

Abstract
The global emergence of Clostridium difficile infection (CDI) has contributed to the recent surge in severe antibiotic-associated diarrhea and colonic inflammation. C. difficile produces two homologous glucosylating exotoxins, TcdA and TcdB, both of which are pathogenic and require neutralization to prevent disease occurrence. However, because of their large size and complex multifunctional domain structures, it has been a challenge to produce native recombinant toxins that may serve as vaccine candidates. Here, we describe a novel chimeric toxin vaccine that retains major neutralizing epitopes from both toxins and confers complete protection against primary and recurrent CDI in mice. Using a nonpathogenic Bacillus megaterium expression system, we generated glucosyltransferase-deficient holotoxins and demonstrated their loss of toxicity. The atoxic holotoxins induced potent antitoxin neutralizing antibodies showing little cross-immunogenicity or protection between TcdA and TcdB. To facilitate simultaneous protection against both toxins, we generated an active clostridial toxin chimera by switching the receptor binding domain of TcdB with that of TcdA. The toxin chimera was fully cytotoxic and showed potent proinflammatory activities. This toxicity was essentially abolished in a glucosyltransferase-deficient toxin chimera, cTxAB. Parenteral immunization of mice or hamsters with cTxAB induced rapid and potent neutralizing antibodies against both toxins. Complete and long-lasting disease protection was conferred by cTxAB vaccinations against both laboratory and hypervirulent C. difficile strains. Finally, prophylactic cTxAB vaccination prevented spore-induced disease relapse, which constitutes one of the most significant clinical issues in CDI. Thus, the rational design of recombinant chimeric toxins provides a novel approach for protecting individuals at high risk of developing CDI.
INTRODUCTION
Clostridium difficile is the most common cause of nosocomial antibiotic-associated diarrhea and is the etiologic agent of pseudomembranous colitis (8, 27, 45). The drug-resistant bacterium causes a wide spectrum of disease symptoms, ranging from mild diarrhea and colitis to fulminant systemic disease and mortality (3, 5). With the recent emergence of hypervirulent strains, the incidence of C. difficile infection (CDI) has increased rapidly worldwide, along with more severe forms of the disease, causing lengthy hospitalization, substantial morbidity, and mortality (37, 42). Two exotoxins (TcdA and TcdB) are the major cause of the disease (30, 41). Both toxins are large, single-chain proteins with similar multidomain structures that include an N terminus catalytic glucosyltransferase domain (GTD), an autoproteolytic cysteine proteinase domain (CPD), a central translocation domain (TM), and a C-terminal, so-called receptor-binding domain (RBD) although its receptor binding function has yet to be confirmed (22). The toxin glucosyltransferase inhibits host Rho GTPases in colonocytes, causing cytoskeletal disruption, barrier dysfunction, diarrhea, and colitis (58). Standard therapy depends on treatment with vancomycin or metronidazole, neither of which is fully effective as up to 35% of patients develop recurring disease within a few weeks (62).
Although fidaxomicin (38) and humanized monoclonal antitoxin IgGs (39) have recently been shown to reduce CDI recurrence in patients, it is generally considered that a vaccine approach targeting the C. difficile toxins is a preferred preventative strategy (15–17, 56). In support of this notion, antibodies against both toxins are protective in hamsters (13, 28, 34), and serum anti-Tcd antibodies in patients correlate with protection against symptomatic disease and recurrence (31, 33). Vaccine candidates that target the C. difficile toxins include toxoids (1, 16, 18, 50, 51, 56) and recombinant TcdA RBD fragments (4, 15, 43, 46–48, 59, 60). Toxoids, generated by formalin inactivation of holotoxins, represent the major focus of vaccine development and have been tested in patients (1, 19, 29, 51). While effective in animal CDI models (17), toxoids have yet to be proven as protective in patients and are associated with several inherent shortcomings, including batch-to-batch variations and potential residual toxicity. In this study, attenuated recombinant toxins were generated that showed superior efficacy over toxoids in protecting mice from C. difficile toxin challenge but failed to confer adequate cross-protection. A toxin chimera constructed subsequently provided concurrent protection against both toxins and C. difficile oral challenge, showing potent efficacy as a new vaccine candidate in experimental CDI models.
MATERIALS AND METHODS
Generation of mutant holotoxins, toxin fragment, and chimeras.
We have previously cloned the full-length TcdA and TcdB genes into a shuttle vector pHis1522 (pHis-TcdA and pHis-TcdB, respectively) and expressed the recombinant holotoxins in Bacillus megaterium (61). Based on this system, point mutations were introduced into conserved amino acids that are responsible for the substrate uridine diphosphoglucose (UDP-Glc) binding in order to generate the glucosyltransferase (GT)-deficient holotoxins (23). To generate GT-mutant holotoxin A, we first designed a unique restriction enzyme (BamHI) site between sequences encoding GT and CPD domains using overlapping PCR. The primer sets used were as follows: pHis-F, 5′-TTTGTTTATCCACCGAACTAAG-3′; Bam-R, 5′-TCTTCAGAAAGGGATCCACCAG-3′; Bam-F, 5′-TGGTGGATCCCTTTCTGAAGAC-3′; and Bpu-R, 5′-ACTGCTCCAGTTTCCCAC-3′.
The final PCR product was digested with BsrGI and Bpu10I and used to replace the corresponding sequence in pHis-TcdA. The resulting plasmid was designated pH-TxA-b. Sequences encoding triple mutations (W101A, D287N, and W519A) in the GT were synthesized by Geneart (Germany) and cloned into pH-TxA-b through BsrGI/BamHI digestion. To generate the mutant holotoxin B construct, the sequence between BsrGI and NheI containing two point mutations (W102A and D288N) was synthesized and inserted into pHis-TcdB at the same restriction enzyme sites, leading to a new plasmid, pH-aTcdB. The fragment with TcdB-RBD was generated by PCR amplification using the following primer set: 5′-GGTTGCTGGATCCGCAAATAAGCTATCTTTTAACTTTAGTGATAAACAAGATGTACC-3′ and 5′-CCATGCTGAGCTCGCTTCACTAATCACTAATTGAGCTGTATCAGGATCAAAATAATAC-3′. The fragment was expressed using pET32 vector (Novagen, NJ).
