

Abstract
The SDHD gene (subunit D of succinate dehydrogenase) has been shown to be involved in the generation of paragangliomas and pheochromocytomas. Loss of heterozygosity of the normal allele is necessary for tumor transformation of the affected cells. As complete SdhD deletion is lethal, we have generated mouse models carrying a “floxed” SdhD allele and either an inducible (SDHD-ESR strain) or a catecholaminergic tissue-specific (TH-SDHD strain) CRE recombinase. Ablation of both SdhD alleles in adult SDHD-ESR mice did not result in generation of paragangliomas or pheochromocytomas. In contrast, carotid bodies from these animals showed smaller volume than controls. In accord with these observations, the TH-SDHD mice had decreased cell numbers in the adrenal medulla, carotid body, and superior cervical ganglion. They also manifested inhibited postnatal maturation of mesencephalic dopaminergic neurons and progressive cell loss during the first year of life. These alterations were particularly intense in the substantia nigra, the most affected neuronal population in Parkinson's disease. Unexpectedly, TH+ neurons in the locus coeruleus and group A13, also lacking the SdhD gene, were unaltered. These data indicate that complete loss of SdhD is not sufficient to induce tumorigenesis in mice. They suggest that substantia nigra neurons are more susceptible to mitochondrial damage than other catecholaminergic cells, particularly during a critical postnatal maturation period.
INTRODUCTION
Mitochondrial complex II (MCII; succinate-ubiquinone oxidoreductase [Sdh]) is composed of four nucleus-encoded subunits (A, B, C, and D) that couple oxidation of succinate to fumarate in the Krebs cycle to the mitochondrial electron transport chain (ETC). This is achieved by transferring electrons from the flavin moiety in SdhA to iron-sulfur clusters in SdhB and then to ubiquinone bound to SdhC and SdhD. These last subunits also serve to anchor the whole complex to the inner mitochondrial membrane (21, 58). Genetic defects in MCII generate several human diseases (for a review, see reference 43). Mutations in Sdh subunits, particularly in SdhB, -C, and -D, commonly produce familial pheochromocytomas and paragangliomas. These are highly vascularized, mostly benign, tumors occurring in the adrenal gland and the carotid body (CB) but also in other catecholaminergic neural-crest-derived tissues (3, 34). Cell lines with reductions in Sdh activity caused by mutations in SdhB or SdhC show signs of oxidative damage and apoptosis, although mutant cells escaping apoptosis may undergo tumor transformation (19, 24, 25). Indeed, spontaneous loss of heterozygosity (LOH) in adult humans carrying a mutant SDHD allele (SDHD+/−) can induce tumorigenesis in the affected cells (5, 6). The tumorigenic potential of MCII mutations has been suggested to derive from the accumulation of succinate, which inhibits α-ketoglutarate-dependent prolyl hydroxylases (PHDs), thus causing a “pseudohypoxic” condition with hypoxia-inducible factor 1α (HIF-1α) stabilization and nuclear translocation in normoxia (8, 41, 51). This would result in constitutive activation of genes that favor tumor growth (42). Impairment of electron transfer from reduced flavin to ubiquinone in SdhC- or SdhD-deficient cells can lead to excessive production of reactive oxygen species (ROS) (21, 58). Hence, it has also been proposed that oxidative stress could contribute to tumorigenesis due to DNA damage (25) and/or ROS-dependent HIF-1α upregulation (12, 19).
The molecular effects of MCII dysfunction in vivo are practically unknown, as bi-allelic genetic deletion of any of the Sdh genes tested so far (SdhB and SdhD knockouts) produce embryonic lethality (4, 31, 40). Moreover, heterozygous SdhD-deficient mice up to 2 years of age do not present tumors or any other obvious pathology, although they seem to have subtle CB alterations (4, 40). The objective of the current research was to develop an SdhD conditional knockout mutant mouse that could recapitulate the LOH required in humans for tumor formation in peripheral paraganglia. To this end, we generated mouse models carrying a “floxed” SdhD allele and either a ubiquitously expressed tamoxifen-inducible CRE recombinase (SDHD-ESR mouse) or a CRE recombinase under the control of the tyrosine hydroxylase (TH) promoter (TH-SDHD mouse), the rate-limiting enzyme for catecholamine synthesis. Our goals were to ascertain whether ablation of the SdhD gene induces either cell death or tumor transformation in vivo and to compare the vulnerability of peripheral and central catecholaminergic neurons to primary mitochondrial ETC dysfunction. In this regard, we were particularly interested in the analysis of dopaminergic neurons in the substantia nigra pars compacta (SNpc), the most important neuronal population affected in Parkinson's disease (PD), as mitochondrial impairment has long been associated with the pathogenesis of this neurodegenerative disorder (14, 15, 48). Herein, we report that deletion of the SdhD floxed allele in adult heterozygous (SdhDflox/−) mice did not result in generation of paragangliomas or pheochromocytomas. Moreover, germ line deletion of the SdhD allele restricted to TH+ tissues did not induce tumor transformation of the catecholaminergic cells, despite the fact that the animals survived for up to a year. In contrast, these last mice showed a selective degeneration of catecholaminergic cells in the peripheral and central nervous system and a pronounced and progressive parkinsonian phenotype. Interestingly, neuronal loss preferentially affected the SNpc and other structures that reach maturation during early postnatal life. Catecholaminergic nuclei, such as the locus coeruleus, that seem to be mature at birth were unaffected.
MATERIALS AND METHODS
Generation of the SDHD-ESR and TH-SDHD mouse strains.
To obtain both the inducible and tissue-specific SdhD mouse mutant strains, we engineered a floxed allele, SdhDflox, which contains two LoxP sites flanking exons 2 to 4 and a NEO cassette for cell selection (Fig. 1A). This construct was targeted to the SdhD genomic locus by homologous recombination in 129SvJ background R1 mouse embryonic stem (ES) cells. Proper targeting was tested by Southern blotting of genomic DNA digested with HindIII and hybridized against an external 5′ probe (Fig. 1C). To test the excision of the SdhDflox allele (Fig. 1B), targeted ES clones were electroporated with a plasmid containing the CRE recombinase gene. DNA from these cells was digested with EcoRV and analyzed by Southern blotting (Fig. 1D) against a NEO probe. ES clones carrying the SdhDflox allele were used for blastocyst injection and chimera generation. Germ line transmission of the SdhDflox allele yielded heterozygous animals, which were subsequently mated with SdhD+/− mice (40) carrying either the tamoxifen-inducible CRE recombinase (20) or the Th-IRES-Cre transgenes (29) to generate the experimental SDHD-ESR and TH-SDHD mouse lines, respectively. Littermates with SdhDflox/+ and SdhDflox/− genotypes lacking CRE recombinase were used as control individuals. Unless otherwise specified, results from both control groups were pooled since no differences between them was found for the phenotypes tested. Routine genotyping was performed for the SdhD alleles by PCR with the primers 5′-AATTGTGCAGAAGTGAG-3′, 5′-GCTGCATACGCTTGATC-3′, and 5′-CATCAAGGCTCACAGTC-3′.
