

Abstract
Activating transcription factor 3 (ATF3) is a common mediator of cellular stress response signaling and is often aberrantly expressed in prostate cancer. We report here that ATF3 can directly bind the androgen receptor (AR) and consequently repress AR-mediated gene expression. The ATF3-AR interaction requires the leucine zipper domain of ATF3 that independently binds the DNA-binding and ligand-binding domains of AR, and the interaction prevents AR from binding to cis-acting elements required for expression of androgen-dependent genes while inhibiting the AR N- and C-terminal interaction. The functional consequences of the loss of ATF3 expression include increased transcription of androgen-dependent genes in prostate cancer cells that correlates with increased ability to grow in low-androgen-containing medium and increased proliferative activity of the prostate epithelium in ATF3 knockout mice that is associated with prostatic hyperplasia. Our results thus demonstrate that ATF3 is a novel repressor of androgen signaling that can inhibit AR functions, allowing prostate cells to restore homeostasis and maintain integrity in the face of a broad spectrum of intrinsic and environmental insults.
INTRODUCTION
The androgen receptor (AR) mediates androgen signaling essential for male sex differentiation and the male reproductive function (48). It is generally believed that defective androgen signaling contributes to various human male urogenital disorders including androgen-insensitivity syndrome and hypospadias (5, 24). Of particular interest, abnormal androgen signaling due to aberrant expression, mutations, or dysregulation of the AR gene has been linked to prostate tumorigenesis and progression of prostate cancer into advanced, castration-resistant disease (50, 51).
Following activation by androgen binding to its C-terminal ligand-binding domain (LBD), AR is translocated to the nucleus. There, the central DNA-binding domain (DBD) of the receptor binds to androgen responsive elements (ARE) and subsequently regulates expression of a plethora of genes that drive cell differentiation and proliferation (28). The transcriptional activity of AR is mainly carried by its N-terminal domain (NTD) and is regulated by various proteins that interact with AR through distinct mechanisms (21, 60). These AR regulators include components of chromatin remodeling/modifying complexes (e.g., ARIP4 and p300/CBP) that predispose permissive chromatin environments for AR binding and molecular adapters (e.g., SRC/p160 family members) that function to recruit basal transcriptional machinery or other transcriptional regulators to AR target promoters (21). Transcriptional factors (e.g., Foxa1) can also interact with AR and bind to DNA sequences in close proximity to ARE, thereby cooperating with AR to regulate gene expression (11). Since the transcriptional activity of AR is also regulated by an intermolecular interaction between its N terminus and C terminus (N-C interaction) (18, 19), AR-associated proteins like SMRT and caspase-8 repress AR-mediated gene expression by disrupting the N-C interaction (33, 47). Given the importance of AR-binding proteins in regulating AR activity, it is not surprising that aberrant expression or malfunction of AR regulators has often been causally related to androgen insensitivity syndrome, prostate cancer, and other urogenital disorders (1, 20). However, only a few of these AR regulators have been validated for their effects on androgen signaling using genetically engineered mouse models.
Activating transcription factor 3 (ATF3) is a member of the ATF/CREB family of transcription factors and can regulate gene expression by binding to the consensus ATF/CREB cis-regulatory element via a basic-region leucine zipper domain (bZIP) (14). A unique feature distinguishing ATF3 from other ATF/CREB members is that ATF3 is a common stress response mediator and can be rapidly induced by a broad spectrum of cellular stresses, including DNA damage, cell injury, oxidative stress, and endoplasmic reticulum stress (13). Due to its frequent induction by cellular stresses, ATF3 has been considered to play a crucial role in the maintenance of cell integrity and homeostasis under stressful conditions (14, 15). Indeed, whereas ATF3 has been found to play pivotal roles in regulating important cellular signaling pathways mediated by p53, transforming growth factor β (TGF-β), Toll-like receptor 4, or eukaryotic factor 2 kinase (12, 22, 25, 64), aberrant expression of the ATF3 gene is frequently associated with various human diseases including hypospadias and prostate cancer (3, 31, 56). However, details of the function of this common stress response mediator remain largely unknown. Although ATF3 can bind to promoters and repress expression of some genes while activating expression of other genes (14), the findings that ATF3 can interact with other proteins (e.g., p53, Smad3, and E6) via its bZIP domain suggest that ATF3 may regulate cellular functions independent of its transcriptional activity (25, 59, 64). Indeed, the binding of ATF3 to the tumor suppressor p53 can activate the latter protein by protecting it from ubiquitin-mediated degradation (64). Since the bZIP structural motif is a major scaffold for protein-protein interaction (29, 46), exploration of the ATF3 interactome might provide a key to a better understanding of its diverse and context-dependent functions in human diseases. In support of this notion, a recent report showed that ATF3 induces expression of the metastasis suppressor KAI1 gene when it interacts with JunB, whereas the binding of ATF3 to the NF-κB p50 subunit represses expression of the same gene in prostate cancer cells (37).
Here, we sought to explore the potential role of ATF3 in regulating AR-mediated signaling. We report that ATF3 is a novel AR-binding protein. The ATF3-AR interaction not only prevented AR from binding to ARE but also disrupted the N-C interaction of AR, leading to repression of androgen signaling in cultured cells as well as in animals. These findings thus link a common stress response mediator to androgen signaling, suggesting that the ATF3-mediated cellular stress response could serve as a mechanism defending against prostate cancer.
MATERIALS AND METHODS
Cell culture and transfections.
LNCaP, VCaP, and PC3 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum. For androgen treatments, cells were cultured in medium containing 10% charcoal-stripped fetal bovine serum for 2 days, followed by addition of R1881 (methyltrienolone; Perkin Elmer) into the medium. Total cellular lipid levels were measured by AdipoRed assays (Lonza) as described by the manufacturer. Transfections were carried out using Lipofectamine 2000 (Invitrogen).
GST pulldown and co-IP assays.
Glutathione S-transferase (GST) pulldown assays were carried out as described previously (64). Essentially, GST or GST fusion proteins were immobilized on glutathione-agarose (Sigma) and then incubated with in vitro-translated proteins or recombinant proteins at 4°C overnight. After extensive washes, bound proteins were eluted and detected by fluorography or Western blotting. For coimmunoprecipitation (co-IP) assays, cell lysates (1 mg) were incubated with the AR (H280; Santa Cruz Biotechnology), ATF3 (C-19; Santa Cruz), or FLAG antibody (Sigma) at 4°C overnight. Thirty microliters of protein A- or protein G-agarose was added, followed by extensive washes of the agarose. Precipitated proteins were then detected by Western blotting (64).
shRNA knockdown and retroviral infections.
Knockdown of ATF3 expression was carried out using a lentivector-based short hairpin RNA (shRNA) system (pSIH-H1 shRNA cloning and lentivector expression system; System Biosciences) following the manufacturer's protocol. The ATF3-targeted sequence was 5′-GCA AAG TGC CGA AAC AAG A-3′ or 5′-GAG AAA CCT CTT TAT CCA A-3′, based on our earlier reports (59). For negative controls, a luciferase-targeted sequence (5′-CTT ACG CTG AGT ACT TCG A-3′) was cloned into the lentivector. For retroviral infections, the ATF3 cDNA was cloned into pBabe-puro and packaged into retroviral particles as previously described (59).
Quantitative reverse transcription-PCR (qRT-PCR).
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) and then reverse transcribed and subjected to real-time PCR assays as previously described (63). The sequences of primers used for amplifying AR and AR target genes are available on request. To determine expression of ATF3 and AR target genes in human prostate cancer, we purchased a TissueScan Prostate Cancer qPCR array from Origene (HPRT102) and subjected the cDNA samples to real-time PCR assays. Only cDNA samples derived from tissues consisting of at least 90% prostate cancer cells were used for analysis. All prostate cancer samples were collected by the manufacturer under protocols approved by the Institutional Review Board (Origene, Rockville, MD).
Gel shift assays.