To generate the chimera TxB-Ar, a unique restriction endonuclease (RE) AgeI site was created at a position between the transmembrane domain (TMD) and RBD without changing the amino acid sequence of expressed pHis-TcdB. Then, the gene encoding the RBD of TcdA was amplified using the primers TxA-Ar-F (5′-AATTACCGGTTTTAACTTAGTAACTGGATGGC-3′) and TxA-Ar-R (5′-AATTGCATGCTGGTACCCTCCATATATCCCAGGGGCTTTTACTCC-3′), and the RBD sequence of TcdB was replaced with that of TcdA through AgeI/KpnI digestion, generating a plasmid (pH-TxB-Ar). To generate the chimera cTxAB, the XhoI/Bpu10I fragment in pH-TxB-Ar was replaced by the fragment carrying W102A and D288N mutations from pH-aTcdB. The resultant constructs carrying full-length mutant toxin and chimera genes were used to transform B. megaterium, and mutant holotoxins and chimeras (Fig. 1A to D) were expressed and purified using methods described previously (61).
Fig 1.

Generation of glucosyltransferase (GT)-mutant holotoxins. aTcdA (A) and aTcdB (B) represent mutant TcdA with triple mutations (W101A, D287N, and W519A) and TcdB with double mutations (W102A and D288N) in their respective GT domains. (C) TxB-Ar is TcdB with its RBD replaced with that of TcdA. (D) cTxAB is TxB-Ar with two mutations (W102A and D288N) in its glucosyltransferase domain. CPD, cysteine protease domain; TMD, transmembrane domain; RBD, receptor-binding domain; His6, six-histidine tag. (E) Vero cell lysates were exposed to wild-type or mutant toxin proteins for 30 min. Rac1 glucosylation was analyzed by immunoblotting using monoclonal antibody (clone 102) that binds only to nonglucosylated Rac1. β-Actin was used as an equal loading control. CT26 cells in a 96-well plate were exposed to aTcdA or TcdA (F) or to aTcdB or TcdB (G) at different concentrations for 72 h. MTT assays were performed, and cell viability is expressed as the percentage of surviving cells compared to cells without toxin exposure. (H) BALB/c mice were i.p. challenged with either 100 ng/mouse wild-type TcdA or TcdB or with 100 μg/mouse of mutant aTcdA or aTcdB. Mouse survival was monitored, and the data show the Kaplan-Meier survival curves (n = 10; P < 0.001, between wild-type and mutant toxin groups).
Glucosyltransferase activity of the toxins.
GT activity of holotoxins and mutant toxins was measured by their ability to glucosylate Rho GTPase Rac1 in a cell-free assay. Vero cell pellets were resuspended in glucosylation buffer (50 mM HEPES, pH 7.5, 100 mM KCl, 1 mM MnCl2, and 2 mM MgCl2) and lysed by passage through a syringe (25 gauge). After centrifugation (at 167,000 × g for 3 min), the supernatant was used as a postnuclear cell lysate. To perform the glucosylation assay, the cell lysates were incubated with TcdA, TcdB, or their mutants (5 μg/ml final concentration) at 37°C for 30 min. The reaction was terminated by heating the sample at 100°C for 5 min in SDS sample buffer. To measure Rac1 glucosylation, lysates were separated on a 12% SDS-PAGE gel and transferred onto a nitrocellulose membrane. Antibodies that specifically recognize the nonglucosylated form of Rac1 (clone 102; BD Bioscience), anti-β-actin (clone AC-40; Sigma), and horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Amersham Biosciences) were used for detection using standard Western blotting with enhanced chemiluminescence.
CD spectroscopy of wild-type and mutant toxins.
Circular dichroism (CD) spectra were recorded on an Aviv 62 spectropolarimeter in the wavelength range of 190 to 260 nm, with a bandwidth of 1.0 nm and scan step of 0.5 nm using a 0.1-cm path length in a 1-cm quartz cell at 22°C. Protein concentrations were in the range of 50 to 200 μg/ml. In each case at least five spectra were accumulated, smoothed, averaged, and corrected for the contribution of solutes.
Cytopathic and cytotoxicity assay.
Cytopathic and cytotoxic activities of the toxins were assayed as described previously (20). CT-26 or Vero cells seeded in 96-well plates were treated with wild-type or mutant toxins. To evaluate cytopathic effects of the toxins on cells, the morphological changes of cells were observed using phase-contrast microscopy. MTT [3-(4,5-dimethylthiazol-2-yl)2 2,5-diphenyl tetrazolium bromide] assays were also performed to measure the cytotoxic activities of the toxins. After 72 h of incubation, 10 μl of MTT (5 mg/ml) was added to each well, and plates were incubated at 37°C for 4 h. The formazan was solubilized with acidic isopropanol (0.4 N HCl in absolute isopropanol), and absorbance was measured at 570 nm using a 96-well enzyme-linked immunosorbent assay (ELISA) plate reader. Cell viability was expressed as the percentage of survival compared cells in untreated control wells. The experiments were repeated three times, and triplicate wells were assessed for cytopathic changes and cytotoxicity in each experiment.
Cytokine production by dendritic cells.
To assess the proinflammatory activities of the chimeric toxin TcdB-Ar, mouse bone marrow-derived dendritic cells (BMDCs) were collected as described previously (11, 12). Cells were either untreated or exposed to TxB-Ar (200 ng/ml) alone or together with goat anti-TcdA and TcdB polysera (Techlab, Inc.) for 24 h, and cell supernatants were harvested. The presence of the cytokine/chemokine interleukin-1β (IL-1β), IL-6, and CXCL1 (keratinocyte-derived chemokine [KC]) in the supernatants was measured by standard ELISA following the manufacturer's directions with murine cytokine quantification kits (Invitrogen and R&D Systems).
Mouse immunizations.
C57BL/6, BALB/c, and CD1 mice (5 to 6 weeks old) were purchased from Jackson Laboratory. All mice used in the experiments were housed in groups of 5 animals per cage under the same conditions. Food, water, bedding, and cages were autoclaved. All animals were handled and cared for according to Institutional Animal Care and Use Committee guidelines and in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. BALB/c or C57BL/6 mice were immunized intraperitoneally (i.p.) or intramuscularly (i.m.) with 5 μg of purified mutant toxins in phosphate-buffered saline (PBS) with alum as an adjuvant for each injection. Control mice received PBS with alum. When aTcdA mixed with aTcdB (5 μg each) or cTxAB was used as the immunogen, a total of 10 μg of protein per injection was used, and mice were given three immunizations with an interval of 10 to 14 days. For passive immunization, 50 μl of polysera collected from alpacas immunized with either aTcdA or aTcdB or with the mixed polysera (100 μl total) was injected i.p. into naïve C57BL/6 mice at 4 h after C. difficile challenge. Control mice were administered the same amount of serum collected from animals prior to toxin immunization (preserum).