Fig 1.
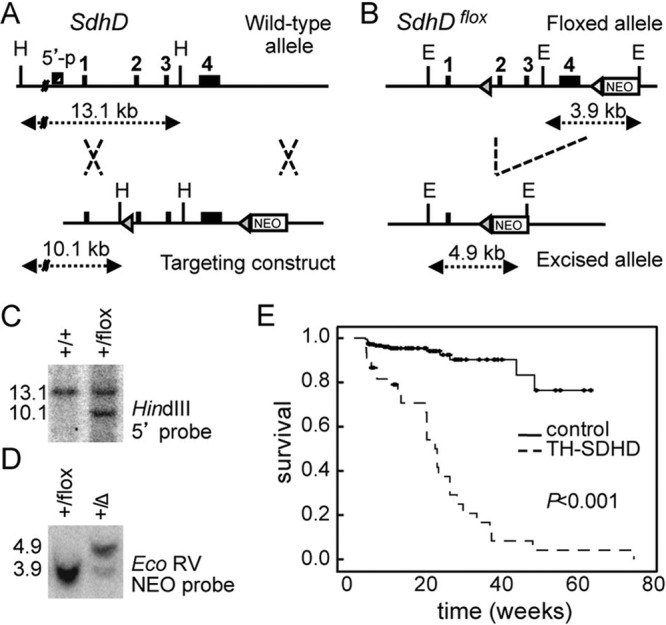
Generation of the TH-SDHD mutant mouse. (A) Scheme of the 16-kb targeting construct engineered to contain two loxP sites (gray triangles) that flank exons 2 to 4 (black boxes) of the SdhD gene, and a neomycin-resistance (NEO) cassette for clone selection. This construct was targeted to the wild-type SdhD genomic locus by homologous recombination. (B) CRE recombinase-mediated excision of the SdhDflox allele. H, HindIII; E, EcoRV. Distances between relevant sites are indicated with dotted arrows. (C) Southern blot of HindIII-digested DNA from neomycin-resistant ES clones, hybridized with a probe against a 5′ region. (D) Southern blot of EcoRV-digested DNA from clones expressing the CRE recombinase, hybridized with a probe against the NEO gene. Numbers on the left are band sizes, in kilobase pairs. The Δ symbol represents the excised SdhD allele. (E) Kaplan-Meier survival curves of control and TH-SDHD mice.
Mouse husbandry and pharmacological treatments.
Mice were housed under temperature-controlled conditions (22°C) in a 12-h light/12-h dark cycle and provided with food and water ad libitum. Either high (100 μg/g of body weight for 4 days) or low (50 μg/g for 2 days) doses of tamoxifen dissolved in corn oil were administered by daily i.p. injections. The antioxidant tempol (Sigma) was given to pregnant females in drinking water protected from light at 2 mM, starting 1 to 2 days before delivery of pups and continuing until the sacrifice of littermates (day P15). All experiments were performed in accordance with institutional guidelines approved by the ethics committee of the Hospital Universitario Virgen del Rocio.
SdhD mRNA level.
Adrenal medullas or striata were dissected and stored frozen at −80° until processing. Total RNA was prepared by using an RNeasy microkit (Qiagen) according to the manufacturer's directions. Reverse transcription of mRNA was performed with a Superscript II reverse transcriptase kit (Invitrogen), and SdhD cDNA was amplified by quantitative PCR with the primers 5′-CCAGCACATTCACCTGTCA-3′ and 5′-ATCAGCCCCAAGAGCAGAA-3′ in the presence of SYBR green. The Arbp housekeeping gene was used for normalization.
Mitochondrion isolation and mitochondrial complex II activity.
Mitochondria were isolated from mouse kidneys as reported elsewhere (40). Succinate-ubiquinone oxireductase activity was determined as described in reference 40, with slight modifications. Briefly, 30 to 50 μg of protein was assayed at 30°C. Samples were diluted 1:4 in the assay reaction buffer (25 mM KH2PO4 [pH 7.2], 5 mM MgCl2, 3 mM potassium cyanide, 2.5 mg/ml bovine serum albumin) and freeze-thawed in liquid nitrogen three times before the essay. Enzymatic activity was measured for a period of 2 min as the decrease in the absorbance at 600 nm due to the reduction of 50 μM 2,6-dichlorophenol-indophenol (DCPIP) coupled to reduction of 130 μM ubiquinone-1. The reaction was carried out in the presence of 3.6 μM antimycin, 5 μM rotenone, and 10 mM succinate.
ATP level.
Adrenal medullas were dissected and stored frozen at −80° until processing. For ATP determination, total extracts from thawed adrenal medullas were prepared by grinding them in 50 μl of 100 mM Tris-HCl, 4 mM EDTA with quartz sand and a plastic pestle followed by boiling for 3 min. After cooling on ice, samples were spun for 1 min at 10,000 × g. Supernatant was collected for ATP determination with the ATP bioluminescence kit HSII (Roche). Between 1 and 0.01 μg of protein was assayed, as in this range the assay was linear. Protein concentration was determined by absorbance at 280 nm.
Oxidative damage.
Mitochondrial DNA oxidative damage (44) and lipid peroxidation (35) were determined according to published protocols. Protein oxidation was analyzed with an OxyBlot protein oxidation detection kit and an OxyIHC oxidative stress detection kit (Millipore), according to manufacturer's directions.
Tissue preparation and histochemistry.
Histological analyses and stereological cell counts were done by following previously described protocols (23, 32, 38). In brief, mice were anesthetized and killed by decapitation. Dissected tissues were fixed in paraformaldehyde and embedded in OCT (Tissue-Tek), for carotid body and superior cervical ganglion, or paraffin, for adrenal gland and brain, before sectioning. Peripheral tissue sections 10 μm thick were used for tyrosine hydroxylase (TH) immunostaining. Nuclei were detected by DAPI (4′,6′-diamidino-2-phenylindole) staining. Coronal mouse brain sections 20 μm thick were used for tyrosine hydroxylase (TH), NeuN, parvalbumin, or DARP32 immunostaining. Antibodies and the dilution factors used were as follows: polyclonal antibody against TH (Novus Biologicals), 1:5,000; monoclonal antibody against NeuN (Chemicon), 1:200; polyclonal antibody against parvalbumin (Swant), 1:5,000; polyclonal antibody against DARP32 (Millipore), 1:5,000. For 3,3′-diaminobenzidine-based detection, an Envision+ kit (Dako) was used according to the manufacturer's recommended protocol. For fluorescence detection, Alexa-Fluor 568–anti-rabbit IgG was used. The size of the adrenal medulla was estimated with the ImageJ software on photographs. In brain, unbiased stereological counts of TH and NeuN immunoreactive neurons were performed in one of every six brain sections covering the complete structure of interest by using the CAST-Grid system coupled to an Olympus microscope with an automated platform. The two hemispheres were considered.
In situ hybridization.
