCCGCCGTTGCTC; SP-LRP-1-R, TTAATTGGTACCAGCGTAGTCCGGGACGTCGTACGGGTAAGGGGCGTCGATAGC. The PCR product was cloned in the pEGFP-N1 vector (Clontech laboratories, Mountain View, USA) using the XhoI and KpnI restriction sites inserted in the primer sequences, and the plasmid construct was named SP-pEGFPN1. Subsequently, the coding region for hLRP-1 β-chain was cloned using the following primers: CT-LRP-1-F, ATATATGGTACCATTAAGCTTGGCTCTGAGTACCAGGTCCTGTACATCG; CT-LRP-1-R, TTAATTCCGCGGTGCCAAGGGGTCCCCTATCTCGTCCT. The PCR product was cloned in frame in the SP-pEGFPN1 plasmid using the KpnI and SacII restriction sites. The corresponding plasmid construct was named SPCT (signal peptide carboxy-terminal). Finally, the coding regions for hLRP-1 D2 and D4 ligand binding clusters were amplified using the following primer pairs: D2-LRP-1-F, ATATATGGTACCCGGGTGAACAATGGCGGCT; D2-LRP-1-R, TTAATTAAGCTTCTGGTCGCACTTTTGGCTGCATT; D4-LRP-1-F, ATATATGGTACCTGCACGGCTAGCCAGTTTGTATGC; D4-LRP-1-R, TTAATTAAGCTTTTCGGCCTTGCAGGTGTTGT. The PCR products were cloned in frame in the SPCT plasmid using the HindIII and KpnI restriction sites, and the plasmid constructs were named mini-LRP-II and mini-LRP-IV, respectively. All the constructs were fully sequenced on both strands.
Cell surface expression of minireceptors was controlled by immunoblotting. Internalization assays were conducted to control their ability to mediate endocytosis by using FITC-labeled human α2 M. LRP-1 knockdown was achieved by RNA interference using a previously validated small interfering RNA (siRNA) approach (12). The transient knockdown of CD44 was performed using the ON-TARGET plus SMART pool siRNA against human CD44 (Dharmacon, distributed by Thermo Fisher Scientific). Nontargeting siRNA used as controls was also purchased from Dharmacon.
Purification of recombinant RAP.
Histidine-tagged RAP was expressed in Escherichia coli BL21 pLysS (Promega, Charbonnières-les-Bains, France) using the pT7H6FX-RAP construct kindly provided by M. S. Nielsen (Department of Medical Biochemistry, University of Aarhus, Denmark). Recombinant RAP was purified by gravity-flow chromatography using a nickel-charged resin (Ni-nitrilotriacetic acid [NTA]- agarose from Qiagen, Courtaboeuf, France) and then controlled after SDS-PAGE by Coomassie blue and immunoblotting using both anti-HA tag and anti-RAP antibodies. The LRP-1 binding capacity was confirmed using a BIAcore X system (BIAcore AB, Uppsala, Sweden), as already reported (49).
Cell culture and transfection.
The FTC-133 human follicular thyroid carcinoma cell line was grown in Dulbecco's modified Eagle medium–Ham's F-12 medium (Dutscher, Brumath, France) with 10% fetal bovine serum, as previously described (50). Plasmids were transiently transfected using Lipofectamine 2000 (Invitrogen), as already reported (12). siRNA sequences were transfected by the same method using the manufacturer's recommendations.
RNA isolation, RT-PCR, and real-time PCR.
Total RNAs were isolated and purified with an Extract-All kit (Eurobio Laboratories, Courtaboeuf, France). Reverse transcription (RT) and real-time PCR were performed with Verso SYBR 2-Step QRT Rox kit (AB-4113/A) and Absolute QPCR SYBR green Rox (AB-1162/B), respectively (Thermo Fisher Scientific). Quantitative PCR was carried out on a Chromo4 real-time detector (Bio-Rad Laboratories, Marne-la-Vallée, France), and β-actin was used for normalization. Primers for LRP-1 (12) and β-actin (28) were previously described. Primers for CD44 were synthesized as follows: CCACATTCTGCAGGTTCCTT and GTGATCAACAGTGGCAATGG (only standard CD44) and ACACATATTGCTTCAATGCTTCAGC and GATGCCAAGATGATCAGCCATTCTGGA (standard CD44s, 482 bp; CD44 variants, >482 bp). Detection of standard CD44 and CD44 variants was performed as reported elsewhere (1). All primers were synthesized by Eurogentec France (Angers, France). Results shown are representative of three independent experiments.
Protein extraction and Western blot analysis.
Whole-cell extracts were prepared by scraping cells in ice-cold lysis buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 5 mM EDTA, 0.5 mM EGTA, 1 mM phenylmethylsulfonylfluoride (PMSF), and 1 mM Na3VO4 supplemented with proteinase inhibitor cocktail from Sigma-Aldrich). After sonication, the remaining pellet was separated by centrifugation (10,000 × g for 10 min at 4°C) and discarded. The protein concentration was quantified by the Bradford method (Bio-Rad Laboratories). Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto a nitrocellulose membrane (Amersham Biosciences), and incubated overnight at 4°C under gentle agitation with anti-LRP-1 (1/1000; 5A6 or 8G1), anti-CD44 (1/1, 000; A020), anti-PTRF (1/2,000), anti-caveolin-1 (1/500), or anti-transferrin receptor (1/100). Membranes were then incubated for 1 h at room temperature with the corresponding horseradish peroxidase-coupled antibody. Tris-buffered saline (TBS)-Tween buffer was used for all washes. Immunoreactive bands were revealed using the ECL Plus chemiluminescence kit (Amersham Biosciences) by using a ChemiDoc-XRS imaging station from Bio-Rad laboratories. Ponceau red staining solution and β-actin antibodies were used to ensure equal loading of the protein samples and for normalization. Immunoblots presented are representative of at least three independent experiments.
Membrane protein isolation by cell surface biotinylation.
FTC-133 cells were washed twice with ice-cold phosphate-buffered saline (PBS), and cell surface proteins were biotinylated with PBS containing 0.5 mg/ml of EZ-Link sulfo-NHS-LC-biotin for 30 min at 4°C. After three washes, cells were incubated with 100 mM glycine in PBS for 30 min at 4°C to limit nonspecific binding. Cells were washed three times before protein extraction in ice-cold lysis buffer as described above. Cell extracts were pelleted at 10,000 × g for 10 min at 4°C, and protein quantification was performed. Solubilized biotinylated proteins (750 μg) were then specifically affinity purified using 40 μl of monomeric avidin-agarose beads. Incubation was performed overnight at 4°C with gentle orbital agitation (5 rpm), followed by five washes with lysis buffer to remove nonspecific binding. For immunoblotting experiments, 40 μl of 2× SDS-containing Laemmli buffer was added, and samples were heated at 100°C during 5 min, centrifuged (10,000 × g for 10 min at 4°C), and resolved by SDS-PAGE followed by immunoblotting analysis.
Raft isolation.
Raft isolation was performed according to a previously validated detergent-free method by using discontinuous sucrose density gradient ultracentrifugation (44). Briefly, tumor cells were scrapped with a detergent-free lysis buffer (TBS [pH 8.1], 1 mM PMSF, 5 mM NaF, 1 mM MgCl2, 1 mM CaCl2, 1 mM Na3VO4, and proteinase inhibitor cocktail). The homogenate was sheared through a 20-gauge needle with 20 complete passes and then centrifuged at 1,000 × g for 10 min at 4°C. The supernatant volume was adjusted to 750 μl with TBS. All steps were performed at 4°C with precooled solution and tubes. Samples were mixed with an equal volume of 85% sucrose–TBS and loaded in SW40 ultracentrifugation tubes. Ten milliliters of 35% sucrose–TBS was carefully overlaid, followed by 2 ml of 5% sucrose–TBS. Tubes were centrifuged at 200,000 × g at 4°C for 18 h. Thirty sequential fractions of 1 ml (each) were gently removed from the top of the tube, aliquoted, and eventually stored at −80°C. Efficiency of raft isolation was checked by Western blot analysis using 50 μl of each fraction. Immunoprecipitations were performed using 300 μl of each fraction.
Cholesterol assay.
FTC-133 cells were cholesterol depleted by direct extraction with 5 mM MβCD in serum-free medium for different times. Cells were harvested in reaction buffer (0.1 M potassium phosphate, pH 7.4, 50 mM NaCl, 5 mM cholic acid, and 0.1% Triton X-100) and sonicated. Cholesterol content was then quantified by using the Amplex Red cholesterol assay kit (Invitrogen), as recommended by the manufacturer. Reactions proceeded for 20 min at 37°C.