Antibody titers and in vitro neutralizing assay.
Antibody titers were measured using a standard ELISA against purified native or recombinant wild-type holotoxins. To assess in vitro neutralizing activities of serum samples, we used mouse intestinal epithelial CT26 cells as these are sensitive to both TcdA and TcdB. The neutralizing titer is defined as the maximum dilution of the samples that blocks cell rounding induced by toxin at a given concentration. This given concentration is four times the minimum dose of the toxin that causes all CT26 cells to round after a 24-h exposure to the toxin, i.e., 5 and 0.25 ng/ml for TcdA and TcdB, respectively. To assess neutralizing epitopes, sera from aTcdB-immunized mice were preincubated with excessive amounts (50 ng/ml) of toxoid B or recombinant TcdB-RBD fragment on ice for 30 min before being added to CT26 cells, and serum neutralizing titers were determined. Wells without serum were included as controls, and neither toxoid B nor the fragment at 50 ng/ml inhibited TcdB cytotoxicity.
Measurement of antitoxin IgG isotypes.
IgG1, IgG2a, IgG2b, IgG2c, and IgG3 anti-TcdB concentrations in the sera of aTcdB- or toxoid B-immunized mice were determined by ELISA using biotinylated anti-mouse IgG subclass antibodies.
Primary and recurrent CDI models.
C57BL/c mice were treated with an antibiotic cocktail followed by oral inoculation of C. difficile as described previously (7). Ten days after the third immunization, mice were given 105 CFU of vegetative bacteria (laboratory VPI10463 strain) via oral gavage. To minimize oxygen exposure, the vegetative cell aliquots in airtight vials were prepared in an anaerobic chamber, and the vials were opened right before inoculation. To assess long-term immunity, mice were orally challenged with 106 CFU of vegetative bacteria 3 months after the third immunization. In some experiments, immunized mice were challenged with 106 spores of UK1 (027/B1/NAP1 strain kindly provided by D. Gerding, VA Chicago Health Care System). To induce CDI relapse, surviving mice were given antibiotic cocktail treatment followed by oral gavage of C. difficile spores (106/mouse) 30 days after the primary infection (55).
Hamster immunization and challenge.
Hamsters were i.p. immunized with 10 μg of cTxAB in 100 μl of PBS with alum as an adjuvant three times with 10-day intervals. Control hamsters were immunized with an equal volume of PBS-alum. Serum samples were collected before and 9 days after each immunization, and anti-TcdA and anti-TcdB IgG titers were measured by standard ELISA. Five days after the third immunization, the hamsters were injected i.p. with clindamycin (30 mg/kg) and then orally challenged with C. difficile UK1 spores (100 CFU/hamster) 5 days later. The animals were monitored daily for diarrhea and other signs of disease, and moribund animals were euthanized.
Statistical analysis.
Data were analyzed by Kaplan-Meier survival analysis with a log rank test of significance, by analysis of variance (ANOVA), and by one-way or two-way ANOVA followed with Bonferroni posttests using the Prism statistical software program. Results are expressed as means ± standard errors of means.
RESULTS
Generation of attenuated C. difficile holotoxins.
TcdA and TcdB are large clostridial toxins that possess complex structural conformations (22, 44). Formalin cross-linking of these toxins is likely to alter conformational epitopes, which can impact immunogenicity. To demonstrate this point, attenuated recombinant TcdA and TcdB were generated with point mutations in the highly conserved amino acids that regulate substrate binding in glucosyltransferases (23), designated aTcdA and aTcdB, respectively (Fig. 1A and B). The mutant holotoxins were essentially devoid of glucosyltransferase activity (Fig. 1E), cytotoxicity (Fig. 1F and G), and in vivo toxicity (Fig. 1H). In contrast to toxoids which demonstrate significant conformational changes compared with native toxins, mutant holotoxins maintained a structure similar to that of native toxins, as determined by circular dichroism spectrometry (Fig. 2A to C). Thus, structurally intact glucosyltransferase-deficient holotoxins are essentially nonpathogenic, making them eminently suitable vaccine candidates.
Fig 2.

Circular dichroism (CD) structural analysis of wild-type and mutant toxins. For secondary structural analysis of wild-type TcdA and mutant aTcdA (A) and of TcdB, aTcdB, and toxoid B (B), far-UV CD spectra were recorded at 22°C. This structural analysis demonstrates that wild-type and GT-mutant toxins are structurally similar since CD spectra are virtually superimposed, but a significant shift occurs in toxoid B. Secondary structural composition elements are illustrated in panel C.
The mutant holotoxin is more immunogenic and protective against C. difficile toxin challenge than toxoid.
Toxoids TcdA and TcdB, generated by formalin inactivation of holotoxins, represent the major focus of vaccine development and have been tested in patients (1, 19, 29, 50, 51). Recent studies in patients (36) and in animal models (30, 41) have highlighted the relative importance of TcdB as one of the primary virulence factors in CDI. We therefore compared the immunogenicity of aTcdB with toxoid TcdB (toxoid B). Both immunogens generated a Th2-type response with serum antitoxin IgG1 and IgG2b in systemically immunized mice (Fig. 3A). However, immunization with aTcdB induced a stronger IgG response and significantly higher neutralizing activity than immunization with toxoid B (Fig. 3B and C). Preincubation of polysera from aTcdB-immunized mice with toxoid B did not significantly reduce their neutralizing activity (Fig. 3C), indicating that the highly neutralizing epitopes were largely absent in toxoid B. As a consequence, aTcdB-immunized mice were fully protected against lethal wild-type TcdB challenge, whereas toxoid B-immunized mice all developed systemic toxemia, with 70% lethality (Fig. 3D). Thus, aTcdB immunization conferred protection against systemic toxin challenge that was superior to that of toxoid B.
Fig 3.