Paraformaldehyde-fixed brains were embedded in gelatin and sectioned in 50-μm coronal slices with a vibratome. Slices were incubated with a UTP-digoxigenin-labeled antisense RNA probe obtained by T7 promoter-driven transcription of an SdhD cDNA containing plasmid (Integrated Molecular Analysis of Genomes and Their Expression Consortium [IMAGE] clone 3989833) overnight at 57°C. After extensive washing, slices were incubated with an antidigoxigenin antibody conjugated with alkaline phosphatase (Roche) in a 1:1,000 dilution overnight at 4°C, followed by incubation in 6 μl/ml nitroblue tetrazolium (NBT) and 4.6 μl/ml 5-bromo-4-chloro-3-indolylphosphate (BCIP) (Roche) for 7 h at room temperature for detection.
X-Gal staining and confocal microscopy.
Expression of CRE recombinase in catecholaminergic tissues of the TH-SDHD mouse was evaluated by mating these mice with floxed Rosa26-lacZ (R26R) mice (52). 5-Bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) staining was done as reported before (37, 56). Images of TH-immunostained sections containing X-Gal-stained precipitates were acquired with a TCS SP2 Leica confocal microscope. Lateral projection images were generated from stacks of optical sections spaced 0.8 μm apart using the Leica software package.
HPLC.
Striata were dissected from brains in ice-cold phosphate-buffered saline (PBS) and sonicated on ice. Homogenates were filtered by Ultrafree-MC centrifuge filter units (Millipore) and kept at −80°C until use. For high-pressure liquid chromatography (HPLC) analysis of dopamine and related metabolites, samples were passed through a chromatographic ALB-215 column (ANTEC Leyden) according to the manufacturer's directions.
Open field tests.
To assess the motor dysfunction phenotype, mice were subjected to an open-field test in a 22.5-cm by 22.5-cm arena for 1 h. Individual animals were monitored using an automatic tracking system (SMART, Panlab). Traveled distance and resting time parameters were calculated using the SMART software (version 2.5.14) (38).
Statistical analysis.
Data are presented as means ± standard errors of the means (SEM). Statistical significance was assessed by Student's t test with a Levene test for homogeneity of variances, in cases of normal distribution, or by the nonparametric Mann-Whitney U test, in cases of nonnormal distribution. PASW18 software was use for statistical analysis.
RESULTS
Survival of SdhD-deficient mice and loss of peripheral catecholaminergic cells.
Heterozygous SdhDflox/− animals with complete germ line ablation of SdhD in catecholaminergic cells (TH-SDHD mice) appeared healthy at birth, and although smaller than their littermates, they seemed to develop normally during the first postnatal months. However, most mutant mice died before the end of the first year of life (Fig. 1E), probably due to the progressive and extensive cell loss occurring in some catecholaminergic organs and brain nuclei (see below).
TH-SDHD mice, even those that survived for up to 1 year, did not show any indication of tumor transformation in the peripheral catecholaminergic tissues. On the contrary, quantitative histological analyses showed a marked decrease of catecholaminergic cells. We studied in detail the adrenal medulla (AM), a relatively large organ almost exclusively formed by compacted strands of chromaffin cells that allowed us to carry out biochemical analyses. AM volume was approximately similar in newborn wild-type and SdhD-deficient animals; however, maturation and growth of the chromaffin tissue during the first postnatal weeks were markedly altered in TH-SDHD mice (Fig. 2A and B). AM cells also showed a significant decrease of intracellular ATP content, indicating an alteration of ETC (Fig. 2C). We used several methods (see Materials and Methods) to ascertain whether SdhD deletion induced an oxidative stress in the surviving AM tissue, with inconclusive results. However, we did see some evidence of selective increase of lipid peroxidation in chromaffin cells of mutant animals (Fig. 2D). Like the AM, other TH+ structures, such as the carotid body (CB) and neighboring superior cervical ganglion (SCG), of juvenile animals also presented a clear decrease in the number of catecholaminergic cells (Fig. 2E to H).
Fig 2.

Histological analyses of peripheral catecholaminergic organs of control and TH-SDHD mice. (A) Representative photographs of adrenal glands from P75 mice. (B) Postnatal increase of volume of the adrenal medulla. (C) ATP levels in adrenal medulla of P75 to P105 mice. (D) Lipid peroxidation in AM of P75 mice for both separate controls (flox/+ and flox/- without CRE) and mutant groups. Similar analysis performed in liver tissue showed no difference between control and TH-SDHD mice (data not shown). (E and F) Representative photographs and numbers of TH+ cells in carotid bodies (CB) from P75 mice. (G and H) Representative photographs and density of TH+ cells in superior cervical ganglion (SCG) from P75 mice. All tissues were immunostained for tyrosine hydroxylase (TH, red). Nuclei were stained with DAPI dye. AG, adrenal gland; AM, adrenal medulla; ca, carotid artery. Bars, 100 μm (E) and 200 μm (A and G). There were 4 to 8 individuals per group and age. *, P ≤ 0.05; **: P ≤ 0.01; ***, P ≤ 0.001 (control versus TH-SDHD groups).
As it could be possible that complete maturation of the catecholaminergic organs is a prerequisite for tumor induction after SdhD deletion, we also generated a time-inducible SdhD mutant model by breeding the SdhD floxed mouse line with a strain containing a tamoxifen-inducible CRE (20). These animals (designated SDHD-ESR mice) carried a paternally inherited SdhD-null allele and a maternal SdhD floxed allele to be excised after CRE activation, thus mimicking the human scenario for familiar paraganglioma. Measurement of SdhD mRNA levels in adrenal medulla and kidney soon after tamoxifen treatment confirmed deletion of the maternally inherited SdhD allele in adult mutant mice (Fig. 3A). In accord with these data, a parallel decrease of succinate-ubiquinone oxireductase activity was measured in isolated kidney mitochondria (Fig. 3B). Analyses of AM and CB in the SDHD-ESR strain performed before the death of the mice (occurring ∼3 weeks after tamoxifen treatment) indicated the absence of tumorigenesis in these organs and a clear trend toward degeneration of the carotid body (Fig. 3C and D). SDHD-ESR animals treated with lower doses of tamoxifen (see Materials and Methods) survived longer (∼8 to 9 weeks) but showed no signs of tumor transformation or hypertrophy in either the AM or CB (data not shown).
Fig 3.

Lack of carotid body hypertrophy in SDHD-ESR mice. (A) Levels of SdhD mRNA in kidney and adrenal gland of separate control (flox/+ and flox/− without CRE) groups and mutant individuals 5 days after tamoxifen injection (at P50), demonstrating general loss of SdhD. r. u., relative units. There were 3 to 7 individuals per group. (B) Succinate ubiquinone oxireductase activity (MCII) in mitochondria isolated from kidney cells 3 weeks after tamoxifen injection. There were 3 to 6 individuals per group. (C) Carotid body (CB) immunostained for tyrosine hydroxylase (TH) 3 weeks after tamoxifen injection. Bar, 100 μm. (D) Total and TH+ fraction volumes of carotid bodies from control (n = 3) and SDHD-ESR (n = 4) animals 3 weeks after tamoxifen injection (at P50). *, P < 0.05; ***, P ≤ 0.001.
Impaired SNpc postnatal maturation and maintenance in TH-SDHD mice.