Immunoprecipitation.
Whole-cell extracts or plasma membrane extracts (corresponding to cell surface biotinylated proteins) were subjected to immunoprecipitation assay with anti-LRP-1 (clone 8G1 or 5A6), anti-CD44 (F10-44-2), or nonspecific IgGs. For immunoprecipitation onto biotinylated proteins, a concentration of 10 mM D-biotin in PBS was first used for competitive elution of biotinylated proteins from avidin-agarose beads. Immunoprecipitation was performed with protein G-Sepharose (Amersham Biosciences) for 4 h at 4°C on an orbital agitator. Samples were washed three times in lysis buffer containing 150 mM or 300 mM NaCl for standard and high-stringency conditions, respectively. Then, protein complexes bound to beads were solubilized under nonreducing conditions and analyzed by immunoblotting as described above.
Endocytosis assays.
Internalization assays for FITC-labeled human α2 M were conducted as previously described (12). For endogenous CD44, internalization assays were conducted using a biochemical methodology adapted from the method of Wu and Gonias (59). Briefly, FTC-133 cells were transfected with siRNAs or treated with RAP for the corresponding times. Cells were then maintained on ice to prevent further endocytosis and washed three times with ice-cold PBS. Cell surface proteins were labeled with non-membrane-permeating sulfo-NHS-LC-biotin at 4°C for 30 min. Then, cells were incubated at 37°C to allow endocytosis. After 30 min to 1 h in complete medium (with or without RAP), cells were replaced on ice, washed three times, and incubated with or without pronase (1 mg/ml) during 10 min to remove surface-bound proteins. Cell extracts were prepared with lysis buffer and subjected to centrifugation (1,000 × g for 10 min). Biotinylated proteins were recovered by using avidin protein immobilized on agarose beads, subjected to SDS-PAGE, and revealed by immunoblotting. In pronase-treated samples, affinity-precipitated proteins with avidin-agarose were interpreted as being internalized, as previously reported (59). The efficiency of protein stripping mediated by pronase at the cell surface was controlled to be over 90%.
Immunofluorescence and confocal microscopy.
FTC-133 cells were seeded onto gelatin-coated glass slides for 4 h at 37°C and then fixed in 4% paraformaldehyde for 5 min at 4°C. After three washes in ice-cold PBS, cells were incubated for 1 h in PBS containing 3% bovine serum albumin and incubated for 45 min with primary antibodies for LRP-1 (8G1), CD44 (A020), EEA-1 (ab2900), or LAMP-1 (ab24170). Control preparations were incubated without primary antibody. Slides were washed five times in PBS, and cells were incubated with secondary antibodies conjugated to Alexa Fluor 488 (green) and Alexa Fluor 568 (red) for 45 min. Nuclei were counterstained with DAPI. Immunofluorescence-labeled cell preparations were analyzed using a Zeiss LSM 710 confocal laser scanning microscope with the 63× oil-immersion objective and Zeiss operating system associated with the ZEN software program (Carl Zeiss MicroImaging GmbH, Germany). Acquisitions were performed by exciting Alexa Fluor 488, Alexa Fluor 568, and DAPI dye with, respectively, an argon laser, a HeNe laser, and a chameleon infrared laser tuned at 730 nm. Emitted fluorescence was detected through the appropriate wavelength window. Twenty images were captured with a 0.25-μm z-step.
Isosurface representations were realized using the AMIRA software program (v5.2; Visage Imaging, Berlin, Germany). In AMIRA, the isosurface encloses all parts of a volume that are brighter than some user-defined threshold. To compare cell labeling, a same threshold was applied for each channel of RAP-treated cells or untreated cells. For colocalization studies, a multichannel field module was used, followed by a correlation plot treatment (subrange values of 15 to 255; gamma, 0.5). Then, an isosurface representation of the correlation plot was realized using the same threshold for each treatment. The distribution of voxels according to channels can be viewed in a two-dimensional scattergram, useful for estimating the degree of colocalization, as detailed elsewhere (64). The intracellular labeling was specifically observed with the multiplanar view module of AMIRA with a z position corresponding to the intracellular position.
Adhesion and trypsinization assays.
FTC-133 cell adhesion to hyaluronic acid (1 mg/ml) or gelatin-coated surfaces was measured as already described (12). Trypsinization assays were carried out as defined elsewhere (12, 58) by incubating cancer cells with 0.025% (wt/vol) trypsin for 10 min.
Densitometric analysis and statistical evaluation.
Culture assays were normalized on the basis of cell viability by using the CellTiter-Glo assay from Promega. Immunoreactive bands were analyzed using the densitometric PhosphorAnalyst software program (Bio-Rad laboratories). Each value is the mean ± standard deviation (SD) for at least three independent experiments, and data were expressed as means ± standard errors of the means (SEM). Comparisons were performed using Student's t test (Prism software program; GraphPad Inc., San Diego, CA).
RESULTS
FTC-133 carcinoma cell attachment is increased by both RAP treatment and LRP-1 silencing.
We recently characterized the intracellular molecular signaling relays involved in the LRP-1-mediated stimulation of cancer cell invasion (28). We thus identified the LRP-1 β-chain as a main docking site for mitogen-activated protein kinase-containing complexes and focal adhesion components in carcinoma. We now hypothesize that a cell surface partner may cooperate with LRP-1 to regulate cancer cell attachment to the ECM. Cell surface expression of LRP-1 was first assessed in FTC-133 cells (Fig. 1A). Differential interference contrast microscopy combined with confocal imaging highlights cell surface expression of LRP-1, especially at the migration front and at the rear of the cell (Fig. 1A, left panel and insets). Furthermore, cell surface protein biotinylation revealed that LRP-1 is highly expressed at the plasma membrane of FTC-133 cells (Fig. 1A, right panel). Its expression level was comparable to that of epithelioid HT1080 cells, a well-established cell model expressing cell surface LRP-1 at high levels (15, 48). For our purpose, two different strategies were used to inhibit LRP-1-mediated endocytosis: RAP treatment and LRP-1 silencing. The recombinant protein RAP was purified on a nickel-agarose column and controlled after SDS-PAGE by Coomassie blue staining and immunoblotting (Fig. 1B). LRP-1 silencing was conducted by using previously validated short interfering sequences (12) and led to 80% downregulation of endogenous LRP-1 expression (Fig. 1C). LRP-1-mediated endocytosis was then analyzed using FITC-labeled α2 M as a test ligand (12). LRP-1-mediated uptake of labeled ligand was inhibited 2-fold and 4-fold under RAP treatment and with selective siRNA targeting of LRP-1, respectively (Fig. 1D). Carcinoma cell adhesion was then investigated under these experimental conditions. We observed that RAP treatment increased 2-fold the cancer cell attachment, similar to that observed under LRP-1 silencing (Fig. 1E). Additionally, to examine the role of LRP-1-mediated uptake in cell-substrate deadhesion, a trypsin deadhesion assay was performed, as previously described (12, 58). As shown in Fig. 1F, LRP-1-silenced cells were two times more resistant to trypsin detachment than siCTRL-overexpressing cells. Similar results were obtained with RAP-treated cells compared to nontreated cells.
Fig 1.