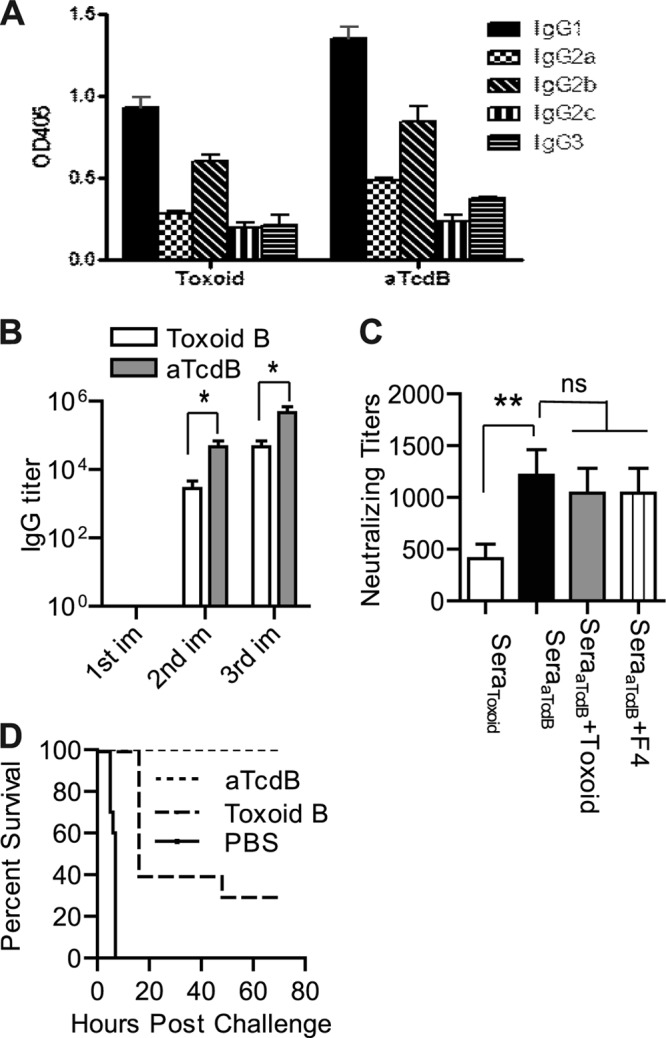
Mouse antibody response and neutralizing titers after aTcdB or toxoid B immunization. Mice were immunized with aTcdB or toxoid B three times, and serum samples were collected. (A) The antitoxin IgG isotypes of the serum samples were measured using standard ELISAs. OD405, optical density at 405 nm. (B) TcdB-specific antibody titers after each immunization (im) with aTcdB or toxoid B (*, P < 0.05). (C) Neutralizing titers of sera from toxoid B-immunized mice (SeraToxoid) and from aTcdB-immunized mice (SeraaTcdB). In some groups, the sera were preincubated with 50 ng/ml of toxoid B (SeraaTcdB + Toxoid) or recombinant fragment TcdB-RBD (SeraaTcdB + F4) before being added to the cells (**, P < 0.01; ns, not significant). (D) Kaplan-Meier survival curves of the data of immunized mice challenged with a lethal dose (100 ng/mouse) of wild-type TcdB (aTcdB versus toxoid B, P = 0.001). All data are representative of at least three independent experiments (error bars indicate standard errors of the means; the data were analyzed by Kaplan-Meier survival analysis or by ANOVA).
Attenuated holotoxins show limited cross-protection against CDI.
Because most pathogenic C. difficile isolates produce TcdA and TcdB, which are highly homologous to each other (58), we examined cross-protection between the two toxins. Consistent with a previous report (35), antibodies generated by aTcdB immunization showed little cross-protection against TcdA and vice versa (Fig. 4A and B). aTcdB immunization protected mice from lethal challenge of TcdB (Fig. 3C) but not of TcdA (Fig. 4C). When mice were immunized with both aTcdA and aTcdB, potent neutralizing antibodies were evident against both toxins (Fig. 4A and B), and animals were fully protected against lethal systemic challenge with the two toxins (Fig. 4D). Moreover, immunization with aTcdA or aTcdB alone only partially protected mice from oral challenge with C. difficile strain VPI10463 (Fig. 4E and F), whereas simultaneous administration of aTcdA and aTcdB induced complete protection against the lethality and diarrhea of CDI (Fig. 4E and F). These data demonstrate a lack of cross-immunogenicity between the two toxins and a requirement for neutralizing antibodies against both toxins for full protection.
Fig 4.

Cross-protection of polysera from mice immunized with mutant toxins. Groups of mice were immunized with the indicated immunogens three times. (A and B) After three immunizations with the indicated immunogens, serum neutralizing titers against TcdA (A) and TcdB (B) were determined (*, P < 0.01 compared to aTcdB in panel A or aTcdA in panel B). (C and D) Kaplan-Meier survival curves of aTcdB-immunized mice (C) or mice immunized with both aTcdA and aTcdB (D) and challenged with a lethal dose of TcdA (panel C, n = 10; P = 0.535) or of TcdA and TcdB (panel D, n = 10; P < 0.001), respectively. (E and F) After oral challenge with the C. difficile VPI10463 strain, mouse survival (E) (aTcdA plus aTcdB versus PBS, P = 0.04) and the percentage of mice developing diarrhea (F) are illustrated. All data are representative of at least three independent experiments.
Generation of the active toxin chimera TxB-Ar and glucosyltransferase mutant cTxAB.
Because of the insufficient cross-protection elicited by the individual antitoxins, we sought to generate a single immunogen capable of inducing potent neutralizing antibodies against both toxins. The RBD is the immunodominant toxin region of TcdA and possesses potent adjuvant activity due to its lectin-like structural repeats (6, 57). In contrast, the neutralizing epitopes in TcdB are primarily confined to the N terminus since preincubation of polysera from aTcdB-immunized mice with recombinant TcdB-RBD did not significantly reduce the serum neutralizing activity (Fig. 3C). Moreover, the N terminus in TcdB is more highly conserved between historical and hypervirulent strains than its RBD (32, 52). These observations prompted us to substitute the RBD of TcdB with that of TcdA, generating an active toxin chimera designated TxB-Ar (Fig. 1C). TxB-Ar retained potent glucosylating activity (Fig. 1E), cytotoxicity (Fig. 5A), and proinflammatory activity (Fig. 5B) equal to that of the wild-type TcdA and TcdB (14). Thus, TxB-Ar functions as a true clostridial glucosylating toxin and likely maintains a toxin-like conformation similar to that of the native C. difficile toxins. To establish a glucosyltransferase-deficient chimera and retain the toxin-like conformation, two point mutations (W101A and D288N) were introduced as described for aTcdB (Fig. 1B and 2B and C), and the resulting chimera was designated cTxAB (Fig. 1D). cTxAB displayed undetectable glucosyltransferase activity in a cell-free glucosylation assay (Fig. 1E) and essentially lost its cytotoxicity (Fig. 5A).
Fig 5.