In parallel with the catecholaminergic cell loss in the peripheral nervous system, TH-SDHD animals showed a marked and progressive postnatal reduction in the number of ventral mesencephalic TH+ neurons (Fig. 4A). Vulnerability of dopaminergic neurons of the substantia nigra pars compacta (SNpc) and ventral tegmental area (VTA) to mitochondrial SdhD deficiency was evaluated by comparative cell count analyses in control and in TH-SDHD mice during the first year of life (Fig. 4B to D). Although neonatal animals already had a small population of TH+ ventral mesencephalic cells, most SNpc and approximately half of VTA dopaminergic neurons in normal mice acquired their postmitotic TH+ phenotype during the first postnatal month, and after reaching a maximum, their number remained constant in adulthood (9, 26). This pattern of ontogenic development was altered in TH-SDHD animals. These mice were born with a slight, nonsignificant decrease of TH+ cell number in the SNpc and similar cell number in VTA in comparison with their respective controls but showed a characteristic inhibition of postnatal neuron maturation and progressive death of TH+ cells upon reaching adulthood. These processes, impaired maturation and accelerated degeneration of dopaminergic cells, were quite aggressive in the SNpc, resulting in an almost total disappearance of TH+ cells in this structure between 6 and 12 months of animal age (Fig. 4B). In the same period, ∼30% of TH+ cells in VTA remained (Fig. 4C). These observations indicate that, similar to what occurs in PD patients (14, 15), SNpc neurons are far more sensitive to mitochondrial dysfunction than VTA. Staining of ventral mesencephalic neurons with NeuN, a panneuronal marker, in 3.5-month-old animals further demonstrated the differential neuronal death in the SNpc and VTA of TH-SDHD mice rather than a loss of TH expression (Fig. 4D). The molecular mechanism of dopaminergic mesencephalic neuronal death in the TH-SDHD mice is unknown, as we could not demonstrate any indication of either caspase-3 activation or nucleus fragmentation in tissue samples from these animals.
Fig 4.

Loss of ventral mesencephalic dopaminergic neurons in TH-SDHD mice. (A) TH immunostaining of neurons in the SNpc and VTA of control and TH-SDHD mice at postnatal days (P) 0, 30 and 180. Bar, 200 μm. (Insets) Neurons at a higher magnification (2.5× the main figure). Number of TH+ neurons in the SNpc (B) and VTA (C) at different animal ages. (D) NeuN+ cell number in the SNpc and VTA of P105 mice. There were 3 to 8 individuals per point. *, P < 0.05; **, P < 0.01; ***, P ≤ 0.001 (control versus TH-SDHD groups). #, P < 0.05; ##, P ≤ 0.001 (P30 versus subsequent ages).
Ablation of the SdhD gene in catecholaminergic cells of the TH-SDHD mouse is expected to increase the production of ROS in mitochondria, as mutations in Sdh proteins have been demonstrated to increase oxidative stress in both in vivo and in vitro systems (19, 25). In accord with this concept, we also observed an increase of lipid peroxidation in AM tissue from TH-SDHD animals (Fig. 2D). Hence, we tested the effect of the superoxide dismutase mimetic tempol (57) upon the postnatal maturation of SNpc neurons. To this end, we added the antioxidant to the drinking water at late stages of gestation and until sacrifice of littermates 15 days after birth. Tempol induced a 48% increase in the number of TH+ SNpc neurons in TH-SDHD animals (4,998 ± 261 neurons in treated mice versus 3,370 ± 486 neurons in nontreated animals [mean ± SEM]; n = 7 and 4, respectively; P = 0.01). However, the effect of tempol in control animals was smaller (13%) and nonsignificant (8,740 ± 647 neurons in treated mice versus 7,743 ± 349 in nontreated mice; n = 5; P = 0.21).
Brain catecholaminergic neurons matured at birth were unaffected by SdhD ablation.
The analysis of catecholaminergic cell death in the TH-SDHD mouse was extended to brainstem nuclei, such as the noradrenergic locus coeruleus (LC) or the dopaminergic group A13, in which most of the neurons had already acquired the catecholaminergic phenotype at birth. In contrast with the accentuated neuronal loss seen in the SNpc, and to a lesser extent in VTA, of TH-SDHD animals, neuron numbers in LC and group A13 were unaltered by the mutation during the first year of life (Fig. 5). As no antibodies for SdhD protein immunodetection are available, we attempted in situ histochemical determination of succinate-ubiquinone oxireductase activity in brain sections. This technique did not provide conclusive results, probably because affected neurons (without oxireductase activity) were intermingled with unaffected cells.
Fig 5.

(A and B) TH immunostaining of noradrenergic neurons in the locus coeruleus (A) and dopaminergic nucleus A13 of the zona incerta (B) of control and TH-SDHD mice at postnatal day 180. (Insets) Neurons at higher magnification (3× the main figure). The axonal bundle of the nigrostriatal pathway (NSP) is shown. Bar, 200 μm. (C and D) Numbers of TH+ neurons in LC (C) and A13 (D) at different ages. There were 3 to 8 individuals per point.
Therefore, to confirm that CRE-mediated recombination had taken place in the unaffected LC and A13 neurons, we checked the expression of functional CRE recombinase, as well as the deletion of the SdhDflox allele in the TH+ neurons of these regions. To this end, we generated a TH-SDHD/R26R mouse line (see Materials and Methods) in which we demonstrated colocalization in every TH+ neuron of X-Gal staining (indicative of CRE recombinase-dependent DNA deletion at the floxed R26R locus) with TH immunoreactivity in brain sections, including LC and A13 nuclei (Fig. 6A). In addition, in situ hybridization analyses directly indicated that SdhD mRNA expression was abolished in LC neurons of TH-SDHD mice to an extent similar to that seen in dopaminergic SNpc or VTA cells, whereas SdhD mRNA expression remained unaltered in noncatecholaminergic cells (Fig. 6B). LC neurons seem to have an extraordinary resistance to mitochondrial complex II dysfunction, as both CRE mediated R26R expression and SdhD ablation (as determined by in situ hybridization analysis) were tested to occur in LC neurons of juvenile TH-SDHD animals (one month old or younger). In contrast, LC TH+ neuron numbers remained unaltered in 1-year old mutants (Fig. 5C). Altogether, these data unequivocally demonstrate that SNpc neurons have special sensitivity to genetic mitochondrial SdhD inhibition in comparison with cells in other catecholaminergic nuclei.
Fig 6.