Recombinant ATF3 and AR protein containing the DBD region (amino acids [aa] 537 to 644 [AR-DBD]) were purified using nitrilotriacetic acid (NTA)-Ni+-agarose as described previously (36, 64). An oligonucleotide containing the ARE in the PSA enhancer (5′-TGGAGGAACATATTGTATTGATTGT-3′) or a mutated oligonucleotide (5′-TGGAGGAATATATTATATTGATTGT-3′; mutations are underlined) (58) was labeled with [α-32P]ATP and incubated with 500 ng of AR-DBD protein in a buffer containing 25 mM HEPES, pH 7.5, 0.5 mM EDTA, 0.5 mM dithiothreitol (DTT), 2.5% glycerol, 50 mM NaCl, and 1 μg of poly(dI-dC) in the absence or presence of recombinant ATF3 (500, 1,000, or 2,000 ng) or bovine serum albumin (BSA) at room temperature for 20 min and then resolved by nondenatured polyacrylamide electrophoresis followed by autography. Nuclear extracts were prepared from LNCaP cells, and 20 μg of nuclear extract was used for binding assays as described previously (65).
Androgen binding assays.
The ligand-binding affinity of AR was determined using a whole-cell binding assay as described previously (30, 42). Briefly, PC3 cells (5 × 104) cultured in charcoal-stripped medium (CSM) were transfected with 0.1 μg of pcDNA3-FLAG-AR with or without 0.1 μg of pCG-ATF3 in 48-well plates for 2 days, followed by incubation with various concentrations of [3H]R1881 (Perkin Elmer) for 2 h. Cells were then extensively washed with phosphate-buffered saline (PBS). The total bound [3H]R1881 was extracted with 100 μl of cold ethanol for 20 min and measured using a scintillation counter. Nonspecific binding at each concentration was measured by adding 1 μM unlabeled R1881 and then subtracted from the total binding to generate values for specific binding. The maximal binding (Bmax) and binding affinity (Kd) values were calculated using SigmaPlot according to the following equation: specific binding = Bmax [L]/(Kd + [L]), where [L] is the concentration of [3H]R1881.
ChIP.
Chromatin immunoprecipitation (ChIP) assays were carried out as descried previously with modifications (63). Essentially, cells cultured in 150-mm dishes were cross-linked with 1% formaldehyde and then lysed for sonication. Cell lysates were incubated with an AR antibody (H-280; Santa Cruz) at 4°C overnight, followed by addition of 30 μl of single-stranded DNA (ssDNA)-saturated protein A-agarose (Upstate). The protein-DNA complexes were eluted, and cross-link was reversed at 65°C overnight. DNA fragments were then purified using a QIAquick PCR purification kit (Qiagen) for real-time PCR assays using primers described previously (2).
Animal experiments and immunohistochemistry.
Animal experiments were carried out according to a protocol approved by the Institutional Committee of Animal Care and Use of the Albany Medical College. For castration, testes of 8-week-old of ATF3 wild-type (WT) and knockout (KO) mice were surgically removed. These mice were housed for 21 days and then injected subcutaneously with 40 mg/kg of testosterone (Sigma) dissolved in corn oil every day for a total of 14 days. Mice in groups of three or four were sacrificed on days 3, 7, 14, and 21 postcastration or on days 3, 7, and 14 after testosterone replenishment, respectively. Prostate lobes were separated from these mice by microdissection and embedded in paraffin for sectioning and histological examination. For immunohistochemistry (IHC), antigens were retrieved in hot citrate buffer. Sections were then blocked in 5% normal horse serum and 1% normal goat serum, incubated with primary antibodies, and stained using an ABC Elite Kit and a DAB (3,3′-diaminobenzidine) Kit (Vector) according to the manufacturer's recommendations. The Ki-67 antibody (1:400) and AR antibody (1:200) were purchased from Abcam (ab15580) and Santa Cruz (N-20), respectively.
RESULTS
ATF3 interacts with AR via its ZIP domain.
We carried out GST pulldown assays to determine whether ATF3 interacts with AR. Immobilized GST-ATF3, but not GST, pulled down in vitro-translated AR protein (Fig. 1A, lane 3), supporting a direct interaction between ATF3 and AR. To corroborate this finding, we coexpressed ATF3 and AR (tagged with FLAG) in PC3 cells, which are null for AR and express a low level of ATF3 (see Fig. S1, lane 2, in the supplemental material), and performed reciprocal coimmunoprecipitation (co-IP) assays using anti-FLAG (Fig. 1B) and anti-ATF3 antibody (Fig. 1C), respectively. Indeed, the FLAG antibody precipitated ATF3 only in the presence of AR but failed to do so in the absence of AR (Fig. 1B, lane 2 versus lane 1). Similarly, the ATF3 antibody precipitated AR only in the presence of ATF3 (Fig. 1C, lane 2). We also examined the interaction between endogenous ATF3 and AR proteins using LNCaP cells, which express high levels of both proteins (see Fig. S1, lane 1). In line with the notion that ATF3 interacts with AR, the ATF3 antibody, but not IgG, precipitated both ATF3 and AR from the cell lysates (Fig. 1D, lane 3 versus lane 2). It is unlikely that the ATF3-AR interaction is affected by the binding of androgen to AR since GST-ATF3 pulled down AR in the presence of R1881 as efficiently as it did without androgen treatments (Fig. 1E, lane 5 versus lane 3). Further GST-pulldown experiments using truncated ATF3 proteins indicated that the ATF3-AR interaction was mediated by the ATF3 C terminus (amino acids [aa] 81 to 181) (Fig. 1F, lane 5). Moreover, an ATF3 mutant lacking the ZIP region (a deletion of residues 102 to 139 [Δ102-139]) failed to pull down AR in GST pulldown assays (Fig. 1G, lane 4 versus lane 3) and was not coprecipitated with AR by an AR-specific antibody (Fig. 1H, lane 8 versus lane 6). It is unlikely that the inability of ATF3 Δ102-139 to bind AR was caused by inappropriate folding of the mutant protein as we previously showed that ATF3 Δ102-139 retained the ability to dimerize with wild-type ATF3 through the N terminus (64). These results thus demonstrate that ATF3 binds AR and that this interaction requires the ZIP domain of ATF3.
Fig 1.

ATF3 interacts with AR via its ZIP domain. (A) GST-ATF3 or GST was immobilized onto glutathione-agarose and incubated with AR labeled with [35S]methionine by in vitro translation. After extensive washes, bound proteins were eluted and visualized by electrophoresis followed by fluorography. (B) PC3 cells were transfected with the indicated plasmids and then subjected to co-IP using anti-FLAG antibody. Precipitated proteins were detected by Western blotting. (C) AR was coexpressed with or without ATF3 in PC3 cells by transfections. Cell lysates were subjected to co-IP using anti-ATF3 antibody followed by Western blotting. (D) LNCaP cell lysates were incubated with anti-ATF3 antibody or rabbit IgG at 4°C overnight and then precipitated with protein A-agarose. Bound proteins were eluted and subjected to Western blotting. (E) In vitro-translated AR was preincubated with or without 100 nM R1881 for 1 h and then subjected to GST pulldown assays to examine the ATF3-AR interaction. (F) The indicated GST-ATF3 fusions were incubated with 35S-labeled AR and subjected to GST pulldown assays. (G) The full-length ATF3 and a mutant lacking the ZIP domain (Δ102–139) were fused to GST and incubated with in vitro-translated AR for GST pulldown assays. Bound proteins were detected by Western blotting with the anti-AR antibody. (H) ATF3 or ATF3 Δ102–139 was coexpressed with or without AR in PC3 cells and then subjected to co-IP assays with anti-AR antibody, followed by Western blotting. α, anti.
ATF3 represses the transactivation activity of AR.