Blockade of LRP-1-mediated endocytosis increases FTC-133 cell attachment. (A) Cell surface expression of LRP-1 was assessed in FTC-133 cells. In the left panel, differential interference contrast (DIC) microscopy was combined with confocal microscopy imaging for LRP-1. In the merged image, insets (zoomed in 300%) highlight cell surface expression of LRP-1 (green) at the leading edge (star and double star) and at the rear of the cell (arrowhead). Bars, 10 μm. In the right panel, cell surface biotinylated proteins were obtained from FTC-133, fibroblastoid HT1080 (f-HT1080), or epithelioid HT1080 (e-HT1080) cells and subjected to immunoblot analysis using anti-LRP-1 antibodies (8G1). (B) Purified HA-tagged RAP was assessed by SDS-PAGE followed by Coomassie blue staining (CBS) and immunoblotting using anti-HA tag antibodies (IB). The gel is shown in its full length. (C) Total RNAs were purified from FTC-133 cells transfected with nonsilencing siRNA (siCTRL) or siRNA targeting LRP-1 (siLRP-1). The transcriptional level of LRP-1 was assessed by RT-PCR, and β-actin primers were used as a normalization control. Numbers under the gel indicate the fold change compared to results for siCTRL cells, used as a reference. (D) Wild-type FTC-133 cells (WT) were treated with RAP (500 nM) or not for 2 h or transfected with siRNA sequences (siCTRL and siLRP-1) and then incubated for 30 min in serum-free medium containing FITC-labeled human α2 M. Endocytosis of nonlabeled ligand was used as a competition experiment (compet.) to ensure the selectivity of the assay. The intracellular fluorescence corresponding to the endocytosis activity was determined as described elsewhere (12) and is expressed as relative units (R.U.), by comparison with signal from WT cells. (E) FTC-133 cells were pretreated with RAP (500 nM) for 24 h or transfected with nonsilencing siRNA (siCTRL) or LRP-1-silencing sequences (siLRP-1). Then, cells were seeded onto gelatin-coated plates, and the nonadherent cells were discarded after 30 min. Results are expressed as percentages of adherent cells compared to WT cells. (F) FTC-133 cells treated or not with RAP (500 nM, 24 h), siRNA control cells (siCTRL), and LRP-1-silenced cells (siLRP-1) were grown in gelatin-coated dishes for 24 h and subjected to trypsinization assay by addition of 0.025% trypsin (wt/vol) for 10 min. Results are expressed as percentages of detached cells. (G) FTC-133 cell adhesion assay was performed on hyaluronic-acid-coated plates (HA) or on gelatin-coated plates for 10 or 30 min. Results are expressed as optical density (OD) measured at 600 nm. Each value is the mean ± SD for at least three independent experiments, each performed in triplicate. n.s., not significant; *, P < 0.05; *∗, P < 0.01.
The adhesion molecule CD44, a major receptor for the glycosaminoglycan hyaluronan, is mainly engaged in cell-ECM interactions during tumorigenesis and appeared to be overexpressed in thyroid cancers (13, 18, 34). To test whether CD44 may also contribute to controlling the adhesive behavior of FTC-133 cells, cell attachment to hyaluronic-acid-coated plates was measured and compared to cell attachment to gelatin-coated plates (Fig. 1G). We observed that hyaluronic acid increased the tumor cell attachment compared to that with gelatin, especially in an early time of attachment.
Inhibition of LRP-1-dependent endocytosis leads to accumulated CD44 at the plasma membrane.
We therefore hypothesized that LRP-1 and CD44 may cooperate to control FTC-133 tumor cell adhesion. Since cancer cells may exhibit different isoforms of CD44 resulting from alternative splicing, PCR experiments were conducted to determine which isoforms are expressed in our carcinoma environment (Fig. 2A). The results revealed that only the standard CD44 (CD44s) was abundantly expressed, whereas the CD44 variants containing various numbers of exon insertions (v1 to v10) remained undetectable. The specificity of the band was controlled by siRNA targeting CD44. CD44 protein expression at the plasma membrane was then examined by immunoblotting from cell surface biotinylated proteins (Fig. 2B). The 85-kDa band corresponds to the standard form of CD44. A minor 120-kDa isoform was also detected, probably attributable to glycosylated CD44. Although the 170-kDa band could reflect a chondroitin sulfate-modified form of CD44, it is more likely that it reveals dimers of the 85-kDa CD44s, since the band is not detected under reducing conditions (52). Altogether, these data indicate that standard CD44 is predominantly expressed in our cell system.
Fig 2.

Analysis of CD44 expression in FTC-133 cells in basal conditions and under LRP-1 inhibition. (A) Total RNAs were purified from FTC-133 cells and subjected to RT-PCR using primers for the standard CD44 (CD44s, 482 bp) and the variant isoforms of CD44 (>482 bp). The gel is shown in its full length. siRNA sequences targeting CD44 were used to control the specificity of the band. β-Actin primers were used as a normalization control. (B) CD44 expression at the plasma membrane (PM) was assessed after cell surface biotinylation of proteins at 4°C by SDS-PAGE in the presence or absence of β-mercaptoethanol (β-ME) and immunoblotting of CD44. The 85-kDa band corresponds to the standard CD44 (CD44s); the arrowhead represents the glycosylated form of CD44, and the star indicates the CD44 dimer. (C) Total RNAs were purified from wild-type carcinomas (treated or not with RAP for 24 h) and from siRNA-transfected cells (siCTRL and siLRP-1). CD44 mRNA expression was quantified using real-time RT-PCR and expressed as relative units (R.U.). β-Actin was used for normalization. Nontreated wild-type (WT) and siCTRL cells served as a reference set to 1. (D) Plasma membrane (PM) extracts from cell surface biotinylated proteins or whole-cell extracts were obtained from FTC-133 cells treated or not with RAP (500 nM, 1 h). Immunoblot analysis was performed using anti-CD44 antibodies (A020). The β-actin levels in the whole-cell extract (left) and in the intracellular fraction (right) served as a loading control. (E) Quantification of CD44 expression at the protein level. (F) FTC-133 cells were treated with or without RAP (500 nM, 1 h) and with DMSO or dynasore (120 μM) for 30 min. DMSO served as a control for dynasore treatment. Plasma membrane extracts were subjected to immunoblot analysis for CD44. Expression of β-actin in the corresponding intracellular fraction served as a loading control. (G) Quantification of cell surface CD44. Each value is the mean ± SD for at least three independent experiments. n.s., not significant; *∗, P < 0.01.
We then studied whether inhibition of LRP-1-dependent endocytosis may impact CD44 expression. CD44 mRNA levels did not vary under RAP treatment or LRP-1 silencing, as revealed by real-time PCR assay (Fig. 2C). However, we found a substantial increase in CD44 expression at the protein level under RAP treatment (Fig. 2D, left panel, and 2E) that seems attributable to CD44 accumulation at the cell surface (Fig. 2D, right panel, and 2E). A similar result was obtained using FTC-238, a distinct thyroid carcinoma cell line described elsewhere (50) (data available on request). Dynasore, a cell-permeating inhibitor of dynamin, was then used to block endocytosis (33). Such treatment led to a comparable accumulation of CD44 and was not affected by RAP treatment (Fig. 2F and G). This suggests that the RAP-induced accumulation of cell surface CD44 was attributable to endocytosis inhibition.
LRP-1 and CD44 coexist in a tight molecular complex at the tumor cell surface.
To test whether LRP-1 and CD44 may participate in a common biomolecular complex in FTC-133 cells, coimmunoprecipitation experiments were carried out under either standard or high-stringency conditions. As shown in Fig. 3A, we were able to successfully immunoprecipitate both LRP-1 α- and β-chains with the 5A6 antibody raised against the extracellular LRP-1 β-chain. CD44 was coimmunoprecipitated with the entire LRP-1 protein (Fig. 3A, left panel), even under high-stringency conditions (Fig. 3A, right panel). Reverse immunoprecipitation experiments with anti-CD44 were also performed with the same cell lysates. The data presented in Fig. 3B confirmed that LRP-1 and CD44 were detected in the same molecular complex (left panel), which persists under high-stringency conditions (right panel). Last, immunoprecipitation assays were conducted with biotinylated proteins to analyze the LRP-1-containing complexes at the cell surface using either anti-LRP-1 β-chain (Fig. 3C, left panel) or anti-LRP-1 α-chain (Fig. 3C, right panel). Both results revealed that CD44 was coimmunoprecipitated with cell surface LRP-1. Altogether, these data revealed the existence of molecular interactions between LRP-1 and CD44 in FTC-133 cells. This result was also confirmed in FTC-238 cells (data available on request).
Fig 3.