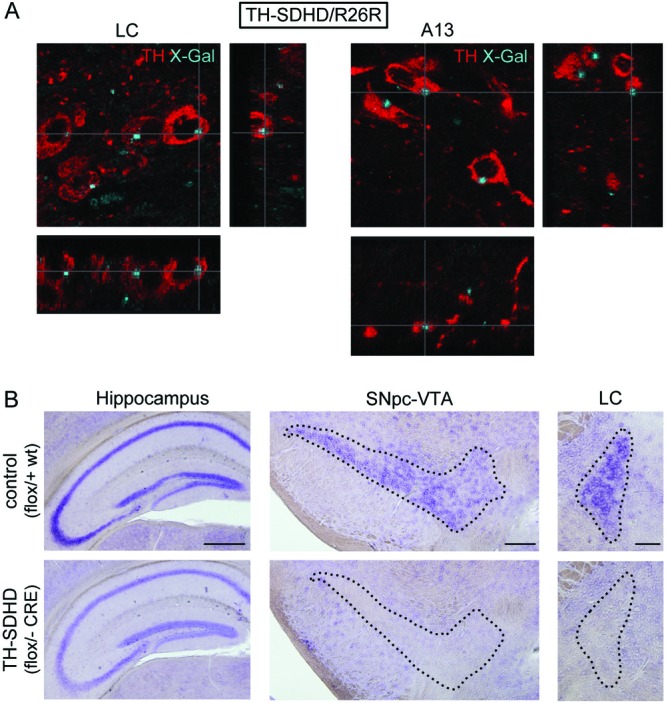
(A) Confocal microscope photographs of the locus coeruleus and dopaminergic nucleus A13 showing colocalization of X-Gal (blue dots) and TH+ (red) neurons in a TH-SDHD mouse carrying the R26R reporter construct. (B) In situ hybridization of SdhD mRNA in the hippocampus, SNpc-VTA, and LC. Note the complete disappearance of mRNA in the catecholaminergic areas (encircled by dotted lines) of the TH-SDHD mouse. Note also that the level of SdhD mRNA expression in the hippocampus of control animals (homozygous for SdhD) was higher than in heterozygous TH-SDHD mice (40). Bars, 500 μm (hippocampus), 200 μm (SNpc/VTA), and 100 μm (LC).
Previous studies with the Th-IRES-Cre mouse (29) suggested that transient TH activation may occur during development in areas that are not TH+ in adults. In our TH-SDHD/R26R animals, we observed a very low level of R26R expression in the striatum, cortex, and hippocampus compared with the SNpc and other TH+ structures. As expected, all the noncatecholaminergic brain areas studied (particularly the cerebral cortex and hippocampus) were normal in TH-SDHD mice (data not shown). In addition, we also checked that the number of DARP32+ medium spiny neurons, the most numerous in the striatum, was normal in TH-SDHD mice (Fig. 7A). Moreover, the number of parvalbumin-positive GABAergic interneurons, which provide most of the neurotropic support for dopaminergic nigrostriatal neurons (23), was also unaffected by SdhD ablation (Fig. 7B). Finally, a quantitative analysis of the level of SdhD mRNA expression in the striatum showed that CRE recombinase-dependent SdhD deletion was not aberrantly activated in noncatecholaminergic striatal cells of TH-SDHD animals (Fig. 7C). Together, these data make it unlikely that CRE expression during development in regions other than the TH+ catecholaminergic areas makes any major contribution to the histological and functional phenotype of TH-SDHD animals.
Fig 7.
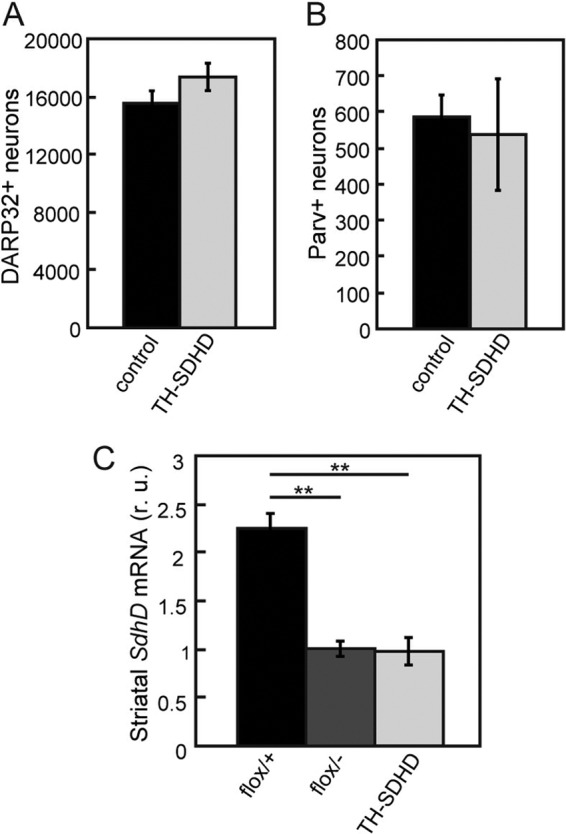
Lack of striatal affects in the TH-SDHD mouse. (A and B) Number of DARP32-positive (A) and parvalbumin-positive (B) neurons in the striata of control and TH-SDHD mice at P75. Immunostaining of striatal neurons was done as described in reference 23. Cell counts were done in 20-μm-thick slices. There were 3 individuals per group. (C) SdhD mRNA levels in striata of control and TH-SDHD mice at P30, as determined by RT-qPCR of total RNA. r. u., relative units. There were 3 to 4 individuals per group. **, P < 0.01 for control versus TH-SDHD groups.
Progressive parkinsonian phenotype in TH-SDHD mice.
As a consequence of the loss of dopaminergic mesencephalic neurons projecting to the striatum, TH-SDHD animals exhibited intense striatal dopaminergic denervation, which seemed to be almost complete at 2 to 3 months of age (Fig. 8A). Measurement of striatal dopamine and its degradation metabolites indicated a profound neurochemical defect in SdhD-deficient animals. In normal mice, a postnatal increase in the number of TH+ neurons of the SNpc resulted in maturation of the nigrostriatal pathway, with a marked rise in the caudate putamen content of dopamine as well as in 3,4-dihydroxyphenylacetic (DOPAC) and homovanillic acid (HVA) levels. The postnatal increases of dopamine and its metabolites were almost abolished in striata from 2.5-month-old TH-SDHD animals (Fig. 8B). Striatal denervation in mutant animals seemed to be complete in 2.5-month-old animals, even though a significant number of SNpc and VTA neurons remained alive (Fig. 4A to C). This suggests that in TH-SDHD mice, the nigrostriatal pathway does not reach full mature development and that the loss of dopaminergic axon terminals could precede death of SdhD−/− neurons. As indicated above, striatal parvalbumin-positive cells, which provide most of the glial-cell-line-derived neurotropic factor (GDNF) needed for trophic maintenance of nigrostriatal neurons (23, 38), were unaltered in TH-SDHD mice (Fig. 7B), ruling out possible noncell autonomous effects of SdhD ablation on dopaminergic nigrostriatal neurons.
Fig 8.

Parkinsonian phenotype of TH-SDHD mice. (A) TH immunostaining of striata of control and TH-SDHD mice at P75. Bar, 500 μm. CPu, caudate putamen; Acb, accumbens nucleus. (B) Dopamine (DA), 3,4-dihydroxyphenylacetic (DOPAC), and homovanillic acid (HVA) levels in striatum at P75, as determined by HPLC. There were 3 to 8 individuals per point. (C) Walking traces of control and TH-SDHD mice in an open-field test for 1 h at the indicated ages. (D) Quantification of distance traveled and resting time (RT). There were 3 to 6 individuals per point. **, P < 0.01; ***, P ≤ 0.001 (control versus TH-SDHD groups); ##, P ≤ 0.001 for P40 versus later ages.