Cytotoxicity and proinflammatory activities of the chimeric toxins. (A) MTT survival assays of CT26 cells exposed to TxB-Ar or cTxAB. Cell viability is expressed as the percentage of surviving cells compared to cells without toxin exposure. (B) Cytokine/chemokine secretion by BMDCs after TxB-Ar (200 ng/ml) exposure. P < 0.01, between TxB-Ar and the other groups. The data were analyzed by ANOVA followed by Bonferroni posttests. Error bars show ± standard errors of the means.
cTxAB vaccination induces potent neutralizing antibodies against both toxins and long-lasting protection in mice.
Intraperitoneal immunization of mice with cTxAB induced a potent systemic antibody response against both toxins (Fig. 6A). A significant IgG response was evident after a single immunization dose (Fig. 6A) that was not measured with aTcdB immunization alone (Fig. 3B) and is likely due to the lack of the RBD adjuvant activity of TcdA (6). After three immunizations, potent serum neutralizing activities with titers over 3,000 were evident against both TcdA and TcdB (Fig. 6B). Consequently, immunized mice were fully protected against lethal systemic challenge with either wild-type TcdA or TcdB (Fig. 6C).
Fig 6.

Chimera cTxAB immunization induces potent neutralizing antibodies against both toxins and protective immunity against CDI. (A and B) Serum anti-TcdA and anti-TcdB IgG titers (A) or neutralizing titers (B) are shown. (C) Mice immunized with PBS (solid lines) or cTxAB (dashed lines) were divided into two groups and challenged with lethal doses of TcdA or TcdB, respectively (P < 0.01 between PBS and cTxAB-immunized groups). (D and E) Mice were challenged with C. difficile VPI10463 vegetative cells 10 days (D) or 3 months (E) after the third immunization with PBS or cTxAB. Mouse mortality (P < 0.05), weight loss, and frequency of diarrhea are illustrated. With the exception of the experiment shown in panel E, all experiments were performed at least three times, and representative results are shown (n = 10 in each experiment; *, P < 0.05).
Next, we evaluated the immunogen cTxAB in a mouse model of CDI (7). After three parenteral immunizations and oral challenge with C. difficile VPI10463, cTxAB-immunized animals failed to show any signs of disease, whereas all vehicle-immunized mice developed diarrhea and weight loss, and approximately 60% of mice succumbed (Fig. 6D). cTxAB vaccination also established long-term protection against CDI, lasting at least 3 months after the third immunization (Fig. 6E).
cTxAB vaccination protects animals from infection with an epidemic C. difficile strain.
Because many different toxin isoforms are evident in clinical isolates, we examined whether cTxAB also confers protection against the most clinically relevant strain, BI/NAP1/027. Immunized mice were challenged with spores of the UK1 clinical isolate BI/NAP1/027 (54). In vehicle-immunized mice, significant disease symptoms (ruffled coat, lethargy, loss of appetite, and severe diarrhea) were evident by day 2 postinfection in all mice, and approximately 40% of mice succumbed by day 4 (Fig. 7A). In contrast, cTxAB-immunized mice were fully protected and showed no signs of disease at any stage (Fig. 7A). We next assessed the potential suitability of cTxAB for clinical intramuscular immunization and demonstrated similar disease protection against spore challenge (Fig. 7B). Furthermore, a single cTxAB dose was sufficient to induce anti-Tcd antibody titers (Fig. 6A) that were protective against CDI. Mice were immunized once on the same day as antibiotic treatment was initiated and were then challenged with spores 6 days later. A single cTxAB immunization dose conferred significant protection against fulminant disease (Fig. 7C) although surviving mice in both groups experienced similar diarrhea (data not shown) and weight loss (Fig. 7D). Finally, we examined the protection of cTxAB vaccination in a traditional hamster CDI model with oral UK1 spore challenge. A potent antibody response against both toxins was induced that conferred significant protection against UK1 spore-induced CDI in hamsters (Fig. 8A to C).
Fig 7.

cTxAB immunization protects mice against infection with an epidemic C. difficile strain. (A and B) After three i.p. (A) or i.m. (B) immunizations, mice were challenged with C. difficile UK1 spores (106/mouse). Mouse mortality (P < 0.05), weight loss, and frequency of diarrhea are illustrated (dotted line, cTxAB; solid line, PBS; dashed line, antibiotic cocktail treatment without spore challenge). Symbols for the weight loss graphs are the same in both panels. (C and D) Mice were immunized with cTxAB once on day −6 when the antibiotic treatment was initiated and then challenged with UK1 spores on day 0. Mouse mortality (C) (P < 0.05) and weight loss (D) are illustrated. With the exception of the experiment shown in panel B, all experiments were performed at least three times, and the representative results are shown (n = 10 in each experiment; *, P < 0.05).
Fig 8.

Protective response of cTxAB vaccination in hamsters. (A) Serum IgG titers after each immunization with cTxAB (10 μg per i.p. injection). (B) Kaplan-Meier survival curves of PBS-immunized (n = 15) or cTxAB-immunized (n = 10) hamsters orally challenged with C. difficile spores (P = 0.0007). (C) Percentage of hamsters in the PBS or cTxAB group that developed diarrhea. All hamsters in the PBS group developed severe diarrhea, whereas only one hamster in the cTxAB group developed diarrhea. The statistics were analyzed by Kaplan-Meier survival analysis or ANOVA.
Prophylactic cTxAB vaccination protects mice from recurrent CDI.
CDI has become increasingly difficult to manage due, in part, to the ineffectiveness of current antibiotic regimens which are associated with high relapse rates (24). We tested the efficacy of cTxAB immunization in preventing disease recurrence in a spore-induced mouse CDI recurrence model (55). The immunization and challenge scheme is illustrated in Fig. 9A. In vehicle-immunized mice, CDI recurrence demonstrated a similar disease course to the primary infection, with severe diarrhea and weight loss being evident and 40% of animals becoming moribund (Fig. 9C). In contrast, cTxAB immunization conferred complete protection against primary and recurrent CDI (Fig. 9C), reflecting long-lasting immunity after vaccination (Fig. 6E). To assess whether cTxAB immunization also protects against disease relapse in naïve animals that recover from CDI, surviving mice were immunized after their recovery from the initial CDI as illustrated (Fig. 9B). cTxAB-immunized animals were completely protected from CDI recurrence, whereas vehicle-immunized mice exhibited profound disease (Fig. 9D).
Fig 9.

cTxAB vaccination protects mice against recurrent CDI. (A and B) Immunization and challenge schemes for CDI relapse models. (C and D) After the second spore challenge illustrated in panel A (C) or panel B (D), mouse mortality, weight loss, and diarrhea were monitored. Symbols for the weight loss graphs are the same in both panels. All experiments were performed at least three times with similar results (n = 8 to 10 in each experiment) Survival figures show pooled data from two experiments and data were analyzed by Kaplan-Meier survival analysis (P is <0.05 in each case. Weight loss was analyzed by ANOVA (*, P < 0.05; error bars indicate standard errors of the means).