Since the transactivation activity of AR is often regulated by its associated proteins (21), we carried out luciferase reporter assays to test whether ATF3 affects the ability of AR to regulate gene expression. As expected, AR promoted transcription of a luciferase reporter driven by a synthetic promoter (ARE-luc) containing four tandem repeats of ARE (5′-AGAACAGCAAGTGCT-3′) (36) (Fig. 2A). Intriguingly, expression of ATF3 repressed AR-mediated transactivation of the reporter in a dose-dependent manner (Fig. 2A). Such repression was not due to a decrease in the AR expression level nor caused by direct repression of the promoter by ATF3 (Fig. 2A). Similar results were obtained using a reporter driven by a native AR target promoter, the probasin (PB) promoter (67) (Fig. 2B). Of note, the synthetic ARE promoter and the PB promoter do not contain any ATF/CREB sequence (5′-TGACGTCA-3′), and thus the repression of AR-mediated transcription was likely independent of the transcriptional activity of ATF3. The repression of the AR transcriptional activity by ATF3 was rather a consequence of binding of ATF3 to AR, as the ATF3 mutant (Δ102–139) deficient in AR binding (Fig. 1G and H) failed to repress AR-mediated activation of the PB promoter (Fig. 2C). These results indicate that the ATF3-AR interaction repressed the transactivation activity of AR. ATF3 also repressed transcription mediated by glucocorticoid receptor (GR) or progesterone receptor (PR) (Fig. 2D), two steroid receptors that are highly homologous to AR in the DNA binding domain (7). However, ATF3 did not decrease the activity of a constitutively active simian virus 40 (SV40) promoter (Fig. 2D), arguing against the possibility that ATF3 repressed gene transcription in general.
Fig 2.

ATF3 represses the transactivation activity of AR. (A) PC3 cells were cotransfected with ARE-Luc, pRL-CMV (where CMV is cytomegalovirus), AR, and/or increasing amounts of ATF3 in charcoal-stripped medium for 1 day and then treated with 1 nM R1881 for dual luciferase activity assays. AR and ATF3 expression levels were determined by Western blotting after normalization using the Renilla luciferase activity. Data are depicted as averages ± standard deviations of three determinations. (B) PC3 cells were cotransfected with PB-Luc, pRL-CMV, AR, and/or increasing amounts of ATF3 for dual luciferase activity assays. Immunoblots show expression levels of AR and ATF3. Data are depicted as averages ± standard deviations of three determinations. (C) PC3 cells were cotransfected with PB-Luc, pRL-CMV, ATF3, or ATF3 Δ102–139 for dual luciferase activity assays. Data are depicted as averages ± standard deviations of three determinations. ns, not significant. (D) In the experiments shown in the left and middle graphs, PC3 cells were cotransfected with ARE-Luc, pRL-CMV, GR, or PR and/or increasing amounts of ATF3 in charcoal-stripped medium for 1 day and then treated with 10 nM dexamethasone (DEX) or 10 μM progesterone (Pg) for 1 day for dual luciferase activity assays. In the experiment shown in the right graph, PC3 cells were cotransfected with pGL3-promoter (containing the SV40 promoter), pRL-CMV, and/or increasing amounts of ATF3 for dual-luciferase activity assays. Data are depicted as averages ± standard deviations of three determinations.
ATF3 represses androgen signaling in prostate cancer cells.
AR-mediated androgen signaling regulates gene expression, leading to multiple outcomes that include increased cell proliferation and de novo synthesis of intracellular lipids (43). To test whether ATF3 can affect androgen signaling in prostate cancer cells, we stably knocked down ATF3 expression with a shRNA (shATF3) (59) in LNCaP cells and determined expression of AR target genes using qRT-PCR. Consistent with the notion that ATF3 represses AR transactivation, downregulation of ATF3 expression increased androgen-induced PSA expression at both the protein (Fig. 3A, lane 4 versus lane 3) and the mRNA level (Fig. 3B). The R1881-mediated induction of TMPRSS2, another well-characterized AR target gene, was also significantly enhanced in shATF3-expressing cells (Fig. 3B). The shATF3 cells appeared to express a higher level of PSA than the control cells expressing shRNA targeting luciferase (shLuc) in the absence of R1881 (Fig. 3A, lane 2 versus lane 1, and B), probably due to the fact that the charcoal-stripped medium (CSM) contained trace amounts of androgens sufficient to promote AR to bind to target genes. Indeed, AR bound to the PSA enhancer at a low level under similar conditions (see Fig. 7E). To confirm these results and ensure that the observation was not a consequence of clonal variation, we transiently knocked down ATF3 expression in LNCaP cells using lentiviral infection (see Fig. S2 in the supplemental material) and then determined mRNA levels of various androgen-dependent genes including PSA, TMPRSS2, Rhou, SLC41A, NKX3.1, and DHCR24 by qRT-PCR. Consistent with the results shown in Fig. 3A and B, transient knockdown of ATF3 expression significantly increased R1881-induced PSA and TMPRSS2 expression (Fig. 3C). Moreover, the androgen-induced expression of other AR target genes was also increased by downregulation of ATF3 expression (Fig. 3C). These results indicate that ATF3 can indeed repress AR-mediated gene expression. To further test this notion, we overexpressed ATF3 in VCaP cells, which normally express low levels of the protein (see Fig. S1, lane 4), using a retroviral vector (pBabe) and determined effects of ATF3 expression on androgen-dependent gene expression. As expected, androgen-induced expression of AR target genes (i.e., PSA and TMPRSS2) was significantly repressed in VCaP cells expressing ectopic ATF3 (Fig. 3D). The androgen-induced decrease in the AR mRNA level in VCaP cells (Fig. 3D) was likely due to self-repression (6). ATF3 expression did not appear to have an effect on this regulation (Fig. 3D). Taken together, our results demonstrate that ATF3 can repress AR-mediated gene expression in prostate cancer cells. In line with this notion, expression levels of PSA and TMPRSS2 in human prostate cancer samples were inversely correlated to the ATF3 expression level (Pearson's correlation r of −0.620 and −0.638, respectively; P < 0.01) (Fig. 3E and F).
Fig 3.

ATF3 represses androgen signaling in prostate cancer cells. (A and B) LNCaP cells were infected with lentiviruses expressing shATF3 or shLuc and selected with puromycin for 4 weeks. A clone stably expressing shATF3 was cultured in CSM for 2 days, treated with 1 nM R1881 for 24 h, and then lysed for Western blotting (A) or qRT-PCR assays (B). Data are depicted as averages ± standard deviations of three determinations. (C) LNCaP cells were infected with shATF3-expressing lentiviruses for 2 days, followed by being cultured in CSM for 2 days and then treated with 1 nM R1881 for 24 h. Cells were lysed for qRT-PCR assays to measure mRNA levels of indicated androgen-dependent genes. Data are depicted as averages ± standard deviations of three determinations. (D) VCaP cells infected with retroviruses expressing ATF3 or empty vector (pBabe) were cultured in CSM for 2 days, followed by treatments with 1 nM R1881 for 1 day. Levels of the indicated mRNAs were measured by qRT-PCR. Data are depicted as averages ± standard deviations of three determinations. (E and F) A qRT-PCR tissue array was used to measure ATF3, PSA, and TMPRSS2 mRNA levels in human prostate cancer samples (n = 20). Relative mRNA levels were converted to logarithms (log2) and plotted for each sample. A linear regression line and Pearson's correlation (r) are shown for each graph. (G) LNCaP cells infected with lentiviruses expressing ATF3-specific shRNA (shATF3-1 and shATF3-2) or shLuc were cultured in CSM for 2 days. Various amounts of R1881 were added on days 0, 3, 6, and 9, and numbers of viable cells were measured on day 10 by MTT [3-(4,5-dimethylthiazol-2-yl)2 2,5-diphenyl tetrazolium bromide] assays. Data are depicted as averages ± standard deviations of three determinations. (H) LNCaP cells expressing shATF3 or shLuc were cultured in CSM for 2 days, followed by treatments with R1881. Levels of cellular lipids were measured by AdipoRed assays. Data are depicted as averages ± standard deviations of three determinations. RFU, relative fluorescence units.
Fig 7.