We also studied whether the anatomical and neurochemical nigrostriatal defects in TH-SDHD animals correlate with a behavioral phenotype by performing open-field tests at different ages. TH-SDHD mice showed a progressive bradykinetic syndrome characterized by marked decrease in the distance traveled in the open field and increase in time spent at rest (Fig. 8C and D).
DISCUSSION
In this paper we describe a new conditional mouse model of SdhD deficiency, which was designed to study tumorigenesis in paraganglionic cells as well as to compare the vulnerability of peripheral and central catecholaminergic neurons to primary mitochondrial ETC dysfunction. In humans, SDHD is considered a tumor suppressor gene, because loss-of-function mutations are the cause of some hereditary paragangliomas, mainly affecting the carotid body (3, 6). Individuals carrying the mutation are heterozygous, and loss of the normal allele in the affected cells (LOH) is required for tumor transformation (5, 6, 22). SdhD−/− mice are nonviable, and heterozygous SdhD+/− animals fail to develop tumors, suggesting that spontaneous LOH of this locus does not take place in mouse paraganglia (4, 40). In our TH-SDHD mice, we achieved complete deletion of both SdhD alleles in TH+ cells, a condition postulated to be the initiating event for tumor transformation. However, far from developing tumors, these animals underwent loss of catecholaminergic cells in the peripheral nervous system. Although TH-SDHD animals had a shortened life span, likely caused by extensive central and peripheral catecholaminergic cell loss, examination of animals that survived even for 1 year never resulted in the detection of tumors.
Genetic and molecular studies have demonstrated that the inherited pattern of paraganglioma involves a parent-of-origin effect (6, 34), which indicates some form of genomic imprinting. Furthermore, it has been suggested that loss of a second imprinted locus in the same chromosome of SdhD is necessary for paraganglioma formation (22). In this regard, a heterozygous mutant with mutations in both SdhD and H19, another imprinted locus proposed to be involved in hereditary paraganglioma, also failed to show any propensity for tumorigenesis (4). It could also be possible that complete maturation of the AM and CB is a prerequisite for tumor appearance. However, heterozygous SdhDflox/− animals, in which we induced loss of the maternally inherited SdhD allele in adulthood (SDHD-ESR mice), also failed to show any tumoral or hypertrophic phenotype. The fact that both the TH-SDHD and SDHD-ESR mice, despite being subjected to loss of the two copies of the gene in catecholaminergic tissues, did not display tumor features further strengthens the idea that a “second hit” is required for paraganglioma pathogenesis. Differences in chromosomal arrangements between humans and mouse, or unknown distinct biochemical features between the two species, could account for the resistance to development of paraganglioma/pheochromocytoma in mouse.
As indicated above, SdhD deficient mice have also been used as a model of mitochondriopathy to investigate the effect of primary mitochondrial ETC inhibition on postnatal maturation of SNpc dopaminergic neurons as well as their maintenance in adult life in comparison with other catecholaminergic cells. Although a vast amount of literature suggests that mitochondria play a central role in PD pathogenesis (14, 39, 47, 48), whether dopaminergic SNpc neurons, the cell group whose destruction is responsible for most of the motor symptoms in PD, are especially susceptible to mitochondrial dysfunction is not definitely established. Surprisingly, there has also been little investigation of whether mitochondrial deficiencies alter the ontogenic maturation of ventral mesencephalic neurons, an elaborate sequence of events occurring during the early postnatal period that involves programmed cell death and acquisition of the dopaminergic phenotype, thus determining the number of TH+ SNpc neurons that reach adulthood (9, 26).
Systemic application of toxins, like MPTP or rotenone, which inhibit mitochondrial complex I has been reported to induce a rather selective dopaminergic SNpc cell loss (7, 28; for reviews, see references 14 and 39). These actions have mostly been observed in adult animals, although previous studies described alterations of adult mesencephalic dopaminergic neurons after application of low doses of MPTP and other mitochondrial inhibitors during the neonatal period (17). A differential protective response of microglia to MPTP treatment between neonatal and adult mice has also been reported (45). However, the effects of systemic toxins depend largely on their distribution and uptake by the different cellular types in the brain and therefore do not unequivocally reflect an inherent sensitivity of the affected neurons to mitochondrial poisoning. Within the context of this discussion, it may be also worth considering that rotenone, a lipophilic substance, can block K+ currents in several cell types (1, 30, 50) and alter global oxygen homeostasis by interfering with carotid body oxygen sensing (36). On the other hand, several genes mutated in familial forms of PD have been shown to encode proteins located in, or capable of interacting with, mitochondria (49, 55). Deletion of these genes does not induce SNpc cell death in mice (27), thus casting doubts on whether mitochondrial bioenergetics alterations per se can lead to neurodegeneration. It has previously been reported that genetic ablation of mitochondrial DNA-encoded genes causing alteration in the function of complexes I, III, IV, and V results in extensive death of ventral mesencephalic dopaminergic neurons. Nonetheless, the selective effect of this mitochondrial damage on SNpc cells, in comparison with other neuronal types, has not been studied in detail (16). In this report we show that mitochondrial dysfunction in mice restricted to catecholaminergic brain areas causes a major inhibition of postnatal mesencephalic dopaminergic neuron phenotypic specification, thus causing a progressive PD-like syndrome due to cell loss predominantly affecting the SNpc. Other catecholaminergic neuronal groups, such as the dopaminergic A13 and noradrenergic LC, that were already mature at birth were practically unaltered. Therefore, these data indicate that SNpc neurons are highly vulnerable to mitochondrial dysfunction. Death of dopaminergic nigrostriatal neurons in TH-SDHD mice appeared to be a cell-autonomous phenomenon, since these mutant animals did not show abnormalities in other areas of the brain or in the striatal neurons innervated by the dopaminergic terminals.