DISCUSSION
A majority of pathogenic C. difficile isolates produce both TcdA and TcdB (58). Thus, neutralizing antibodies elicited against both toxins are ideally required to impart complete protection against CDI. Because the two homologous toxins exhibit 66% overall amino acid sequence similarity (58), it is important to understand cross-protective epitopes that may assist in designing immune-based interventions. It has long been known that immunization with individual C. difficile toxoids generates poor cross-protection (neutralizing titers of <20) (35). Peptide fragments containing full or partial TcdA-RBD sequence, the immunodominant region of the toxin, have been extensively evaluated as vaccine candidates (4, 15, 43, 46, 47, 59, 60) but generally have resulted in poor protective responses (4, 60). A more recent report (43) has shown that immunization with a TcdA-RBD peptide fragment expressed in Bacillus subtilis induced weak cross-protection against TcdB (neutralizing titers ranging from 10 to 100). The findings in our study are consistent with these reports since immunization of mice with individual nonpathogenic holotoxins generated high neutralizing IgG titers against the respective toxin but demonstrated little cross-neutralization (titers of <100) (Fig. 4A and B) and, more importantly, were not completely protective against experimental CDI (Fig. 4E and F). Therefore, there is an urgent need to generate a fully cross-protective Tcd immunogen.
We report a novel clostridial toxin chimera, cTxAB, as a vaccine candidate for CDI. In designing cTxAB, several aspects were taken into consideration: (i) the immunogen should be nontoxic and safe; (ii) the native toxin structure should be maintained, thus preserving conformational epitopes; (iii) the immunogen should retain the major immunodominant domains and neutralizing epitopes from both toxins; and (iv) the immunogen should be conserved, facilitating widespread protection against diverse clinical isolates. cTxAB maintains a native toxin-like conformation but is essentially nontoxic in mice and hamsters. It is expressed in Bacillus megaterium, a nonpathogenic and endotoxin-free production system suitable for clinical use. Additionally, cTxAB retains the major conserved neutralizing epitopes of TcdA and TcdB and thus induces potent neutralizing antibodies against both toxins that protect experimental animals against primary and recurrent CDI induced by both historic and epidemic strains of C. difficile.
Because multiple C. difficile strains expressing different toxin isoforms are isolated from CDI patients, it is desirable for a vaccine to confer broad protection across clinically relevant isolates. Recent reports have shown that TcdA is relatively well conserved between historical and epidemic strains, whereas TcdB shows a higher degree of variability (32, 52). The RBD domain is the most variable region within TcdB, whereas the N terminus encompassing the GTD and CPD domains is more conserved between historical and epidemic strains (32). Consequently, we designed cTxAB to comprise the N terminus from TcdB and the C terminus RBD from TcdA, thus retaining conserved epitopes across toxin isoforms from different clinical strains. In support of this as a successful immunization strategy, cTxAB induced complete protection not only against the historical VPI10463 stain but also, and more importantly, against the most clinically relevant isolate, BI/NAP1/027, in mice. Whether this candidate vaccine indeed provides global protection against all pathogenic C. difficile isolates will ultimately need to be determined in clinical trials.
C. difficile infection causes a wide spectrum of clinical symptoms, ranging from mild diarrhea to fulminant disease and mortality (27, 45). Unlike the hamster model of CDI where animals consistently develop a fulminant and lethal disease course (40), the recently established mouse CDI model develops a wider range of disease symptoms that more accurately resemble major aspects of the human disease (7). In this study, we chose to challenge mice with doses of C. difficile that recapitulate the full clinical spectrum of disease symptoms ranging from a self-limiting diarrhea to fulminant systemic disease (53). We also conducted confirmatory experiments in hamsters in accordance with previous studies that mainly use mortality as a single disease parameter for evaluation (28, 43). In our opinion, while mortality is an important parameter, it is more useful to fully evaluate the efficacy of vaccines against a spectrum of complex disease symptoms, including disease recurrence as is evident in CDI. To illustrate this point in our study, aTcdA-immunized mice were fully protected against mortality after C. difficile challenge (Fig. 4E). This result, although in agreement with several previous findings (28, 43), should not be interpreted as a demonstration of cross-protection against TcdB since aTcdA immunization was still associated with significant intestinal disease in these animals (Fig. 4F). In yet another example, although a single cTxAB immunization dose conferred significant protection against mortality induced by CDI (Fig. 7C), it did not altogether prevent diarrhea or weight loss (Fig. 7D and data not shown). Although repeated immunization is important in conferring complete disease protection in mice, prophylactic immunization may remain an attractive preventative strategy in patients who have previously been exposed to the infection. Thus, it remains to be determined in a clinical trial setting whether a single vaccination dose is sufficient to provide prophylactic protection against CDI.
Disease recurrence is one of the most significant clinical complications associated with CDI, occurring in 25 to 35% of patients with primary infection and rising up to 50% in subsequent infective episodes (24). Using a mouse model of CDI relapse that we have recently described (55), we assessed the efficacy of the protective immunity of cTxAB both before and after the initial infective episode. In all cases, cTxAB vaccination conferred complete protection against disease recurrence. Thus, a regimen of vaccination after the initial episode of CDI may be especially relevant in a clinical setting because such patients are at greater risk of developing recurrent CDI.
It has long been known that systemic antitoxin antibodies are protective against CDI (18, 26), but the underlying mechanisms for mucosal protection are not fully understood. Our data demonstrated that the parenteral cTxAB vaccination induced full disease protection, which may be mediated at both systemic and mucosal levels. Systemic cTxAB immunization induced potent serum neutralizing antibodies, and mice were completely protected against lethal systemic toxin challenge (Fig. 6C). We have previously reported that toxins are released into the bloodstream of piglets experimentally infected with C. difficile (21, 54), and toxemia is associated with systemic and fulminant CDI in both piglets and mice (53). Although toxemia has not yet been definitively demonstrated in CDI patients, preliminary reports indicate that toxins may cause systemic complications and fulminant disease (2, 9, 10, 25, 49). Systemic IgG neutralizing antibodies are therefore likely to play an important role in disease prevention by inactivating toxins released from lumen into submucosa and circulation. As for the protection at the mucosal surface, we failed to identify a detectable amount of antitoxin neutralizing IgA in the intestinal lavage specimen from cTxAB-immunized mice (data now shown). Whether these antitoxin IgG antibodies also mediated the protection of intestinal mucosal surfaces is under investigation.