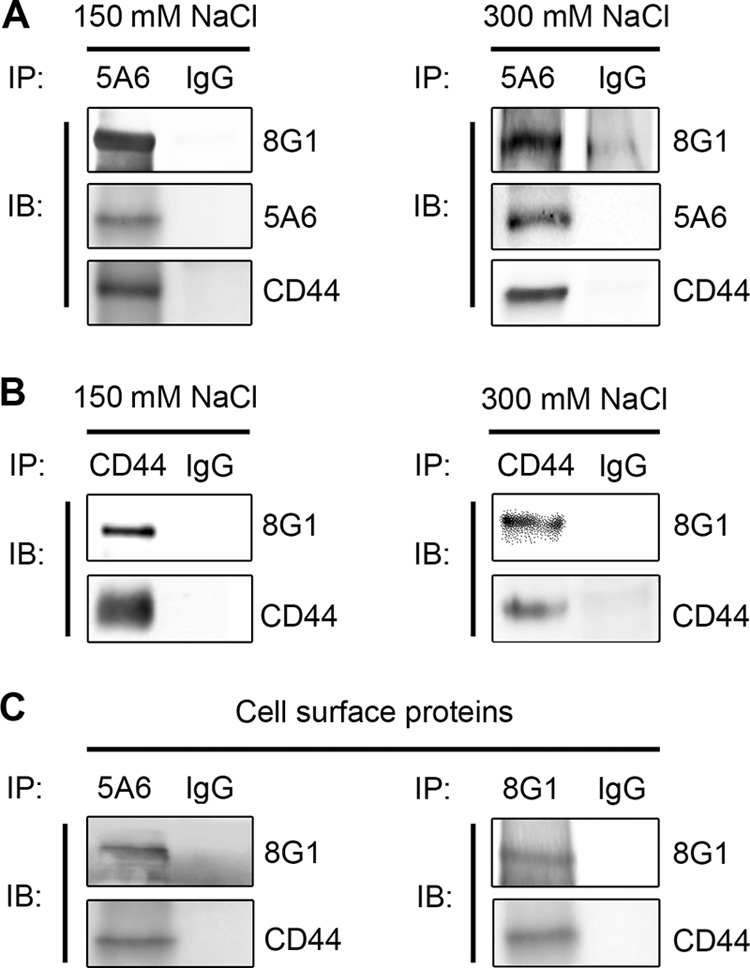
Coimmunoprecipitation of LRP-1 and CD44 in the same molecular complex. (A) LRP-1-containing complexes were immunoprecipitated from FTC-133 whole-cell extracts by using the mouse anti-LRP-1 monoclonal antibody 5A6. Immunocomplexes were then subjected to SDS-PAGE and immunoblotted (IB) by using specific antibodies for LRP-1 β-chain (5A6), LRP-1 α-chain (8G1), and CD44 (A020). Both standard-stringency (150 mM NaCl; left panel) and high-stringency (300 mM NaCl; right panel) conditions were used. (B) Immunoprecipitation assays were performed with FTC-133 cells using anti-CD44 antibody (F10-44-2) under experimental conditions described above. (C) Biotinylation of cell surface proteins was performed at 4°C with FTC-133 cells. Proteins were affinity precipitated with avidin-agarose beads, and then LRP-1-containing complexes were immunoprecipitated by either anti-LRP-1 β-chain (5A6; left panel) or anti-LRP-1 α-chain (8G1; right panel) and analyzed by Western blotting by using anti-LRP-1 (8G1) and anti-CD44 (A020) antibodies. Nonspecific IgGs were used as a negative control of immunoprecipitation assays.
The fourth ligand binding cluster of LRP-1 mediates binding to CD44 at the plasma membrane.
The extracellular LRP-1 α-chain harbors four ligand-binding clusters, two of which (clusters II and IV) are mostly involved in the binding of extracellular ligands (31). The extracellular part of the LRP-1 β-chain was also recently identified as binding pro-cathepsin D in fibroblasts (2). Moreover, the LRP-1 β-chain contains a short cytoplasmic tail able to link a membrane partner by recruiting common molecular adaptors (45). In order to identify regions on LRP-1 able to bind CD44, we overexpressed functional HA-tagged-minireceptors derived from LRP-1. All the recombinant variants of LRP-1 contained the entire LRP-1 β-chain, including the extracellular part. Mini-LRP-II (molecular mass of 153 kDa) and mini-LRP-IV (molecular mass of 164 kDa) contained the second and fourth ligand binding clusters of LRP-1, respectively, whereas the SPCT construct (molecular mass of 106 kDa) exhibited only the LRP-1 β-chain (Fig. 4A). Expression of HA-tagged minireceptors at the cell surface was assessed by HA tag-directed immunoprecipitation of cell surface biotinylated proteins followed by immunoblotting analysis using anti-HA tag antibody, which revealed the expected molecular weights (Fig. 4B, top panel). The data indicated that the LRP-1-derived minireceptor carrying only ligand-binding domain IV is sufficient to bind endogenous CD44 (Fig. 4B, bottom panel).
Fig 4.
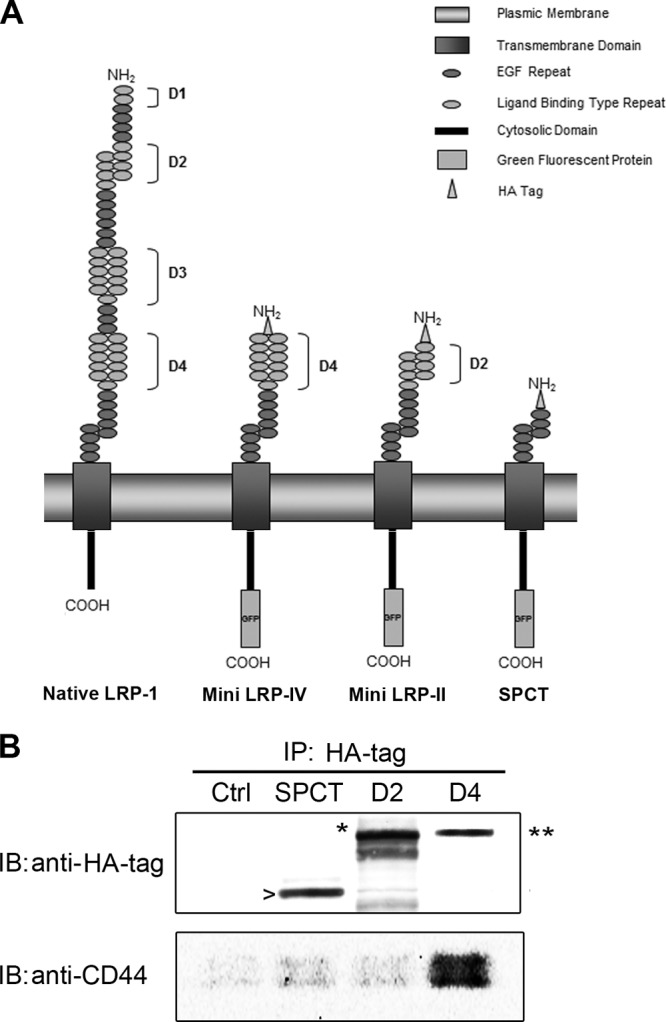
Endogenous CD44 binds to ligand-binding cluster IV of LRP-1. (A) Schematic representation of native LRP-1 and minireceptors derived from LRP-1 carrying extracellular binding domain IV (mini LRP-IV), extracellular binding-domain II (mini LRP-II) or no-ligand-binding cluster (SPCT). (B) FTC-133 cells were transiently transfected with HA-tagged SPCT (SPCT), HA-tagged mini LRP-II (D2), or HA-tagged mini LRP-IV (D4). Nontransfected cells served as a control (Ctrl). After 24 h of transfection, biotinylation of cell surface proteins was performed, followed by an immunoprecipitation (IP) assay with an anti-HA tag antibody. Then, immunoblot (IB) analysis was conducted using both anti-HA tag and anti-CD44 antibodies (A020). Bands revealed by the anti-HA tag antibody correspond to the expected molecular masses of SPCT (106 kDa; arrowhead), mini LRP-II (153 kDa; star), and mini LRP-IV (164 kDa; double star).
Lipid rafts are not required for molecular connections between LRP-1 and CD44.
Although LRP-1 was reported to be located at the plasma membrane in both clathrin-coated pits and lipid rafts, connections with membrane coreceptors were mainly found in caveola-rich fractions (5, 59, 61, 63). Moreover, CD44 was also reported to be associated mostly with lipid rafts (55). Therefore, we next investigated whether LRP-1 binds CD44 in lipid rafts by using a detergent-free method. Lipid rafts were isolated from FTC-133 cells by discontinuous sucrose density gradient ultracentrifugation and identified in fractions 2 and 3 by PTRF and caveolin-1 staining (23, 27), as previously reported (44) (Fig. 5A, top panel). The transferrin receptor, used as a marker of nonraft fractions (20), was detected from fraction 5 and especially at the bottom of the sucrose gradient. Then, a LRP-1 coimmunoprecipitation assay was conducted with each fraction, followed by immunoblotting analyses against both LRP-1 and CD44 (Fig. 5A, bottom panel). Although LRP-1 appeared rather concentrated in lipid rafts, we observed that binding of LRP-1 to CD44 occurs in both raft and nonraft fractions. To sustain this observation, membrane cholesterol depletion was obtained by MβCD treatment. Such a treatment affected the membrane cholesterol content in a time-dependent fashion (Fig. 5B, left panel). Furthermore, PTRF and caveolin-1 staining disappeared in both fractions 2 and 3 under MβCD treatment (Fig. 5B, right panel), thus validating the efficiency of the cholesterol depletion. As shown in Fig. 5C, coimmunoprecipitation experiments were performed with or without MβCD by using either anti-LRP-1 (left panel) or anti-CD44 (right panel) antibodies. The result indicated that LRP-1–CD44 binding was maintained under lipid raft depletion.