We have shown that in adrenal chromaffin cells, SdhD ablation leads to a decrease in ATP content. Therefore, the cell loss, particularly in the dopaminergic SNpc cell group, observed in the TH-SDHD mice could be a consequence of bioenergetic deficiency, possibly aggravated by mitochondria ROS overproduction due to inefficient transference of electrons from Sdh to ubiquinone, similar to what occurs in cells lacking SdhB or SdhC proteins (19, 25). The involvement of mitochondrial ROS in SNpc cell damage was proposed several years ago (46), and a causative link between ROS production and selective SNpc cell loss was recently established. Adult dopaminergic SNpc neurons are autonomous Ca2+-dependent pacemakers that require a proper mitochondrial ETC function for cytosolic Ca2+-buffering homeostasis. Therefore, these cells are normally exposed to an oxidant stress, derived from the particularly active ETC, which not only compromises their long-term survival but also makes them particularly vulnerable to mitochondrial alterations (11, 18). In accord with this idea, systemic antioxidant treatment of TH-SDHD mice with tempol appeared to have a protective role with regard to SNpc neurons and favored catecholaminergic phenotypic specification. However, this observation must be taken as preliminary, since although we have been able to measure a higher level of lipid peroxidation in AM samples from TH-SDHD mice compared to controls, other signs of oxidative stress, such as oxidative DNA damage or increased protein oxidation, were undetectable. In addition, due to technical limitations, we do not have yet any direct measurement of ROS levels in SNpc neurons of the mutant animals. Nevertheless, it is intriguing that other catecholaminergic cells, for example, those in the LC that, like SNpc neurons, posses large terminal axonal fields and exhibit Ca2+-dependent spontaneous action potential firing (10), are resistant to mitochondrial dysfunction. Although Ca2+-dependent pacemaking is surely a major risk for neurodegeneration, our findings suggest that additional metabolic and/or developmental differences between SNpc and other catecholaminergic neurons must exist to explain their distinct sensitivity to mitochondrial ETC inhibition. It is known that adult SNpc and LC neurons are both highly sensitive to glial-cell-line-derived neurotropic factor (GDNF) deficiency (38), and this trophic factor protects LC neurons against oxidants (2). Therefore, it could be that in TH-SDHD animals, LC neurons are unaffected because they have stronger trophic support and antioxidant defense than SNpc cells. Indeed, a peculiar property of SNpc neurons with respect to LC and A13 cells is that the former reach full phenotype specification in postnatal life, which includes TH expression as well as final establishment of synapses at the distant striatum (9). In addition, we have recently shown that GDNF expression by neostriatal interneurons, necessary to support trophically SNpc cells, is not yet fully developed at birth but matures during postnatal weeks 2 to 4 (23). Neurons of LC or A13, which mature during embryonic life, may have reached some of their targets at birth and therefore are provided with the trophic support that endows them with resistance to Sdh deficiency. In contrast, the postnatal development of the nigrostriatal pathway and striatal GDNF-producing interneurons leaves SNpc cells deprived of target-derived trophic support for a period during which they are more susceptible to mitochondrial dysfunction. It is likely that in TH-SDHD animals, the nigrostriatal pathway never reaches full maturation. This would explain why they have complete striatal denervation even at early juvenile stages, despite the fact that numerous SNpc TH+ cells are conserved. Lack of trophic support during postnatal maturation and strict dependence on mitochondrial ETC function are features that together could explain the exceptional vulnerability of the SNpc to stressful conditions.
The data presented here provide unequivocal support for a major selective role of mitochondria in SNpc neuronal loss in PD; however, it must be noted that TH-SDHD mice do not exactly reproduce the neuronal loss seen in PD. For example, TH-SDHD animals had a clear loss of dopaminergic neurons in the VTA, whereas these neurons are spared or only slightly affected in PD patients as well as in the MPTP mouse model of PD. Furthermore, LC neurons, unaltered in TH-SDHD animals, are also affected in PD. The observations in this report reinforce a relatively little explored perspective in PD pathogenesis associated with early postnatal SNpc cell phenotypic specification, which could also be strictly dependent on proper mitochondrial function. In this regard it is worth emphasizing that TH-SDHD mice show death in peripheral sympathetic neurons and paraneurons, as is also observed in early stages of PD (33, 54). Our results support the long-standing notion (17) that subtle developmental alterations or exposure to environmental pollutants, occurring during the critical perinatal period, could diminish the final number of physiologically mature SNpc neurons. In accord with the proposed “multiple-hit” hypotheses for PD (13, 53), the affected individuals would surely be more susceptible to other hits damaging SNpc neurons later in life, thus predisposing them to suffer the disease in adulthood.
ACKNOWLEDGMENTS
Support was obtained from the Marcelino Botín Foundation, the Spanish Ministries of Science and Health (TERCEL), and the Andalusian Government.
We thank José Antonio Rodríguez-Gómez, María Hidalgo-Figueroa, and Alberto Castejón for technical assistance. Confocal analyses were performed with software provided by Konstantin Levitsky (Optical Microscopy and Confocal Unit of IBiS; patent no. P201100749).
Footnotes
Published ahead of print 18 June 2012
REFERENCES
- 1. Archer SL, Huang J, Henry T, Peterson D, Weir EK. 1993. A redox-based O2 sensor in rat pulmonary vasculature. Circ. Res. 73:1100–1112 [DOI] [PubMed] [Google Scholar]
- 2. Arenas E, Trupp M, Akerud P, Ibanez CF. 1995. GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron 15:1465–1473 [DOI] [PubMed] [Google Scholar]
- 3. Bardella C, Pollard PJ, Tomlinson I. 2011. SDH mutations in cancer. Biochim. Biophys. Acta 1807:1432–1443 [DOI] [PubMed] [Google Scholar]
- 4. Bayley JP, et al. 2009. Sdhd and SDHD/H19 knockout mice do not develop paraganglioma or pheochromocytoma. PLoS One 4:e7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baysal BE. 2008. Clinical and molecular progress in hereditary paraganglioma. J. Med. Genet. 45:689–694 [DOI] [PubMed] [Google Scholar]
- 6. Baysal BE, et al. 2000. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287:848–851 [DOI] [PubMed] [Google Scholar]
- 7. Betarbet R, et al. 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 3:1301–1306 [DOI] [PubMed] [Google Scholar]
- 8. Briere JJ, et al. 2005. Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum. Mol. Genet. 14:3263–3269 [DOI] [PubMed] [Google Scholar]
- 9. Burke RE. 2003. Postnatal developmental programmed cell death in dopamine neurons. Ann. N. Y. Acad. Sci. 991:69–79 [DOI] [PubMed] [Google Scholar]
- 10. Chan CS, Gertler TS, Surmeier DJ. 2010. A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson's disease. Mov. Disord. 25 (Suppl. 1):S63–S70 [DOI] [PubMed] [Google Scholar]
- 11. Chan CS, et al. 2007. ‘Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature 447:1081–1086 [DOI] [PubMed] [Google Scholar]
- 12. Chandel NS, et al. 1998. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. U. S. A. 95:11715–11720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Collier TJ, Kanaan NM, Kordower JH. 2011. Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non-human primates. Nat. Rev. Neurosci. 12:359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dauer W, Przedborski S. 2003. Parkinson's disease: mechanisms and models. Neuron 39:889–909 [DOI] [PubMed] [Google Scholar]