In summary, attenuated chimeric C. difficile toxins capable of inducing potent neutralizing antitoxins and protection against CDI can be manufactured inexpensively using a reproducible and safe, endotoxin-free expression system in B. megaterium and may provide a novel prophylactic and therapeutic approach to combat primary and recurrent CDI.
ACKNOWLEDGMENTS
These studies were supported by grants NO1-30050, R01AI088748, R01DK084509, K01DK076549, R01AI10094001, 1UL1RR029876-01, and R21-DK078032-01 and by the John S. Dunn Gulf Coast Consortium for Chemical Genomics Robert A. Welch Collaborative Grant Program.
We thank Abraham L. Sonenshein and Charles B. Shoemaker (Tufts University) for critical review of the manuscript.
We report that we have no conflicts of interest.
Footnotes
Published ahead of print 21 May 2012
REFERENCES
- 1. Aboudola S, et al. 2003. Clostridium difficile vaccine and serum immunoglobulin G antibody response to toxin A. Infect. Immun. 71:1608–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abougergi MS, Broor A, Cui W, Jaar BG. 2010. Intravenous immunoglobulin for the treatment of severe Clostridium difficile colitis: an observational study and review of the literature. J. Hosp. Med. 5:E1–E9 [DOI] [PubMed] [Google Scholar]
- 3. Bartlett JG. 2002. Clinical practice. Antibiotic-associated diarrhea. N. Engl. J. Med. 346:334–339 [DOI] [PubMed] [Google Scholar]
- 4. Belyi IF, Varfolomeeva NA. 2003. Construction of a fusion protein carrying antigenic determinants of enteric clostridial toxins. FEMS Microbiol. Lett. 225:325–329 [DOI] [PubMed] [Google Scholar]
- 5. Borriello SP. 1998. Pathogenesis of Clostridium difficile infection. J. Antimicrob. Chemother. 41(Suppl C):13–19 [DOI] [PubMed] [Google Scholar]
- 6. Castagliuolo I, et al. 2004. Clostridium difficile toxin A carboxyl-terminus peptide lacking ADP-ribosyltransferase activity acts as a mucosal adjuvant. Infect. Immun. 72:2827–2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen X, et al. 2008. A mouse model of Clostridium difficile-associated disease. Gastroenterology 135:1984–1992 [DOI] [PubMed] [Google Scholar]
- 8. Cloud J, Kelly CP. 2007. Update on Clostridium difficile associated disease. Curr. Opin. Gastroenterol. 23:4–9 [DOI] [PubMed] [Google Scholar]
- 9. Cunney RJ, Magee C, McNamara E, Smyth EG, Walshe J. 1998. Clostridium difficile colitis associated with chronic renal failure. Nephrol. Dial. Transplant. 13:2842–2846 [DOI] [PubMed] [Google Scholar]
- 10. Dobson G, Hickey C, Trinder J. 2003. Clostridium difficile colitis causing toxic megacolon, severe sepsis and multiple organ dysfunction syndrome. Intensive Care Med. 29:1030. [DOI] [PubMed] [Google Scholar]
- 11. Feng H, Zeng Y, Whitesell L, Katsanis E. 2001. Stressed apoptotic tumor cells express heat shock proteins and elicit tumor-specific immunity. Blood 97:3505–3512 [DOI] [PubMed] [Google Scholar]
- 12. Feng H, et al. 2005. Listeria-infected myeloid dendritic cells produce IFN-beta, priming T cell activation. J. Immunol. 175:421–432 [DOI] [PubMed] [Google Scholar]
- 13. Fernie DS, Thomson RO, Batty I, Walker PD. 1983. Active and passive immunization to protect against antibiotic associated caecitis in hamsters. Dev. Biol. Stand. 53:325–332 [PubMed] [Google Scholar]
- 14. Flegel WA, et al. 1991. Cytokine response by human monocytes to Clostridium difficile toxin A and toxin B. Infect. Immun. 59:3659–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gardiner DF, Rosenberg T, Zaharatos J, Franco D, Ho DD. 2009. A DNA vaccine targeting the receptor-binding domain of Clostridium difficile toxin A. Vaccine 27:3598–3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghose C, et al. 2007. Transcutaneous immunization with Clostridium difficile toxoid A induces systemic and mucosal immune responses and toxin A-neutralizing antibodies in mice. Infect. Immun. 75:2826–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giannasca PJ, Warny M. 2004. Active and passive immunization against Clostridium difficile diarrhea and colitis. Vaccine 22:848–856 [DOI] [PubMed] [Google Scholar]
- 18. Giannasca PJ, et al. 1999. Serum antitoxin antibodies mediate systemic and mucosal protection from Clostridium difficile disease in hamsters. Infect. Immun. 67:527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greenberg RN, Marbury TC, Foglia G, Warny M. 2012. Phase I dose finding studies of an adjuvanted Clostridium difficile toxoid vaccine. Vaccine 30:2245–2249 [DOI] [PubMed] [Google Scholar]
- 20. He X, et al. 2009. Antibody-enhanced, Fc gamma receptor-mediated endocytosis of Clostridium difficile toxin A. Infect. Immun. 77:2294–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He X, et al. 2009. An ultrasensitive rapid immunocytotoxicity assay for detecting Clostridium difficile toxins. J. Microbiol. Methods 78:97–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jank T, Aktories K. 2008. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 16:222–229 [DOI] [PubMed] [Google Scholar]
- 23. Jank T, Giesemann T, Aktories K. 2007. Clostridium difficile glucosyltransferase toxin B-essential amino acids for substrate binding. J. Biol. Chem. 282:35222–35231 [DOI] [PubMed] [Google Scholar]
- 24. Johnson S. 2009. Recurrent Clostridium difficile infection: causality and therapeutic approaches. Int. J. Antimicrob. Agents 33(Suppl 1):S33–S36 [DOI] [PubMed] [Google Scholar]
- 25. Johnson S, et al. 2001. Fatal pseudomembranous colitis associated with a variant Clostridium difficile strain not detected by toxin A immunoassay. Ann. Intern. Med. 135:434–438 [DOI] [PubMed] [Google Scholar]
- 26. Katchar K, et al. 2007. Association between IgG2 and IgG3 subclass responses to toxin A and recurrent Clostridium difficile-associated disease. Clin. Gastroenterol. Hepatol. 5:707–713 [DOI] [PubMed] [Google Scholar]
- 27. Kelly CP, LaMont JT. 2008. Clostridium difficile–more difficult than ever. N. Engl. J. Med. 359:1932–1940 [DOI] [PubMed] [Google Scholar]
- 28. Kim PH, Iaconis JP, Rolfe RD. 1987. Immunization of adult hamsters against Clostridium difficile-associated ileocecitis and transfer of protection to infant hamsters. Infect. Immun. 55:2984–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kotloff KL, et al. 2001. Safety and immunogenicity of increasing doses of a Clostridium difficile toxoid vaccine administered to healthy adults. Infect. Immun. 69:988–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuehne SA, et al. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713 [DOI] [PubMed] [Google Scholar]