ATF3 prevents AR from binding to ARE both in vitro and in vivo. (A) The purified AR-DBD protein (aa 537 to 644; 500 ng) was incubated with 32P-labeled oligonucleotide containing ARE derived from the PSA enhancer (lanes 1 to 4) or a mutated oligonucleotide (lanes 5 and 6) and subjected to gel shift assays. For competition assays (lanes 3 and 4), 50-fold and 100-fold amounts of unlabeled oligonucleotide were mixed with labeled oligonucleotide and AR-DBD protein. The arrow indicates the AR-ARE binding complex. (B) Increasing amounts of purified ATF3 proteins (100, 200, and 500 ng) or BSA were preincubated with AR-DBD protein at 4°C for 30 min and then subjected to gel shift assays. The arrow indicates the AR-DNA complex. (C) An equal amount (500 ng) of ATF3 or the ATF3 Δ102–139 mutant was preincubated with the AR-DBD protein for gel shift assays. The arrow indicates the AR-DNA complex. (D) LNCaP cells stably expressing shATF3 or shLuc were cultured in CSM for 2 days and then treated with 1 nM R1881 for 24 h. Nuclear extracts were prepared and incubated with 32P-labeled oligonucleotide as described for panel A. The main AR-DNA binding band is indicated by the arrow. (E) LNCaP cells expressing shATF3 or shLuc were treated as described for panel D and then fixed with formaldehyde for ChIP assays using anti-AR antibody or control IgG. Real-time PCR was used to quantify amounts of DNA fragments spanning the ARE in the TMPRSS2 enhancer, the PSA enhancer, or the PSA proximal promoter, as indicated. For specificity control, a random fragment in the GAPDH coding region was also amplified and quantified by real-time PCR. Data are depicted as averages ± standard deviations of three determinations. The P values were calculated using the Student t test.
AR-mediated gene expression promotes prostate cancer cell growth and intracellular lipid synthesis (43). We therefore tested whether ATF3 expression affects these androgen signaling events in prostate cancer cells. Consistent with the increased expression of AR target genes, knockdown of ATF3 expression with two independent shRNAs (59) sensitized LNCaP cells to respond to androgen stimulation (Fig. 3G and H). Both cell growth in response to low concentrations of R1881 (Fig. 3G) and androgen-stimulated total lipid accumulation (Fig. 3H) were significantly increased in the shATF3 cells compared to levels in control shLuc cells. Our results thus indicate that ATF3 is a novel repressor of androgen signaling in prostate cancer cells.
ATF3 deficiency promotes prostate epithelial proliferation in mice.
Transgenic AR gene expression in mice results in increased cell proliferation in the prostate (53), in line with the notion that androgen signaling functions to sustain epithelial proliferation in adult prostates. To determine whether ATF3 represses androgen signaling under physiological conditions, we examined proliferation of prostate epithelial cells in adult ATF3 knockout (KO) mice by immunohistochemical (IHC) staining for Ki-67 expression. The ATF3 KO mice were developed and characterized previously (16). As expected, proliferating (Ki-67-positive) cells were rare in the wild-type (WT) prostates (53). Interestingly, although the AR expression levels were similar between the WT and KO cells (Fig. 4A), we found that numbers of Ki-67-positive cells in ATF3-deficient prostates were significantly increased (Fig. 4A and B). Consistent with the increased cell proliferation, benign epithelial hyperplasia was frequently found in prostates of the ATF3 KO mice starting at the age of 2 months (Fig. 4C). The anterior (AP), ventral (VP), and dorsal-lateral (DLP) prostates of KO mice (8 weeks of age) exhibited increased numbers of epithelial cells, enhanced epithelial infolding and focal tufting, decreased secretion, and a thickened smooth-muscle layer in stroma around epithelial hyperplasia (Fig. 4C). In comparison, the WT glands showed a secretory duct-acinus system composed of cuboidal luminal epithelial cells with no apparent infolding or tufting morphology (Fig. 4C). We did not detect any overt malignant lesions in the prostates of these young ATF3 KO mice, an observation which is not unexpected as AR-transgenic mice developed prostatic lesions only after a long latency (>1 year) (53). These results thus suggest that the AR activity was likely increased in the ATF3 KO mice.
Fig 4.

ATF3 deficiency promotes prostate epithelial proliferation in mice. (A) Anterior prostates of ATF3 wild-type (WT) and knockout (KO) mice (8 weeks of age) were embedded in paraffin, sectioned, and stained for Ki-67 or AR expression. Arrows indicate Ki-67-positive cells. (B) At least 1,000 luminal cells of anterior prostates (AP), ventral prostates (VP), or dorso-lateral prostates (DLP) of ATF3 WT and KO mice were counted for Ki-67 positivity under a microscope (random ×20 fields). Data are depicted as averages ± standard deviations of three determinations. *, P < 0.05, Student t test (n = 3). (C) Prostates of WT and ATF3 KO mice were subjected to hematoxylin and eosin staining. (D, E, and F) ATF3 WT and KO mice were castrated for 21 days, followed by subcutaneous injections of 40 mg/kg testosterone for 14 days. Prostates were sectioned and stained for Ki-67 expression (D). Ki-67-positive epithelial cells in anterior prostates were counted, and the results are shown in panel E. Hematoxylin and eosin staining of representative AP sections is shown in panel F. Data are depicted as averages ± standard deviations of three determinations. *, P < 0.05, Student t test (n = 3). (G) Ventral prostates were dissected for RNA preparation and used for qRT-PCR assays for AR target gene expression. Data are depicted as averages ± standard deviations of three determinations. ***, P < 0.001, Student t test.
Histopathological changes of prostate glands caused by castration and androgen replacement are often used to evaluate androgen signaling in rodents. To confirm that ATF3 deficiency promotes androgen signaling in mouse prostates, we castrated the WT and KO mice for 21 days and then replenished these mice with testosterone for 14 days. ATF3 expression in prostate epithelial cells of wild-type mice was increased by castration (see Fig. S3A in the supplemental material), similar to the response of LNCaP cells upon androgen deprivation (see Fig. S3B). This result is consistent with the notion that prostate tissues are subjected to acute cellular stresses (e.g., oxidative stress) during the dramatic remodeling that occurs subsequent to castration (54). As expected, androgen deprivation by castration resulted in almost complete loss of proliferation (Fig. 4D and E; see also Fig. S4A and B in the supplemental material) and epithelial atrophy in anterior prostates of the WT mice (see Fig. S3C). In contrast, significant numbers of Ki-67-positive cells were detected in the prostates of the ATF3 KO mice even 21 days after castration (Fig. 4D and E; see also Fig. S4A and B), suggesting that ATF3 deficiency could sensitize AR signaling to respond to the postcastration androgen level. However, the ATF3-deficient prostates still underwent atrophy after castration (see Fig. S4C). Testosterone replenishment subsequent to castration dramatically stimulated proliferation of prostate epithelial cells (Fig. 4D and E), resulting in luminal epithelial repopulation in the WT mice (Fig. 4F). In contrast, the epithelium of prostates of ATF3 KO mice was hyperplastic by 7 days after androgen replenishment (Fig. 4F), consistent with a significant increase in the numbers of Ki-67-positive luminal cells in the KO animals (Fig. 4D and E; see also Fig. S4A and B). Most importantly, testosterone-induced expression of AR target genes (i.e., Pbsn and Msmb) was significantly increased in ATF3-deficient prostates compared to levels in WT prostates (Fig. 4G). These results thus support the notion that ATF3 deficiency enhances androgen signaling in regrowing mouse prostates. Of note, ATF3 deficiency had negligible effects on AR expression under these experimental conditions (see Fig. S4D).
ATF3 does not block AR nuclear translocation.