- 15. Dawson TM, Dawson VL. 2003. Molecular pathways of neurodegeneration in Parkinson's disease. Science 302:819–822 [DOI] [PubMed] [Google Scholar]
- 16. Ekstrand MI, et al. 2007. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. U. S. A. 104:1325–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fredriksson A, Fredriksson M, Eriksson P. 1993. Neonatal exposure to paraquat or MPTP induces permanent changes in striatum dopamine and behavior in adult mice. Toxicol. Appl. Pharmacol. 122:258–264 [DOI] [PubMed] [Google Scholar]
- 18. Guzman JN, et al. 2010. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468:696–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. 2008. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 28:718–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hayashi S, McMahon AP. 2002. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244:305–318 [DOI] [PubMed] [Google Scholar]
- 21. Hederstedt L. 2003. Structural biology. Complex II is complex too. Science 299:671–672 [DOI] [PubMed] [Google Scholar]
- 22. Hensen EF, et al. 2004. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene 23:4076–4083 [DOI] [PubMed] [Google Scholar]
- 23. Hidalgo-Figueroa M, Bonilla S, Gutierrez F, Pascual A, Lopez-Barneo J. 2012. GDNF Is predominantly expressed in the PV+ neostriatal interneuronal ensemble in normal mouse and after injury of the nigrostriatal pathway. J. Neurosci. 32:864–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ishii T, et al. 2011. Mitochondrial reactive oxygen species generation by the SDHC V69E mutation causes low birth weight and neonatal growth retardation. Mitochondrion 11:155–165 [DOI] [PubMed] [Google Scholar]
- 25. Ishii T, et al. 2005. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 65:203–209 [PubMed] [Google Scholar]
- 26. Jackson-Lewis V, et al. 2000. Developmental cell death in dopaminergic neurons of the substantia nigra of mice. J. Comp. Neurol. 424:476–488 [DOI] [PubMed] [Google Scholar]
- 27. Kitada T, Tong Y, Gautier CA, Shen J. 2009. Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J. Neurochem. 111:696–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langston JW, Ballard PA., Jr 1983. Parkinson's disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 309:310. [DOI] [PubMed] [Google Scholar]
- 29. Lindeberg J, et al. 2004. Transgenic expression of Cre recombinase from the tyrosine hydroxylase locus. Genesis 40:67–73 [DOI] [PubMed] [Google Scholar]
- 30. Lopez-Barneo J, Ortega-Saenz P, Piruat JI, Garcia-Fernandez M. 2006. Oxygen-sensing by ion channels and mitochondrial function in carotid body glomus cells. Novartis Found. Symp. 272:54–64 [DOI] [PubMed] [Google Scholar]
- 31. Maher LJ, III, et al. 2011. Mouse models of human familial paraganglioma. In Martin JF. (ed), Pheochromocytoma—a new view of the old problem. Intech, Rijeka, Croatia: http://www.intechopen.com/books/pheochromocytoma-a-new-view-of-the-old-problem/mouse-models-of-human-familial-paraganglioma [Google Scholar]
- 32. Mejias R, et al. 2006. Neuroprotection by transgenic expression of glucose-6-phosphate dehydrogenase in dopaminergic nigrostriatal neurons of mice. J. Neurosci. 26:4500–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Minguez-Castellanos A, et al. 2007. Do alpha-synuclein aggregates in autonomic plexuses predate Lewy body disorders?: a cohort study. Neurology 68:2012–2018 [DOI] [PubMed] [Google Scholar]
- 34. Muller U. 2011. Pathological mechanisms and parent-of-origin effects in hereditary paraganglioma/pheochromocytoma (PGL/PCC). Neurogenetics 12:175–181 [DOI] [PubMed] [Google Scholar]
- 35. Ohkawa H, Ohishi N, Yagi K. 1979. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 95:351–358 [DOI] [PubMed] [Google Scholar]
- 36. Ortega-Saenz P, Pardal R, Garcia-Fernandez M, Lopez-Barneo J. 2003. Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. J. Physiol. 548:789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pardal R, Ortega-Saenz P, Duran R, Lopez-Barneo J. 2007. Glia-like stem cells sustain physiologic neurogenesis in the adult mammalian carotid body. Cell 131:364–377 [DOI] [PubMed] [Google Scholar]
- 38. Pascual A, et al. 2008. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat. Neurosci. 11:755–761 [DOI] [PubMed] [Google Scholar]
- 39. Perier C, Bove J, Vila M. 2012. Mitochondria and programmed cell death in Parkinson's disease: apoptosis and beyond. Antioxid. Redox Signal. 16:883–895 [DOI] [PubMed] [Google Scholar]
- 40. Piruat JI, Pintado CO, Ortega-Saenz P, Roche M, Lopez-Barneo J. 2004. The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Mol. Cell. Biol. 24:10933–10940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pollard PJ, et al. 2005. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 14:2231–2239 [DOI] [PubMed] [Google Scholar]
- 42. Ruan K, Song G, Ouyang G. 2009. Role of hypoxia in the hallmarks of human cancer. J. Cell Biochem. 107:1053–1062 [DOI] [PubMed] [Google Scholar]
- 43. Rustin P, Munnich A, Rotig A. 2002. Succinate dehydrogenase and human diseases: new insights into a well-known enzyme. Eur. J. Hum. Genet. 10:289–291 [DOI] [PubMed] [Google Scholar]
- 44. Santos JH, Mandavilli BS, Van Houten B. 2002. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods Mol. Biol. 197:159–176 [DOI] [PubMed] [Google Scholar]
- 45. Sawada H, et al. 2007. Activated microglia affect the nigro-striatal dopamine neurons differently in neonatal and aged mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Neurosci. Res. 85:1752–1761 [DOI] [PubMed] [Google Scholar]
- 46. Schapira AH. 2008. Mitochondrial dysfunction in neurodegenerative diseases. Neurochem. Res. 33:2502–2509 [DOI] [PubMed] [Google Scholar]
- 47. Schapira AH, Jenner P. 2011. Etiology and pathogenesis of Parkinson's disease. Mov. Disord. 26:1049–1055 [DOI] [PubMed] [Google Scholar]
- 48. Schapira AH, et al. 1992. Mitochondrial function in Parkinson's disease. The Royal Kings and Queens Parkinson's Disease Research Group. Ann. Neurol. 32(Suppl):S116–S124 [DOI] [PubMed] [Google Scholar]
- 49. Schon EA, Przedborski S. 2011. Mitochondria: the next (neurode)generation. Neuron 70:1033–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Searle GJ, Hartness ME, Hoareau R, Peers C, Kemp PJ. 2002. Lack of contribution of mitochondrial electron transport to acute O2 sensing in model airway chemoreceptors. Biochem. Biophys. Res. Commun. 291:332–337 [DOI] [PubMed] [Google Scholar]
- 51. Selak MA, et al. 2005. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7:77–85 [DOI] [PubMed] [Google Scholar]
- 52. Soriano P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21:70–71 [DOI] [PubMed] [Google Scholar]
- 53. Sulzer D. 2007. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 30:244–250 [DOI] [PubMed] [Google Scholar]
- 54. Tolosa E, Gaig C, Santamaria J, Compta Y. 2009. Diagnosis and the premotor phase of Parkinson disease. Neurology 72:S12–20 [DOI] [PubMed] [Google Scholar]
- 55. Vila M, Ramonet D, Perier C. 2008. Mitochondrial alterations in Parkinson's disease: new clues. J. Neurochem. 107:317–328 [DOI] [PubMed] [Google Scholar]
- 56. Villadiego J, et al. 2005. Selective glial cell line-derived neurotropic factor production in adult dopaminergic carotid body cells in situ and after intrastriatal transplantation. J. Neurosci. 25:4091–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wilcox CS, Pearlman A. 2008. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol. Rev. 60:418–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yankovskaya V, et al. 2003. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299:700–704 [DOI] [PubMed] [Google Scholar]