- 31. Kyne L, Warny M, Qamar A, Kelly CP. 2001. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 357:189–193 [DOI] [PubMed] [Google Scholar]
- 32. Lanis JM, Barua S, Ballard JD. 2010. Variations in TcdB activity and the hypervirulence of emerging strains of Clostridium difficile. PLoS Pathog. 6:e1001061 doi:10.1371/journal.ppat.1001061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leav BA, et al. 2010. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 28:965–969 [DOI] [PubMed] [Google Scholar]
- 34. Libby JM, Jortner BS, Wilkins TD. 1982. Effects of the two toxins of Clostridium difficile in antibiotic-associated cecitis in hamsters. Infect. Immun. 36:822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Libby JM, Wilkins TD. 1982. Production of antitoxins to two toxins of Clostridium difficile and immunological comparison of the toxins by cross-neutralization studies. Infect. Immun. 35:374–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Loo VG, et al. 2011. Host and pathogen factors for Clostridium difficile infection and colonization. N. Engl. J. Med. 365:1693–1703 [DOI] [PubMed] [Google Scholar]
- 37. Loo VG, et al. 2005. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N. Engl. J. Med. 353:2442–2449 [DOI] [PubMed] [Google Scholar]
- 38. Louie TJ, et al. 2011. Fidaxomicin versus vancomycin for Clostridium difficile infection. N. Engl. J. Med. 364:422–431 [DOI] [PubMed] [Google Scholar]
- 39. Lowy I, et al. 2010. Treatment with monoclonal antibodies against Clostridium difficile toxins. N. Engl. J. Med. 362:197–205 [DOI] [PubMed] [Google Scholar]
- 40. Lyerly DM, Saum KE, MacDonald DK, Wilkins TD. 1985. Effects of Clostridium difficile toxins given intragastrically to animals. Infect. Immun. 47:349–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lyras D, et al. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McDonald LC, et al. 2005. An epidemic, toxin gene-variant strain of Clostridium difficile. N. Engl. J. Med. 353:2433–2441 [DOI] [PubMed] [Google Scholar]
- 43. Permpoonpattana P, et al. 2011. Immunization with Bacillus spores expressing toxin A peptide repeats protects against infection with Clostridium difficile strains producing toxins A and B. Infect. Immun. 79:2295–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pruitt RN, Chambers MG, Ng KK, Ohi MD, Lacy DB. 2010. Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc. Natl. Acad. Sci. U. S. A. 107:13467–13472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7:526–536 [DOI] [PubMed] [Google Scholar]
- 46. Ryan ET, et al. 1997. Protective immunity against Clostridium difficile toxin A induced by oral immunization with a live, attenuated Vibrio cholerae vector strain. Infect. Immun. 65:2941–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sauerborn M, Leukel P, von Eichel-Streiber C. 1997. The C-terminal ligand-binding domain of Clostridium difficile toxin A (TcdA) abrogates TcdA-specific binding to cells and prevents mouse lethality. FEMS Microbiol. Lett. 155:45–54 [DOI] [PubMed] [Google Scholar]
- 48. Seregin SS, Aldhamen YA, Rastall DP, Godbehere S, Amalfitano A. 2012. Adenovirus-based vaccination against Clostridium difficile toxin A allows for rapid humoral immunity and complete protection from toxin A lethal challenge in mice. Vaccine 30:1492–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Siarakas S, Damas E, Murrell WG. 1995. Is cardiorespiratory failure induced by bacterial toxins the cause of sudden infant death syndrome? Studies with an animal model (the rabbit). Toxicon 33:635–649 [DOI] [PubMed] [Google Scholar]
- 50. Siddiqui F, et al. 2012. Vaccination with parenteral toxoid B protects hamsters against lethal challenge with toxin A-negative, toxin B-positive clostridium difficile but does not prevent colonization. J. Infect. Dis. 205:128–133 [DOI] [PubMed] [Google Scholar]
- 51. Sougioultzis S, et al. 2005. Clostridium difficile toxoid vaccine in recurrent C. difficile-associated diarrhea. Gastroenterology 128:764–770 [DOI] [PubMed] [Google Scholar]
- 52. Stabler RA, et al. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 10:R102 doi:10.1186/gb-2009-10-9-r102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steele J, et al. 2012. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J. Infect. Dis. 205:384–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Steele J, Feng H, Parry N, Tzipori S. 2010. Piglet models of acute or chronic Clostridium difficile illness. J. Infect. Dis. 201:428–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sun X, et al. 2011. Mouse relapse model of Clostridium difficile infection. Infect. Immun. 79:2856–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Torres JF, Lyerly DM, Hill JE, Monath TP. 1995. Evaluation of formalin-inactivated Clostridium difficile vaccines administered by parenteral and mucosal routes of immunization in hamsters. Infect. Immun. 63:4619–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. von Eichel-Streiber C, Zec-Pirnat I, Grabnar M, Rupnik M. 1999. A nonsense mutation abrogates production of a functional enterotoxin A in Clostridium difficile toxinotype VIII strains of serogroups F and X. FEMS Microbiol. Lett. 178:163–168 [DOI] [PubMed] [Google Scholar]
- 58. Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18:247–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ward SJ, Douce G, Dougan G, Wren BW. 1999. Local and systemic neutralizing antibody responses induced by intranasal immunization with the nontoxic binding domain of toxin A from Clostridium difficile. Infect. Immun. 67:5124–5132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ward SJ, Douce G, Figueiredo D, Dougan G, Wren BW. 1999. Immunogenicity of a Salmonella typhimurium aroA aroD vaccine expressing a nontoxic domain of Clostridium difficile toxin A. Infect. Immun. 67:2145–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang G, et al. 2008. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol. 8:192 doi:10.1186/1471–2180-8-192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zar F, Bakkanagari S, Moorthi KMLST, Davis M. 2007. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin. Infect. Dis. 45:302–307 [DOI] [PubMed] [Google Scholar]