Having shown that ATF3 represses androgen signaling in both cultured cells and a genetically engineered animal model, we sought to explore the underlying mechanism. Upon androgen stimulation, AR translocates from the cytoplasm to the nucleus and binds to ARE to transactivate gene expression. AR-interacting proteins may interfere with AR nuclear translocation and consequently repress AR signaling (60). To test whether ATF3 affected AR nuclear translocation, we coexpressed ATF3 (fused to mCherry) and AR (fused to green fluorescent protein [GFP]) in PC3 cells and examined cells under a fluorescence microscope. As expected (41, 64), ATF3 was predominantly localized in the nucleus, and this subcellular localization pattern was not altered in the presence of androgen (Fig. 5A). Conversely, AR was mainly localized in the cytoplasm in the absence of the androgen but rapidly translocated to the nucleus after androgen stimulation (Fig. 5A). Coexpression of ATF3 with AR did not change the percentage of cells with nuclear AR following androgen supplementation (Fig. 5A and B), indicating that ATF3 did not block AR nuclear translocation.
Fig 5.
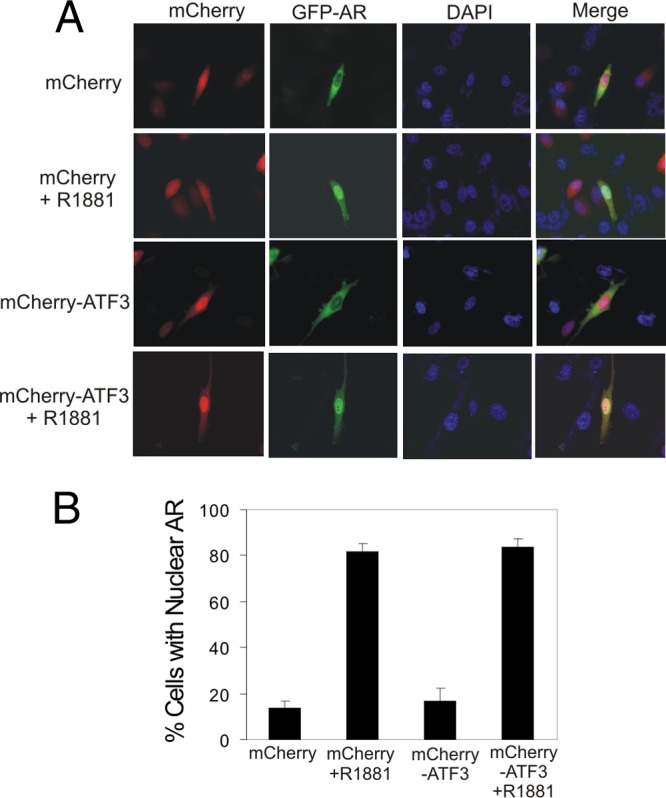
ATF3 does not affect nuclear translocation of AR. PC3 cells were cotransfected with GFP-AR with mCherry-ATF3 or mCherry and then cultured in CSM for 2 days followed by treatments with 1 nM R1881 for 1 h. Cells were then fixed with paraformaldehyde, stained with DAPI (4′,6′-diamidino-2-phenylindole), and observed under a fluorescence microscope (A). At least 300 GFP/mCherry-positive cells were counted for nuclear AR localization (B). Data are depicted as averages ± standard deviations of three determinations.
ATF3 binds AR at its DBD and LBD regions.
To gain an insight into the mechanism by which ATF3 represses AR signaling, we carried out GST pulldown assays to identify the AR region(s) responsible for ATF3 binding using various truncated AR proteins. The results indicate that ATF3 bound two AR regions: (ii) aa 537 to 662 (Fig. 6A, lane 3), which contains the DBD and hinge regions, and (ii) aa 663 to 919 (Fig. 6A, lane 4), which coincides with the LBD region. The interaction between ATF3 and aa 537 to 662 was stronger than the interaction with the LBD. Further experiments showed that ATF3 did not bind the hinge region (aa 624 to 662) or the neighboring region within the LBD (aa 624 to 804) (Fig. 6A, lanes 7 and 8), indicating that ATF3 binds AR at the DBD and the C terminus of LBD. To corroborate these results, we incubated a purified, histidine-tagged, recombinant AR protein that contains the DBD region (aa 537 to 644) (AR-DBD) (36) with GST-ATF3 for GST pulldown experiments. Indeed, the immobilized GST-ATF3, but not GST, could pull down the recombinant AR-DBD protein (Fig. 6B, lane 3 versus lane 2). Since the recombinant LBD C-terminal peptide (aa 804 to 919 of AR [AR804–919]) was insoluble in our bacterial lysates, we constructed a vector expressing a FLAG-tagged C-terminal fragment (FLAG-AR804–919) and carried out co-IP assays to confirm the ATF3-LBD interaction. ATF3 was efficiently immunoprecipitated by the FLAG antibody in the presence of the AR C-terminal fragment (Fig. 6C, lane 2), confirming that ATF3 indeed bound the C terminus of the LBD.
Fig 6.

ATF3 binds AR at the DBD and LBD regions. (A) Indicated GST-AR fusion proteins were incubated with in vitro-translated ATF3 and subjected to GST pulldown assays. (B) A histidine-tagged AR fragment (aa 537 to 644) containing the DBD was purified by NTA-Ni+-agarose and incubated with immobilized GST-ATF3 or GST for GST pulldown assays. (C) ATF3 was coexpressed with or without FLAG-AR804–919 in PC3 cells by transfections. Cell lysates were subjected to co-IP assays with anti-FLAG antibody.
ATF3 blocks AR binding to its target promoters/enhancers.
The finding that ATF3 directly bound the AR DBD region suggests a possibility that ATF3 represses AR-mediated gene expression by preventing AR from binding to ARE. To explore this possibility, we carried out gel shift assays using a recombinant ATF3 protein (59) and the recombinant AR-DBD protein to determine whether ATF3 can affect the DNA binding activity of AR. The recombinant ATF3 protein retained the capability to bind the AR DBD region (see Fig. S5, lane 3, in the supplemental material). As expected, the AR-DBD protein bound and reduced the electrophoretic motility of an oligonucleotide containing an ARE derived from the PSA enhancer region (58) (Fig. 7A, lane 2) but failed to bind an oligonucleotide containing the mutated ARE (Fig. 7A, lane 6). The specificity of the AR DNA binding was further evidenced by results demonstrating that the shifted band was diminished in the presence of the unlabeled oligonucleotide (competitor) (Fig. 7A, lanes 3 to 4). Interestingly, addition of ATF3, but not an AR-unrelated protein (BSA), to the assays significantly decreased AR binding to the oligonucleotide in a dose-dependent manner (Fig. 7B, lanes 3 to 5 versus lane 2). The largest amount of ATF3 almost completely blocked AR from binding to the ARE (Fig. 7B, lane 5). These results were probably not due to competitive binding of the ARE by ATF3 since no additional protein-DNA complex was noted in the assays. Rather, the ATF3-induced loss of DNA binding was due to direct interaction of ATF3 with the DBD region of AR as the recombinant ATF3 Δ102-139 protein deficient in binding to the AR DBD region (see Fig. S5, lane 8, in the supplemental material) failed to inhibit the DNA binding activity of AR (Fig. 7C, lane 4 versus lane 3). These results thus indicate that the ATF3-AR interaction could indeed prevent AR from binding to ARE. To corroborate these results, we carried out gel shift assays using nuclear extracts prepared from LNCaP cells where ATF3 expression was knocked down by shRNA (shATF3) (Fig. 3A). Knockdown of ATF3 expression, which had little effect on AR expression (Fig. 3A), significantly increased the amount of AR bound by the labeled oligonucleotide (Fig. 7D, lane 5 versus lane 4). The observed DNA-binding band(s) was likely specific to AR as the binding was negligible when the cells were cultured in the androgen-deprived medium (CSM) but was strongly induced when the cells were treated with R1881 (Fig. 7D). Thus, our results indicated that ATF3 could affect AR binding to its target genes. To further explore this possibility, we carried out chromatin immunoprecipitation (ChIP) assays to determine enrichment of AR on the genomic regions spanning the functional ARE in the TMPRSS2 enhancer and the PSA enhancer, as well as the PSA proximal promoter, using LNCaP cells expressing a high (shLuc) or decreased (shATF3) ATF3 level. As expected, while very little AR was associated with these ARE-containing regions in the absence of R1881, the androgen strongly promoted AR binding to these promoter/enhancers but not an unrelated genomic region (glyceraldehyde-3-phosphate dehydrogenase [GADPH] coding region) (Fig. 7E). Intriguingly, the amounts of AR bound to these AR target genes were significantly increased in the shATF3 cells (Fig. 7E). Of note, trace amounts of androgens existing in CSM appeared sufficient to mediate a low level of AR binding to the promoters/enhancers (Fig. 7E). Taken together, our results demonstrate that ATF3 binds AR and prevents the latter protein from binding to target genes.