Fig 5.

LRP-1 and CD44 are found associated in both raft and nonraft fractions. (A) In the top panel, FTC-133 cell lysates were subjected to a discontinuous sucrose density gradient ultracentrifugation. PTRF and caveolin 1 (cav-1) were used to reveal raft fractions, whereas the transferrin receptor (TfR) was used as a marker of nonrafts. In the bottom panel, immunoprecipitation assays (IP) were conducted with fractions 1 to 9 by using anti-LRP-1 antibody (5A6) followed by immunoblotting revelation using anti-LRP-1 (8G1) and anti-CD44 (A020) antibodies. The fractions used for immunoprecipitation are indicated under the membrane picture. (B) FTC-133 cells were treated with or without 5 mM methyl-β-cyclodextrin (MβCD). In the left panel, cholesterol content was then quantified at different times of incubation (15, 30, and 45 min). Results were expressed as percentages of results for nontreated cells. In the right panel, after 30 min of incubation with or without MβCD, raft fractions (fractions 2 and 3) were isolated by sucrose density gradient ultracentrifugation, as described above, and checked for PTRF and cav-1 expression by immunoblotting. (C) FTC-133 cells were treated or not with MβCD for 30 min. After biotinylation of cell surface proteins, LRP-1-containing complexes (left panel) or CD44-containing complexes (right panel) were immunoprecipitated by either the anti-LRP-1 β-chain (5A6) or anti-CD44 (F10-44-2), respectively. Immunoblotting analysis (IB) was realized with anti-LRP-1 (8G1) and anti-CD44 (A020) antibodies. Nonspecific IgGs were used as a negative control of immunoprecipitation assays.
LRP-1 and CD44 are colocalized in FTC-133 cells.
The cellular localization of LRP-1 and CD44 was further investigated by confocal immunofluorescence microscopy. Merged images of LRP-1 (green) and CD44 (red) revealed that both receptors highly colocalize in FTC-133 cells, particularly at the migration front and at the rear of the cell (Fig. 6, upper line and insets). To gain further insight, we applied a correlation plot treatment and an isosurface representation to estimate the number of the colocalized voxels (64). The isosurface representation and the colocalization scattergram indicated a strong colocalization between LRP-1 and CD44, (Fig. 6, middle line). Moreover, we examined the position of the colocalized voxels by using a multiplanar display that allows simultaneous visualization of three orthogonal planes. Using a z value corresponding to an intracytoplasmic position (Fig. 6, lower line), we also detected colocalization between LRP-1 and CD44, particularly at the nucleus periphery and at the trailing edge. This showed that the LRP-1–CD44 complex exists at the cell surface and also in the cytoplasmic compartment, suggesting that such a molecular complex could be internalized.
Fig 6.

Confocal imaging reveals that LRP-1 and CD44 are colocalized in FTC-133 cells. FTC-133 cells were plated onto gelatin-coated coverslips for 4 h at 37°C, fixed, washed, and stained with Alexa Fluor 488 for LRP-1 (green) and Alexa Fluor 568 for CD44 (red) and analyzed by confocal microscopy. Nuclei were counterstained with diaminido phenylindole (DAPI) (blue). Images were treated with AMIRA software, as indicated in Materials and Methods. In the upper panel, LRP-1 labeling (left), CD44 labeling (middle), and a merged image (right) are shown. In the merged image, insets from the leading edge (star and double star) and the rear of the cell (arrowhead) highlight the colocalization of LRP-1 and CD44 (zoomed in 300%). The middle panel displays the isosurface representation of LRP-1–CD44 colocalization (yellow spots) and the two-dimensional scattergram, used to estimate the degree of LRP-1–CD44 colocalization. The lower panel represents the intracellular colocalization of LRP-1–CD44 (green spots) revealed by one single cross section (xy axis at left; yz axis at right). The rear of the cell is indicated by the arrow. Images are representative of three distinct sets of culture. Bars, 10 μm.
CD44 is found in early endosomes and lysosomal vesicles in a LRP-1-dependent pathway.
To examine whether cell surface CD44 undergoes endocytosis through LRP-1 and to assess the fate of internalized CD44, carcinoma cells were treated or not with RAP to inhibit LRP-1-mediated endocytosis. Then, CD44 was costained either with EEA1 (early endosome-associated protein 1) (Fig. 7A), a marker of early endosomes (36), or with LAMP-1 (lysosome-associated membrane protein 1) (Fig. 7B), a marker of lysosomes (9). Double staining with antibodies against CD44 and EEA-1 indicates that CD44 was mainly present in early endosome compartments (Fig. 7A, top panel and insets). This location was drastically disturbed by RAP, especially at the leading edge (Fig. 7A, top panel, star in insets). Representation of the intracellular compartment sustains these observations (Fig. 7A, bottom panel). Furthermore, we investigated whether CD44 may be delivered to LAMP-1-positive vesicles by using double staining with anti-CD44 and anti-LAMP-1 antibodies. Labeling revealed that internalized CD44 is found in the lysosomal compartment (Fig. 7B). The endocytic route of CD44 to lysosomes is dependent on LRP-1, as revealed by RAP treatment. These data revealed that once internalized by LRP-1, CD44 can be routed through early endosomes toward lysosomes.
Fig 7.

CD44 colocalization with EEA-1 and LAMP-1 is LRP-1 dependent. FTC-133 cells were plated on gelatin-coated coverslips for 4 h at 37°C and treated or not with 500 nM RAP for 1 h. (A) Cells were stained with Alexa Fluor 488 for CD44 (green) and Alexa Fluor 568 for EEA-1 (red). Nuclei were counterstained in blue with DAPI. Cells were analyzed by confocal microscopy, and images were processed using the AMIRA software program. In the top panel, CD44 labeling (left), EEA-1 labeling (middle), and isosurface representation (right), without RAP (- RAP) or under RAP treatment (+ RAP), are shown. Insets (zoomed in 200%) from the leading edge (star) and the rear of the cell (arrowhead) highlight that colocalization of CD44 with EEA-1 was drastically disturbed by RAP treatment. In the bottom panel, intracellular colocalization of CD44: EEA-1 (green spots) in FTC-133 cells treated without RAP (- RAP) or with RAP (+ RAP), as revealed by one single cross section (xy axis in left; yz axis in right), is shown. The rear of the cell is indicated by the arrow. (B) Cells were treated without RAP (- RAP) or with RAP (+ RAP) and were stained with Alexa Fluor 488 for CD44 (green) and Alexa Fluor 568 for LAMP-1 (red), and an isosurface representation (yellow spots) is displayed. All images are representative of three independent experiments. Bars, 10 μm.
LRP-1-mediated internalization of CD44 occurs mainly via clathrin-coated pits.
To confirm these results, endocytosis assays were conducted using a validated biochemical approach derived from the method of Wu and Gonias (59) in both control and LRP-1-deficient cells. After cell surface protein labeling with non-membrane-permeating sulfo-NHS-LC-biotin at 4°C, cells were cultured at 37°C for 30 min to allow endocytosis. Internalization was stopped by decreasing the temperature to 4°C, and cells were surface digested with pronase. Efficiency of pronase digestion was controlled in Fig. 8A (left frame, lanes 1 and 2). In the pronase-treated samples, affinity-precipitated CD44 with avidin-agarose was considered the internalized fraction, as already described (59). Immunoblotting analysis against CD44 revealed that CD44 endocytosis was inhibited 4-fold under LRP-1 silencing (Fig. 8A, left frame, lanes 3 and 4, and right frame). The same method was used to estimate the internalized fraction of CD44 under RAP treatment (Fig. 8B). In accordance with the foregoing results, CD44 uptake was reduced by about 3-fold in RAP-treated cells (Fig. 8B, left frame, lanes 1 and 2, and right frame). No change was detected when cells were pretreated with MβCD (Fig. 8B, left frame, lanes 3 and 4, and right frame), suggesting that LRP-1-mediated endocytosis of CD44 occurs mainly through clathrin-coated pits. To sustain this hypothesis, the internalized fraction of CD44 was quantified under hyperosmotic conditions to inhibit clathrin-mediated endocytosis (22) (Fig. 8C). Without sucrose (Fig. 8C, left frame, lanes 1 and 2), CD44 internalization was sharply diminished by RAP treatment, as previously observed in the results shown in Fig. 8B. Endocytosis of CD44 was highly reduced under hyperosmotic conditions (Fig. 8C, left frame, lane 3 versus lane 1, and right frame), revealing that it proceeds mostly through a clathrin-dependent mechanism. The internalized fraction of CD44 under hyperosmotic conditions was poorly affected by RAP treatment (Fig. 8C, left frame, lane 4 versus lane 3, and right frame). In accord with data shown in Fig. 8B, this result indicates that the LRP-1-mediated uptake of CD44 occurs mainly through clathrin-coated pits. The LRP-1-mediated uptake of CD44 was confirmed using FTC-238 cells (data available on request).