ATF3 does not affect androgen binding but disrupts the AR N-C interaction.
Although it bound to the AR C-terminal ligand-binding domain, ATF3 did not appear to interfere with the AR-androgen interaction since R1881 bound to AR in the presence of ATF3 with the same affinity (Kd of 0.158 ± 0.028) as it did without ATF3 expression (Kd of 0.156 ± 0.018) (Fig. 8A). The slight increase (∼27%) in the maximal binding capability (Bmax) of the ATF3-expressing cells was probably because AR was expressed at a slightly high level in these cells (Fig. 8A, inset blots). Since the intermolecular N-C interaction is required for AR binding to ARE and can promote AR transactivation activity (18, 19), we explored, using a mammalian two-hybrid assay (52), whether the binding of ATF3 to the AR C terminus could affect the AR N-C interaction. The binding of the AR N-terminal fragment [VP16-AR(AB)] to the AR C-terminal fragment [Gal4-AR(DE)] (Fig. 8B) recruited the VP16 transactivation domain to a promoter containing Gal4-binding sites (Gal4-Luc) in the presence of androgen, resulting in increased luciferase expression, as expected (Fig. 8C, bar 2 versus bar 1). Expression of ATF3 dramatically decreased the VP16-induced reporter activity in the presence of Gal4-AR(DE) (Fig. 8C, bars 3 to 5) but had little effect on the reporter activity when only one of the AR fragments was present (Fig. 8C, bars 6 to 12), indicating that ATF3 inhibited the AR N-C interaction. Of note, ATF3 had little effect on the transactivation activity of VP16 as measured using a Gal4-VP16 fusion protein (Fig. 8D, bars 3 to 5). The inhibitory effect on the N-C interaction likely required the ATF3-AR interaction, since loss of the AR-binding region (Δ102-139) significantly impaired the effect (Fig. 8E). Therefore, ATF3 bound the AR DBD and LBD regions and likely repressed androgen signaling through two distinct mechanisms: by preventing AR from binding to target promoters/enhancers and by disrupting the AR N-C interaction.
Fig 8.

ATF3 does not affect the AR-androgen interaction but inhibits the AR N-C interaction. (A) PC3 cells cotransfected with AR and/or ATF3 were cultured in charcoal-stripped medium for 2 days and then incubated with the indicated amounts of [3H]R1881 for 2 h. After extensive washing, bound [3H]R1881 was extracted with methanol, and extract solutions were subjected to scintillation counting. Nonlinear regression was used to calculate Bmax and Kd. Inserted blots show AR expression levels. (B) Diagram representing the AR fragments used in the mammalian two-hybrid assay. (C) PC3 cells were transfected with Gal4-Luc, pRL-TK, Gal4-AR(DE), VP16-AR(AB), and/or increasing amounts of ATF3 in charcoal-stripped medium and then treated with 1 nM R1881 for 1 day for dual luciferase activity assays. Data are depicted as averages ± standard deviations of three determinations. (D) PC3 cell were transfected with Gal4-Luc, Gal4-VP16 (in pBIND), and/or increasing amounts of ATF3 as indicated for dual luciferase activity assays. Data are depicted as averages ± standard deviations of three determinations. (E) PC3 cells were transfected with Gal4-Luc, pRL-TK, Gal4-AR(DE), VP16-AR(AB), ATF3, or Δ102–139 for dual luciferase activity assays. Expression of the AR fragments and ATF3 and Δ102–139 was determined by Western blotting. Data are depicted as averages ± standard deviations of three determinations.
DISCUSSION
The cellular stress response is a common mechanism safeguarding cell integrity under various intrinsic and extrinsic insults. The fact that ATF3 expression is rapidly induced by diverse cellular stresses including DNA damage and oxidative stress suggests that ATF3 plays a general role in regulating the cellular stress response (14). Here, we have identified ATF3 as a novel repressor of AR-mediated cell signaling in normal and cancerous prostate epithelial cells. To our knowledge, ATF3 is the only AR regulator known to directly interact with AR and respond to such a broad range of intracellular and environmental cues. Our finding thus has revealed a missing link between androgen signaling and the cellular stress response. In this regard, the repression of AR-mediated gene expression by ATF3 may function as a common mechanism for the cell to repress AR functions (e.g., to inhibit cell proliferation) in order to restore homeostasis under various adverse and stressed conditions. Indeed, whereas AR can drive chromosomal translocations and subsequent expression of fusion genes to promote cellular transformation (34, 38), androgen signaling is repressed by cellular stresses such as DNA damage and oxidative stress (26, 39). Given that a failure to mount an appropriate stress response is often associated with human diseases including cancer (27), our finding suggests that ATF3-mediated repression of AR functions may serve as a general mechanism which can serve to defend against prostate cancer. This contention is supported by the observations that ATF3 expression is often downregulated in prostate cancers (31, 56).
As a DNA-binding protein, ATF3 is generally thought to regulate cellular functions through direct regulation of gene expression. However, our results argue for a mechanism by which ATF3 represses androgen signaling independent of ATF3 transcriptional activity. Indeed, we found no evidence indicating that ATF3 binds ARE and directly represses androgen-induced expression of AR target genes. Rather, ATF3 repressed androgen signaling through direct interaction with AR, as the ATF3 Δ102–139 protein deficient in AR binding failed to counteract AR-mediated gene expression (Fig. 2C) and failed to block the binding of AR to ARE (Fig. 7C). This mechanism is reminiscent of mechanisms utilized by many other AR repressors (60) and is in line with our previous findings that ATF3 can regulate cellular functions independent of its transcriptional activity (59, 64). Whereas both the basic region and the ZIP domain of ATF3 can mediate protein-protein interactions (25, 41, 59, 64), we found that ATF3 directly bound AR through the ZIP domain. c-Jun, a bZIP protein capable of promoting prostate cancer cell growth (10), was previously shown to interact with AR (49), suggesting that the characteristic leucine residues in the ZIP domain might be responsible for AR binding. However, these hydrophobic residues and the coiled-coil ZIP structure (29) are probably insufficient for ATF3 binding to AR as CREB, another bZIP-containing protein, does not interact with AR (23). Moreover, JDP-2, the closest family member whose ZIP domain has 90% homology with ATF3, only weakly bound AR (see Fig. S6 in the supplemental material). Therefore, the ability to bind AR and repress androgen signaling might set ATF3 apart from other bZIP proteins. Although ATF3 can form a heterodimer with c-Jun (14), it is important that ATF3 does not seem to bind AR through c-Jun as recombinant ATF3 and AR proteins could interact (see Fig. S5 in the supplemental material).
The AR transactivation activity is tightly controlled by multiple mechanisms, including regulation of ligand binding, nuclear translocation, and DNA-binding activity (21). Our results suggest that neither androgen-induced AR nuclear translocation nor the binding of androgens to AR was affected by ATF3. Rather, our results indicate that ATF3 bound the DBD region of AR, thereby preventing AR from binding to its target genes, a mechanism similar to that for Daxx and HOXB13 (35, 43). Such an inhibitory effect argues against a possibility that AR recruits ATF3 to DNA, thereby allowing ATF3 to directly regulate expression of androgen-dependent genes. However, given that a large portion of AR-binding sites revealed by genome-wide ChIP-microarray (ChIP-chip) or ChIP with high-throughput sequencing (ChIP-seq) approaches do not seem to contain ARE (40, 61), it would be interesting to explore whether AR could be recruited to these genomic sites through binding to ATF3. Since the AR DBD region has a high degree of similarity with other steroid receptors (62), ATF3 may exert a similar effect on these receptors. Indeed, we found that ATF3 additionally repressed PR- and GR-mediated gene expression (Fig. 2D).
In addition to DBD, ATF3 also bound AR at the LBD C terminus. This region coincides with the second transactivation domain (AF-2) of AR and can mediate an intermolecular interaction with the N terminus of AR (8). Like SMRT (33), the binding of ATF3 to the AR LBD disrupted the N-C interaction. Whereas this effect could dissociate transcriptional coactivators (e.g., SRC-1) from AR, thereby directly impairing AR transactivation (17), it might also inhibit the AR DNA-binding activity (32). However, interference with the N-C interaction is unlikely to be the only mechanism by which ATF3 prevented AR from binding to target genes as ATF3 could significantly decrease the amount of DNA bound by the recombinant AR protein that lacks the LBD (Fig. 7B). Therefore, the disruption of the N-C interaction might serve as an additional mechanism by which ATF3 prevents AR from binding to androgen-responsive promoters. Although it does not directly bind AR, the tumor suppressor p53 was shown to exert a similar effect as ATF3 on the AR N-C interaction (52). Since ATF3 is a p53-associated protein (64), it might be that p53 indirectly regulates AR transactivation through interaction with ATF3. It is important that PC3 cells used in our experiments are null for p53. Regardless of this likelihood, given that ATF3 can activate p53 in the DNA damage response (64), ATF3 might protect prostate epithelial cells from transformation through two distinct mechanisms, i.e., transactivating p53 target genes and repressing AR-mediated gene expression.
Like many other transcription factors, ATF3 plays a complicated, context-dependent role in cancer development (15). A recent model suggests that ATF3 may prevent the onset of a cancer but promote distant dissemination of malignant cells (66). Our finding that ATF3 can repress androgen signaling indicates that ATF3 expression might be detrimental to the growth of prostate cancer cells, a notion supported by several unbiased cDNA microarray analyses demonstrating that ATF3 expression is downregulated in prostate cancer (31, 56). However, these results are not consistent with other studies showing elevated ATF3 expression in prostate cancer cells (45, 55). Whereas this discrepancy might be reflective of the context-dependent nature of the role that ATF3 plays in cancer, it is worth noting that the ATF3 gene can be expressed as distinct splicing variants, many of which lack the ZIP domain (44) and therefore might counteract the activity of the full-length ATF3 through dimerization with the latter (9). Interestingly, whereas ATF3 could bind AR in the absence of androgens (Fig. 6C), this interaction was not altered by R1881 (Fig. 1E), suggesting that ATF3 might inhibit the growth of prostate cancer cells under castration or androgen-deprived conditions. Accordingly, the development of castration resistance in prostate cancer cells might be accompanied by decreased ATF3 expression. Indeed, the ATF3 expression level was lower in castration-resistant C4-2 cells than in the parental LNCaP cells (see Fig. S7A in the supplemental material). Moreover, a recent unbiased study revealed that ATF3 expression was significantly decreased in castration-resistant prostate cancers compared to expression in untreated primary cancers (see Fig. S7B) (4). Our results thus suggest that therapeutic agents such as thapsigargin that can induce ATF3 expression (57) would repress AR signaling and therefore be of benefit to patients with advanced, castration-resistant prostate cancer.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants (R01CA139107 and R01CA164006) and a Department of Defense award (W81XWH-07-1-0095) to C.Y. and NIH grant (R01DK067049) to S.W.H.
We thank Lirim Shemshedini, Elizabeth Wilson, Marianne Sadar, and Ami Aronheim for providing materials. We are also grateful to Robert Matusik and Xiuping Yu for technical support.
Footnotes
Published ahead of print 4 June 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Adachi M, et al. 2000. Androgen-insensitivity syndrome as a possible coactivator disease. N. Engl. J. Med. 343:856–862 [DOI] [PubMed] [Google Scholar]
- 2. Andersen R, et al. 2010. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 17:535–546 [DOI] [PubMed] [Google Scholar]
- 3. Beleza-Meireles A, et al. 2008. Activating transcription 3: a hormone responsive gene in the etiology of hypospadias. Eur. J. Endocrinol. 158:729–739 [DOI] [PubMed] [Google Scholar]
- 4. Best CJM, et al. 2005. Molecular alterations in primary prostate cancer after androgen ablation therapy. Clin. Cancer Res. 11:6823–6834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brinkmann AO. 2001. Molecular basis of androgen insensitivity. Mol. Cell Endocrinol. 179:105–109 [DOI] [PubMed] [Google Scholar]
- 6. Cai C, et al. 2011. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 20:457–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carson-Jurica MA, Schrader WT, O'Malley BW. 1990. Steroid receptor family: structure and functions. Endocr. Rev. 11:201–220 [DOI] [PubMed] [Google Scholar]
- 8. Centenera MM, Harris JM, Tilley WD, Butler LM. 2008. The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol. Endocrinol. 22:2373–2382 [DOI] [PubMed] [Google Scholar]
- 9. Chen BPC, Liang G, Whelan J, Hai T. 1994. ATF3 and ATF3ΔZip: transcriptional repression versus activation by alternatively spliced isoforms. J. Biol. Chem. 269:15819–15826 [PubMed] [Google Scholar]
- 10. Chen SY, et al. 2006. c-Jun enhancement of androgen receptor transactivation is associated with prostate cancer proliferation. Oncogene 25:7212–7223 [DOI] [PubMed] [Google Scholar]
- 11. Gao N, et al. 2003. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcripitonal regulation of prostatic genes. Mol. Endocrinol. 17:1484–1507 [DOI] [PubMed] [Google Scholar]
- 12. Gilchrist M, et al. 2006. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441:173–178 [DOI] [PubMed] [Google Scholar]
- 13. Hai T, Hartman MG. 2001. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene 273:1–11 [DOI] [PubMed] [Google Scholar]
- 14. Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. 1999. ATF3 and stress responses. Gene Expr. 7:321–325 [PMC free article] [PubMed] [Google Scholar]
- 15. Hai T, Wolford CC, Chang Y-S. 2010. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: is modulation of inflammation a unifying component? Gene Exp. 15:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hartman MG, et al. 2004. Role for activating transcription factor 3 in stress-induced β-cell apoptosis. Mol. Cell. Biol. 24:5721–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. He B, Kemppainen JA, Voegel JJ, Gronemeyer H, Wilson EM. 1999. Activation function 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH2-terminal domain. J. Biol. Chem. 274:37219–37225 [DOI] [PubMed] [Google Scholar]
- 18. He B, Kemppainen JA, Wilson EM. 2000. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem. 275:22986–22994 [DOI] [PubMed] [Google Scholar]
- 19. He B, Lee LW, Minges JT, Wilson EM. 2002. Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal interaction. J. Biol. Chem. 277:25631–25639 [DOI] [PubMed] [Google Scholar]
- 20. Heemers HV, Tindall DJ. 2005. Androgen receptor coregulatory proteins as potential therapeutic targets in the treatments of prostate cancer. Curr. Cancer Ther. Rev. 1:175–186 [Google Scholar]
- 21. Heemers HW, Tindall DJ. 2008. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 28:778–808 [DOI] [PubMed] [Google Scholar]
- 22. Jiang H-Y, et al. 2004. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 24:1365–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jorgensen JS, Nilson JH. 2001. AR suppresses transcription of the α glycoprotein hormone subunit gene through protein-protein interactions with cJun and activating transcription factor 2. Mol. Endocrinol. 15:1496–1504 [DOI] [PubMed] [Google Scholar]
- 24. Kalfa N, Philibert P, Sultan C. 2008. Is hypospadias a genetic, endocrine or environmental disease, or still an unexplained malformation? Int. J. Androl. 32:187–197 [DOI] [PubMed] [Google Scholar]