Fig 8.

Clathrin-coated pits are required for LRP-1-mediated endocytosis of CD44 in FTC-133 cells. FTC-133 cells were transfected with nontargeting siRNA sequences (siCTRL) or siRNA targeting LRP-1 (siLRP-1) for 48 h (A) or treated with 500 nM RAP for 1 h (B and C), followed by treatment with 5 mM MβCD for 45 min (B) or 400 mM sucrose for 1 h (C). Cells were then transferred to ice, and cell surface proteins were biotinylated. Then, tumor cells were transferred to 37°C to permit endocytosis for 30 min in complete medium (with or without RAP, MβCD, or sucrose). Cells were then placed on ice, washed, and incubated with pronase (1 mg/ml) to remove surface-bound proteins. Biotinylated proteins were recovered by avidin protein immobilized to agarose beads, subjected to SDS-PAGE, and revealed by immunoblotting for CD44 and stand for the endocytosis fraction, as previously reported (59). Binding of CD44 to the cell surface and the efficiency of pronase were controlled in panel A (left frame, lanes 1 and 2, respectively). Histograms (right frames) represent the quantification of internalized CD44. Results are expressed as percentages compared to results for control cells (A) or wild-type nontreated cells (B and C), which serve as a reference. n.s., not significant; *∗, P < 0.01.
LRP-1-mediated endocytosis of CD44 controls tumor cell attachment.
To apprehend the biological function of the LRP-1-mediated uptake of membrane-bound CD44 in the tumor context, a cell attachment assay was performed using RAP to block LRP-1-mediated endocytosis in CD44-silenced cells, compared to results in CD44-expressing cells (Fig. 9). As expected, RAP treatment of cells transfected with nonsilencing sequences increases carcinoma cell attachment by about 2-fold. Remarkably, the RAP-induced cell attachment was completely abolished under CD44 silencing. This indicates that the increased cell adhesion observed under LRP-1 blockade was attributable mainly to CD44 accumulation at the cell surface (Fig. 2). Altogether, these data demonstrate that LRP-1-mediated endocytosis of CD44 constitutes a major molecular mechanism to control tumor cell adhesion.
Fig 9.
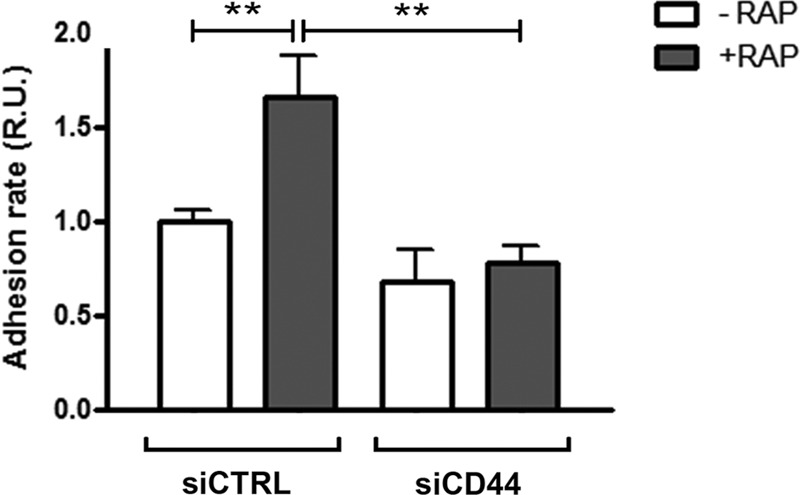
Cell surface accumulation of CD44 is responsible for the stimulation of tumor cell adhesion under LRP-1 blockade. FTC-133 cells were transfected with nontargeting siRNA (siCTRL) or CD44-silencing sequences (siCD44) and treated or not with RAP (500 nM) for 1 h. Cell adhesion was then assayed for 30 min as described for Fig. 1. Results are expressed compared to those for nontreated control cells, which serve as a reference set to 1. Each value is the mean ± SD of at least three independent experiments, each performed in triplicate. *∗, P < 0.01.
DISCUSSION
In this study, we identified the transmembrane glycoprotein CD44 as a novel cell surface partner for LRP-1 and demonstrated that LRP-1 functions as an endocytic receptor for CD44 to regulate tumor cell attachment.
In the field of cancer, the endocytic receptor LRP-1 was first reported to mediate the uptake of various extracellular proteinases, thus reducing excessive remodeling of the extracellular matrix (14). Furthermore, numerous studies indicated that a decreased LRP-1 level correlates with increased invasiveness of tumor cells from various tissues (25, 39). LRP-1 was then widely associated with antitumor properties and considered as an attractive receptor for targeting the invasive behavior of malignant cells. However, the functionalities associated with LRP-1 now appear to be much more complex and multifaceted. Indeed, more-recent data demonstrated that LRP-1 may also exhibit proinvasive properties, depending on tissue specificity (2, 12, 35). From our recent work, LRP-1 emerged as a key coordinator of cell-matrix interactions governing the adhesion-deadhesion balance in cancer cells to promote invasion (11, 12). However, molecular knowledge to explain this contribution was missing. In the present study, the discovery of CD44 as a new transmembrane partner of LRP-1 provides new insight and improves our understanding of how LRP-1 controls cell-matrix interactions in a tumor context. Using both biochemical and imaging approaches, we demonstrated that LRP-1 and CD44 are tightly associated in the same biomolecular complexes at the surface of tumor cells. For the most part, results have been obtained based on endogenously expressed proteins. Moreover, LRP-1 and CD44 are found associated even under high-stringency conditions and without a chemical cross-linker, ensuring the relevance of our discovery.
Up to now, CD44-mediated internalization was poorly understood. Our data suggest that its uptake is mediated mainly by LRP-1 in thyroid carcinoma. Indeed, only about 30% of CD44 remained internalized in LRP-1-silenced cells or under RAP treatment. Interestingly, LRP-1-mediated endocytosis appears to be efficient in regulating the cell surface amount of CD44, especially by targeting CD44 to lysosomes. Although lysosome-mediated catabolism of intracellular CD44 was already evoked, most studies suggested rather the recycling of internalized CD44 back to the plasma membrane (26, 54). In these earlier studies, the authors have focused on the CD44-mediated internalization of hyaluronan. Conversely, we considered CD44 to be not only a motor receptor for endocytosis of hyaluronan but also a ligand for internalization and catabolism. In our cellular environment, the LRP-1-mediated endocytosis of CD44 was indeed found not to be related to ligation of hyaluronan, thereby explaining, at least in part, the apparently contrasting results when considering the fate of internalized CD44.
Data from discontinuous sucrose density gradient ultracentrifugation and MβCD treatment revealed that the LRP-1–CD44 complex is highly located in lipid rafts but also is detected in clathrin-coated pits. Surprisingly, we demonstrated that LRP-1-mediated endocytosis of CD44 occurs mainly through clathrin-coated pits. This was rather unexpected, since CD44 was mostly described as being associated with lipid rafts (16, 26). Thankamony and Knudson have reported that CD44 endocytosis requires membrane cholesterol and palmitic acid incorporation (55). Furthermore, a LRP-1 connection with other membrane coreceptors was found mainly in caveola-rich fractions (5, 59, 63). It is tempting to speculate that in lipid rafts, CD44 might rather function as a novel coreceptor for LRP-1 to trigger intracellular signals, as previously reported for PDGFR (5). Indeed, CD44 is well known to act as a coreceptor for receptor tyrosine kinases, including Met and ErbB family members, to regulate cancer-related events (42). This is consistent with the ability of LRP-1 to control signaling pathways, contributing to the aggressive behavior of tumor cells. We have recently demonstrated that LRP-1 sustains tumor progression by mediating the induction of an ERK cascade and inhibition of JNK phosphorylation in thyroid carcinoma (28). Others have shown that binding of the protease nexin 1 to LRP-1 stimulates the ERK pathway, leading to matrix metalloproteinase 9 (MMP-9) expression and metastasis development (17). The signaling function of both LRP-1 and CD44 sheds light on Src kinase as a prospective common molecular relay. Nevertheless, LRP-1 appeared to control carcinoma cell spreading, adhesion, and migration through a Src-independent activation of ERK involving the recruitment of Shp-2 and p21-activated kinase (28). Additionally, CD44 silencing did not abolish tyrosine phosphorylation within the cytoplasmic tail of LRP-1 (unpublished data). Besides, the coreceptor function of CD44 seems to be mediated through the CD44 splice variants and especially the CD44 v6 variant, rather than the standard CD44 isoform (38, 42). Such isoforms of CD44, resulting from alternative splicing, remained undetectable under our experimental conditions. Altogether, this suggests that LRP-1–CD44 constitutes an endocytic complex rather than a signaling complex in our experimental environment.