- 25. Kang Y, Chen C, Massague J. 2003. A self-enabling TGFß response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial Cells. Mol. Cell 11:915–926 [DOI] [PubMed] [Google Scholar]
- 26. Ketola K, Vainio P, Fey V, Kallioniemi O, Iljin K. 2010. Monensin is a potent inducer of oxidative stress and inhibitor of androgen signaling leading to apoptosis in prostate cancer cells. Mol. Cancer Ther. 9:3175–3185 [DOI] [PubMed] [Google Scholar]
- 27. Kourtis N, Tavernarakis N. 2011. Cellular stress response pathways and ageing: intricate molecular relationships. EMBO J. 30:2520–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lamont KR, Tindall DJ. 2010. Androgen regulation of gene expression. Adv. Cancer Res. 107:137–162 [DOI] [PubMed] [Google Scholar]
- 29. Landschulz WH, Johnson PF, McKnight SL. 1988. The leucine zipper: A hypothetical structure common to a new class of DNA binding proteins. Science 240:1759–1764 [DOI] [PubMed] [Google Scholar]
- 30. Langley E, Kemppainen JA, Wilson EM. 1998. Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J. Biol. Chem. 273:92–101 [DOI] [PubMed] [Google Scholar]
- 31. Lapointe J, et al. 2004. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 101:811–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li J, Fu J, Toumazou C, Yoon H-G, Wong J. 2006. A role of the amino-terminal (N) and carboxyl-terminal (C) interaction in binding of androgen receptor to chromatin. Mol. Endocrinol. 20:776–785 [DOI] [PubMed] [Google Scholar]
- 33. Liao G, et al. 2003. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J. Biol. Chem. 278:5052–5061 [DOI] [PubMed] [Google Scholar]
- 34. Lin C, et al. 2009. Nuclear receptor-induced chromosomal proximity and DNA breaks underlies specific translocations in cancer. Cell 139:1069–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin D-Y, et al. 2004. Negative regulation of androgen receptor transcriptional activity by Daxx. Mol. Cell. Biol. 24:10529–10541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu G-Z, Wang H, Wang Z. 2003. Identification of a highly conserved domain in the androgen receptor that suppresses the DNA-binding domain-DNA interactions. J. Biol. Chem. 278:14956–14960 [DOI] [PubMed] [Google Scholar]
- 37. Liu W, et al. 2011. KAI1 gene is engaged in NDRG1 gene-mediated metastasis suppression through the ATF3-NFκB complex in human prostate cancer. J. Biol. Chem. 286:18949–18959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mani R-S, et al. 2009. Induced chromosomal proximity and gene fusions in prostate cancer. Science 326:1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mantoni TS, Reid G, Garrett MD. 2006. Androgen receptor activity is inhibited in response to genotoxic agents in a p53-independent manner. Oncogene 25:3139–3149 [DOI] [PubMed] [Google Scholar]
- 40. Massie CE, et al. 2007. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 8:871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mo P, Wang H, Lu H, Boyd DD, Yan C. 2010. MDM2 mediates ubiquitination and degradation of activating transcription factor 3. J. Biol. Chem. 285:26908–26915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ni L, Yang C-S, Gioeli D, Frierson H, Toft DO, Paschal BM. 2010. FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Mol. Cell. Biol. 30:1243–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Norris J, et al. 2009. The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol. Cell 36:405–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pan Y, Chen H, Siu F, Kilberg MS. 2003. Amino acid deprivation and endoplasmic reticulum stress induce expression of multiple activating transcription factor-3 mRNA species that, when overexpressed in HepG2 cells, modulate transcription by the human asparagine synthetase promoter. J. Biol. Chem. 278:38402–38412 [DOI] [PubMed] [Google Scholar]
- 45. Pelzer AE, et al. 2006. The expression of transcription factor activating transcription factor 3 in the human prostate and its regulation by androgen in prostate cancer. J. Urol. 175:1517–1522 [DOI] [PubMed] [Google Scholar]
- 46. Phizicky E, Fields S. 1995. Protein-protein interactions: Methods for detection and analysis. Microbiol. Rev. 59:94–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Qi W, Wu H, Yang L, Boyd DD, Wang Z. 2007. A novel function of caspase-8 in the regulation of androgen-receptor-driven gene expression. EMBO J. 26:65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quigley CA, et al. 1995. Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr. Rev. 16:271–321 [DOI] [PubMed] [Google Scholar]
- 49. Sato N, et al. 1997. Androgenic induction of prostate-specific antigen gene is repressed by protein-protein interaction between the androgen receptor and AP-1/c-Jun in the human prostate cancer cell line LNCaP. J. Biol. Chem. 272:17485–17494 [DOI] [PubMed] [Google Scholar]
- 50. Scher HI, Sawyers CL. 2005. Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 23:8253–8261 [DOI] [PubMed] [Google Scholar]
- 51. Shen M, Abate-Shen C. 2010. Molecular genetics of prostate cancer: new prospects for old challenges. Gene Dev. 24:1967–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shenk J, et al. 2001. p53 represses androgen-induced transactivation of prostate-specific antigen by disrupting hAR amino- to carboxyl-terminal interaction. J. Biol. Chem. 276:38472–38479 [DOI] [PubMed] [Google Scholar]
- 53. Stanbrough M, Leav I, Kwan PL, Bubley GJ, Balk SP. 2001. Prostatic intraepithelial neoplasia in mice expressing an androgen receptor transgene in prostate epithelium. Proc. Natl. Acad. Sci. U. S. A. 98:10823–10828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tam NN, Gao Y, Leung YK, Ho SM. 2003. Androgenic regulation of oxidative stress in the rat prostate: Involvement of NAD(P)H oxidases and antioxidant defense machinery during prostatic involution and regrowth. Am. J. Pathol. 163:2513–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Taylor B, et al. 2010. Integrative genomic profiling of human prostate cancer. Cancer Cell 18:11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tomlins SA, et al. 2007. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 39:41–51 [DOI] [PubMed] [Google Scholar]
- 57. Vander Griend DJ, et al. 2009. Amino acid containing thapsigargin analogues deplete androgen receptor protein via synthesis inhibition and induce the death of prostate cancer cells. Mol. Cancer Ther. 8:1340–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang C, et al. 2003. Identification of a novel transcription factor, GAGATA-binding protein, involved in androgen-mediated expression of prostate-specific antigen. J. Biol. Chem. 278:32423–32430 [DOI] [PubMed] [Google Scholar]
- 59. Wang H, Mo P, Ren S, Yan C. 2010. Activating transcription factor 3 activates p53 by preventing E6-associated protein from binding to E6. J. Biol. Chem. 285:13201–13210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang L, Hsu C-L, Chang C. 2005. Androgen receptor corepressors: an overview. Prostate 63:117–130 [DOI] [PubMed] [Google Scholar]
- 61. Wang Q, et al. 2009. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weatherman RV, Fletterick RJ, Scanlan TS. 1999. Nuclear-receptor ligands and ligand-binding domains. Annu. Rev. Biochem. 68:559–581 [DOI] [PubMed] [Google Scholar]
- 63. Yan C, Boyd DD. 2006. Histone H3 acetylation and H3 K4 methylation define distinct chromatin regions permissive for transgene expression. Mol. Cell. Biol. 26:6357–6371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yan C, Lu D, Hai T, Boyd DD. 2005. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 24:2425–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yan C, Wang H, Boyd DD. 2001. KiSS-1 represses 92-kDa type IV collagenase expression by down-regulating NF-κB binding to the promoter as a consequence of IκBα-induced block of p65/p50 nuclear translocation. J. Biol. Chem. 276:1164–1172 [DOI] [PubMed] [Google Scholar]
- 66. Yin X, Dewille JW, Hai T. 2008. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene 27:2118–2127 [DOI] [PubMed] [Google Scholar]
- 67. Zhang J, Thomas TZ, Kasper S, Matusik RJ. 2000. A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology 141:4698–4710 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.