Here we have demonstrated that RAP-stimulated tumor cell attachment is due mainly to CD44 accumulation at the plasma membrane. The same result was obtained in LRP-1-silenced tumor cells, in which cell surface accumulation of CD44 was found to be responsible for the increased cell attachment (data not shown). This is consistent with findings of previous studies indicating that overexpression of CD44 potentiates the adhesion of breast cancer and prostate cancer cells (13). Furthermore, a decreased CD44 level at the cell surface was associated with impaired cell-ECM interactions and attenuated cancer cell adhesion (13, 40). Our results reveal that the LRP-1-mediated endocytosis of CD44 is crucial to regulate the molecular links established between tumor cells and ECM. The ability of LRP-1 to alter the trafficking and functional properties of CD44 toward cell adhesion is reminiscent of what has been observed for β-integrins. Indeed, Spijkers and colleagues have described a complex formation between LRP-1 and β2-integrin by using recombinant proteins and showed that LRP-1 inhibition is associated with impaired β2-dependent adhesion to endothelial cells (51). In our study, the use of LRP-1 minireceptors revealed that the fourth ligand binding cluster of LRP-1 is involved in CD44 binding, as previously mentioned for β2-integrins (46, 51). LRP-1 was also described to mediate the uptake of integrin-containing complexes in macrophages to facilitate the cellular detachment at the rear of the cell (7). Consistent with these data, we found that LRP-1 is highly colocalized with CD44 at the trailing edge and at the migration front of thyroid carcinoma cells. In breast cancer cells, LRP-1 was prominent at the leading edge during migration steps (8), and CD44 was likewise mostly observed with other glycoproteins at the migration front (65). These results are consistent with our recent findings that highlighted LRP-1 involvement in cell polarization from the leading edge to the trailing tail through its ability to control molecular composition and dynamics of focal adhesion (12, 28).
Finally, our results revealed a new cell surface ligand for LRP-1 and characterized a formerly unknown pathway of CD44 internalization, critical to defining the adhesive properties of tumor cells. CD44 isoforms are mainly engaged in cell-ECM interactions during tumorigenesis and appear promising for targeting the aggressive behavior of tumors. However, clinical trials performed with monoclonal anti-CD44 antibodies have been disappointing due to adverse effects and fatal events (56). Targeting the LRP-1-mediated internalization of CD44 might offer a new therapeutic perspective to improve antitumor strategies. Altogether, our data increase our understanding of how LRP-1 contributes to malignant diseases and strengthen the molecular knowledge of the integrated functional relationships between endocytosis and cell-matrix adhesion.
ACKNOWLEDGMENTS
This work was supported by grants from CNRS, Ligue Nationale Contre le Cancer (CCIR-GE, Conférence de Coordination Interrégionale du Grand Est 2012), CPER 2007-2013, and Fonds National pour la Santé ACI 2008 (Cancéropôle Grand-Est Project). G.P. and E.S. were recipients of grants from Région Champagne-Ardenne (2008-2011 and 2010-2011, respectively). M.D. and M.K. acknowledge support from the Agence National pour la Recherche (ANR-08-MNPS-042-04 and ANR-09-BIOT-015-01, TIMPAD and VECtoBrain projects, respectively).
We acknowledge the expert technical support of C. Terryn (SFR CAP-Santé, Reims, France) and H. Bobichon (FRE CNRS/URCA no. 3481) for confocal microscopy. We thank F. Rabenoelina and L. Parent for technical assistance and S. Wright for editorial assistance. We are grateful to M. S. Nielsen (Department of Medical Biochemistry, University of Aarhus, Denmark) for kindly providing us with the pT7H6FX-RAP vector.
Footnotes
Published ahead of print 18 June 2012
REFERENCES
- 1. Akisik E, Bavbek S, Dalay N. 2002. CD44 variant exons in leukemia and lymphoma. Pathol. Oncol. Res. 8:36–40 [DOI] [PubMed] [Google Scholar]
- 2. Beaujouin M, et al. 2010. Pro-cathepsin D interacts with the extracellular domain of the beta chain of LRP1 and promotes LRP1-dependent fibroblast outgrowth. J. Cell Sci. 123:3336–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. 2003. LRP: role in vascular wall integrity and protection from atherosclerosis. Science 300:329–332 [DOI] [PubMed] [Google Scholar]
- 4. Boucher P, et al. 2007. LRP1 functions as an atheroprotective integrator of TGFbeta and PDFG signals in the vascular wall: implications for Marfan syndrome. PLoS One 2:e448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boucher P, et al. 2002. Platelet-derived growth factor mediates tyrosine phosphorylation of the cytoplasmic domain of the low density lipoprotein receptor-related protein in caveolae. J. Biol. Chem. 277:15507–15513 [DOI] [PubMed] [Google Scholar]
- 6. Cam JA, Zerbinatti CV, Li Y, Bu G. 2005. Rapid endocytosis of the low density lipoprotein receptor-related protein modulates cell surface distribution and processing of the beta-amyloid precursor protein. J. Biol. Chem. 280:15464–15470 [DOI] [PubMed] [Google Scholar]
- 7. Cao C, et al. 2006. Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO J. 25:1860–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chazaud B, et al. 2002. Promigratory effect of plasminogen activator inhibitor-1 on invasive breast cancer cell populations. Am. J. Pathol. 160:237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen JW, Pan W, D'Souza MP, August JT. 1985. Lysosome-associated membrane proteins: characterization of LAMP-1 of macrophage P388 and mouse embryo 3T3 cultured cells. Arch. Biochem. Biophys. 239:574–586 [DOI] [PubMed] [Google Scholar]
- 10. Czekay RP, Kuemmel TA, Orlando RA, Farquhar MG. 2001. Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol. Biol. Cell 12:1467–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dedieu S, Langlois B. 2008. LRP-1: a new modulator of cytoskeleton dynamics and adhesive complex turnover in cancer cells. Cell Adh. Migr. 2:77–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dedieu S, et al. 2008. LRP-1 silencing prevents malignant cell invasion despite increased pericellular proteolytic activities. Mol. Cell. Biol. 28:2980–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Draffin JE, McFarlane S, Hill A, Johnston PG, Waugh DJ. 2004. CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res. 64:5702–5711 [DOI] [PubMed] [Google Scholar]
- 14. Emonard H, et al. 2005. Regulation of matrix metalloproteinase (MMP) activity by the low-density lipoprotein receptor-related protein (LRP). A new function for an “old friend”. Biochimie 87:369–376 [DOI] [PubMed] [Google Scholar]
- 15. Emonard H, et al. 2004. Low density lipoprotein receptor-related protein mediates endocytic clearance of pro-MMP-2.TIMP-2 complex through a thrombospondin-independent mechanism. J. Biol. Chem. 279:54944–54951 [DOI] [PubMed] [Google Scholar]
- 16. Eyster CA, et al. 2009. Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic 10:590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fayard B, et al. 2009. The serine protease inhibitor protease nexin-1 controls mammary cancer metastasis through LRP-1-mediated MMP-9 expression. Cancer Res. 69:5690–5698 [DOI] [PubMed] [Google Scholar]
- 18. Griffith OL, Melck A, Jones SJ, Wiseman SM. 2006. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J. Clin. Oncol. 24:5043–5051 [DOI] [PubMed] [Google Scholar]
- 19. Guttman M, et al. 2009. Interactions of the NPXY microdomains of the low density lipoprotein receptor-related protein 1. Proteomics 9:5016–5028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harder T, Scheiffele P, Verkade P, Simons K. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Herz J, Strickland DK. 2001. LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108:779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heuser JE, Anderson RG. 1989. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J. Cell Biol. 108:389–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hill MM, et al. 2008. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 132:113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu K, Wu C, Mars WM, Liu Y. 2007. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor-related protein 1-mediated integrin signaling. J. Clin. Invest. 117:3821–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kancha RK, Stearns ME, Hussain MM. 1994. Decreased expression of the low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor in invasive cell clones derived from human prostate and breast tumor cells. Oncol. Res. 6:365–372 [PubMed] [Google Scholar]
- 26. Knudson W, Chow G, Knudson CB. 2002. CD44-mediated uptake and degradation of hyaluronan. Matrix Biol. 21:15–23 [DOI] [PubMed] [Google Scholar]
- 27. Lajoie P, Nabi IR. 2007. Regulation of raft-dependent endocytosis. J. Cell Mol. Med. 11:644–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langlois B, et al. 2010. LRP-1 promotes cancer cell invasion by supporting ERK and inhibiting JNK signaling pathways. PLoS One 5:e11584 doi:10.1371/journal.pone.0011584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Y, Lu W, Bu G. 2003. Essential role of the low density lipoprotein receptor-related protein in vascular smooth muscle cell migration. FEBS Lett. 555:346–350 [DOI] [PubMed] [Google Scholar]
- 30. Li Y, Marzolo MP, van Kerkhof P, Strous GJ, Bu G. 2000. The YXXL motif, but not the two NPXY motifs, serves as the dominant endocytosis signal for low density lipoprotein receptor-related protein. J. Biol. Chem. 275:17187–17194 [DOI] [PubMed] [Google Scholar]
- 31. Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. 2008. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88:887–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma Z, et al. 2002. Regulation of Rac1 activation by the low density lipoprotein receptor-related protein. J. Cell Biol. 159:1061–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Macia E, et al. 2006. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10:839–850 [DOI] [PubMed] [Google Scholar]
- 34. Misra S, et al. 2011. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 278:1429–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Montel V, Gaultier A, Lester RD, Campana WM, Gonias SL. 2007. The low-density lipoprotein receptor-related protein regulates cancer cell survival and metastasis development. Cancer Res. 67:9817–9824 [DOI] [PubMed] [Google Scholar]
- 36. Mu FT, et al. 1995. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J. Biol. Chem. 270:13503–13511 [DOI] [PubMed] [Google Scholar]
- 37. Muratoglu SC, Mikhailenko I, Newton C, Migliorini M, Strickland DK. 2010. Low density lipoprotein receptor-related protein 1 (LRP1) forms a signaling complex with the platelet-derived growth factor receptor-β in endosomes and regulates activation of the MAPK pathway. J. Biol. Chem. 285:14308–14317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nestl A, et al. 2001. Gene expression patterns associated with the metastatic phenotype in rodent and human tumors. Cancer Res. 61:1569–1577 [PubMed] [Google Scholar]
- 39. Obermeyer K, et al. 2007. The expression of low density lipoprotein receptor-related protein in colorectal carcinoma. Oncol. Rep. 17:361–367 [PubMed] [Google Scholar]
- 40. Okamoto I, et al. 1999. CD44 cleavage induced by a membrane-associated metalloprotease plays a critical role in tumor cell migration. Oncogene 18:1435–1446 [DOI] [PubMed] [Google Scholar]
- 41. Orian-Rousseau V. 2010. CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 46:1271–1277 [DOI] [PubMed] [Google Scholar]
- 42. Orian-Rousseau V, Chen L, Sleeman JP, Herrlich P, Ponta H. 2002. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 16:3074–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Orr AW, Pallero MA, Xiong WC, Murphy-Ullrich JE. 2004. Thrombospondin induces RhoA inactivation through FAK-dependent signaling to stimulate focal adhesion disassembly. J. Biol. Chem. 279:48983–48992 [DOI] [PubMed] [Google Scholar]
- 44. Persaud-Sawin DA, Lightcap S, Harry GJ. 2009. Isolation of rafts from mouse brain tissue by a detergent-free method. J. Lipid Res. 50:759–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pietrzik CU, et al. 2004. FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J. Neurosci. 24:4259–4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ranganathan S, et al. 2011. Molecular basis for the interaction of low density lipoprotein receptor-related protein 1 (LRP1) with integrin alphaMbeta2: identification of binding sites within alphaMbeta2 for LRP1. J. Biol. Chem. 286:30535–30541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ranganathan S, et al. 2004. Serine and threonine phosphorylation of the low density lipoprotein receptor-related protein by protein kinase Calpha regulates endocytosis and association with adaptor molecules. J. Biol. Chem. 279:40536–40544 [DOI] [PubMed] [Google Scholar]
- 48. Selvais C, et al. 2011. Cell cholesterol modulates metalloproteinase-dependent shedding of low-density lipoprotein receptor-related protein-1 (LRP-1) and clearance function. FASEB J. 25:2770–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Selvais C, et al. 2009. Metalloproteinase-dependent shedding of low-density lipoprotein receptor-related protein-1 ectodomain decreases endocytic clearance of endometrial matrix metalloproteinase-2 and -9 at menstruation. Endocrinology 150:3792–3799 [DOI] [PubMed] [Google Scholar]
- 50. Sid B, et al. 2006. Human thyroid carcinoma cell invasion is controlled by the low density lipoprotein receptor-related protein-mediated clearance of urokinase plasminogen activator. Int. J. Biochem. Cell Biol. 38:1729–1740 [DOI] [PubMed] [Google Scholar]
- 51. Spijkers PP, et al. 2005. LDL-receptor-related protein regulates beta2-integrin-mediated leukocyte adhesion. Blood 105:170–177 [DOI] [PubMed] [Google Scholar]
- 52. Suzuki M, et al. 2002. Kunitz-type protease inhibitor bikunin disrupts phorbol ester-induced oligomerization of CD44 variant isoforms containing epitope v9 and subsequently suppresses expression of urokinase-type plasminogen activator in human chondrosarcoma cells. J. Biol. Chem. 277:8022–8032 [DOI] [PubMed] [Google Scholar]
- 53. Takayama Y, Takahashi H, Mizumachi K, Takezawa T. 2003. Low density lipoprotein receptor-related protein (LRP) is required for lactoferrin-enhanced collagen gel contractile activity of human fibroblasts. J. Biol. Chem. 278:22112–22118 [DOI] [PubMed] [Google Scholar]
- 54. Tammi R, et al. 2001. Hyaluronan enters keratinocytes by a novel endocytic route for catabolism. J. Biol. Chem. 276:35111–35122 [DOI] [PubMed] [Google Scholar]
- 55. Thankamony SP, Knudson W. 2006. Acylation of CD44 and its association with lipid rafts are required for receptor and hyaluronan endocytosis. J. Biol. Chem. 281:34601–34609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tijink BM, et al. 2006. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 12:6064–6072 [DOI] [PubMed] [Google Scholar]
- 57. Webb DJ, Nguyen DH, Gonias SL. 2000. Extracellular signal-regulated kinase functions in the urokinase receptor-dependent pathway by which neutralization of low density lipoprotein receptor-related protein promotes fibrosarcoma cell migration and Matrigel invasion. J. Cell Sci. 113:123–134 [DOI] [PubMed] [Google Scholar]
- 58. Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. 2004. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol. Cell. Biol. 24:8113–8133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu L, Gonias SL. 2005. The low-density lipoprotein receptor-related protein-1 associates transiently with lipid rafts. J. Cell Biochem. 96:1021–1033 [DOI] [PubMed] [Google Scholar]
- 60. Yepes M, et al. 2003. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J. Clin. Invest. 112:1533–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zemskov EA, Mikhailenko I, Strickland DK, Belkin AM. 2007. Cell-surface transglutaminase undergoes internalization and lysosomal degradation: an essential role for LRP1. J. Cell Sci. 120:3188–3199 [DOI] [PubMed] [Google Scholar]
- 62. Zhang G, Cai X, Lopez-Guisa JM, Collins SJ, Eddy AA. 2004. Mitogenic signaling of urokinase receptor-deficient kidney fibroblasts: actions of an alternative urokinase receptor and LDL receptor-related protein. J. Am. Soc. Nephrol. 15:2090–2102 [DOI] [PubMed] [Google Scholar]
- 63. Zhang H, et al. 2004. Localization of low density lipoprotein receptor-related protein 1 to caveolae in 3T3-L1 adipocytes in response to insulin treatment. J. Biol. Chem. 279:2221–2230 [DOI] [PubMed] [Google Scholar]
- 64. Zinchuk V, Zinchuk O, Okada T. 2007. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem. Cytochem. 40:101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zohar R, et al. 2000. Intracellular osteopontin is an integral component of the CD44-ERM complex involved in cell migration. J. Cell. Physiol. 184:118–130 [DOI] [PubMed] [Google Scholar]