

Abstract
Urinary tract infections (UTI), primarily caused by uropathogenic Escherichia coli (UPEC), are one of the leading bacterial infections due to their high frequency and rate of recurrence. Both type 1 pilus adhesive organelles (fim) and the QseC sensor kinase have been implicated in UPEC virulence during UTI and have been individually reported to be promising drug targets. Deletion of qseC leads to pleiotropic effects due to unregulated activation of the cognate response regulator QseB, influencing conserved metabolic processes and diminishing expression of virulence genes, including type 1 pili. Here, we discern the type 1 pilus-dependent and -independent effects that contribute to the virulence attenuation of a UPEC qseC deletion mutant in a murine model of experimental UTI. We show that although a ΔqseC mutant restored for type 1 pilus expression regains the ability to colonize the host and initiate acute infection up to 16 h postinfection, it is rapidly outcompeted during acute infection when coinoculated with a wild-type strain. As a result, this strain has a diminished capacity to establish chronic infection. A prophylactic oral dose of a FimH small-molecular-weight antagonist (ZFH-02056) further reduced the ability of the qseC mutant to establish chronic infection. Thus, loss of QseC significantly enhances the efficacy of ZFH-02056. Collectively, our work indicates that type 1 pili and QseC become critical in different infection stages, and that dual targeting of these factors has an additive effect on ablating UPEC virulence.
INTRODUCTION
Urinary tract infections (UTI) are one of the most common bacterial infections, afflicting primarily women and accounting for more than 3.5 billion dollars in health care costs in the United States alone (18). Approximately one out of three patients with a UTI will experience recurrence (rUTI) within 6 to 12 months, and in 60% of these cases, rUTI is induced by the same bacterial strain that caused the original infection (15, 28), indicating the presence of a possible intestinal and/or extraintestinal reservoir within the host (14, 36, 38, 39). Nearly 85% of community-acquired UTIs are caused by uropathogenic Escherichia coli (UPEC) (18), which can overcome host defenses in part by invading bladder epithelial cells (14, 35). Numerous bacterial factors have been shown to be important in UPEC UTIs, including capsule, iron acquisition systems, toxins, a virulence plasmid, tRNAs, pathogenicity islands, and colonization factors (1, 7, 10, 20, 22, 23, 34, 50, 51, 53). Adhesive pili assembled by the chaperone/usher pathway (CUP), such as type 1 pili, contain adhesins at their tips that are thought to play an important role in host-pathogen interactions (48). Each sequenced UPEC strain has been found to encode a multitude of CUP operons (7, 23, 49). Some CUP adhesins are known to recognize specific receptors with stereochemical specificity (26, 45). For example, FimH, the type 1 pilus tip adhesin, has been shown to bind mannosylated glycoproteins, as well as N-linked oligosaccharides on β1 and α3 integrins and the pattern recognition receptor TLR4, which are expressed throughout the luminal surface of human and murine bladders (5, 13, 26, 33, 45, 46, 55). These FimH-mediated interactions facilitate bacterial colonization and invasion, triggering apoptosis and exfoliation (35, 38) and inducing elevated levels of cyclic AMP (cAMP) (4). Upon internalization, UPEC are exocytosed in a TLR-4-dependent process (4); however, bacteria can escape into the host cell cytoplasm, where they are able to subvert expulsion and innate defenses by replicating into biofilm-like intracellular bacterial communities (IBCs) (2, 29). Subsequently, UPEC disperse from the IBC, escape into the bladder lumen, and reinitiate the process by binding and invading neighboring uninfected epithelial cells (29). The murine model has revealed two potential outcomes of these acute events, either resolution of infection with the establishment of quiescent intracellular reservoirs that can serve as seeds for recurrence (38) or the establishment of chronic bladder infection (chronic cystitis) (21). Chronic cystitis is characterized by persistent, high-titer bacteriuria (>104 CFU/ml), as well as high-titer bacterial bladder burdens at ≥2 weeks postinfection (wpi) and evidence of chronic bladder inflammation (11, 21). The ability of UPEC to form IBCs appears to be a critical determinant of disease outcome, as a previous study suggests that a higher number of IBCs formed during the acute stages of infection precedes the development of chronic cystitis (43). Thus, differing selection pressures and bacterial population dynamics, as well as niche residence and population bottlenecks, determine the ability of UPEC to survive and cause an acute infection and/or persist long term in the urinary tract as a chronic extracellular infection (chronic cystitis) (21, 43).
The IBC pathogenic cascade, which has been characterized in a murine model of infection (2, 27, 29) and has been extensively documented in samples from human clinical studies (17, 42), is FimH dependent (52). It has been shown that the fimH gene is under positive selection in human clinical isolates of UPEC (6), further supporting its role in human disease. Thus, it has been reported that type 1 pili fulfill the molecular Koch's postulates of microbial pathogenesis (44). Type 1 pili are encoded by the fim gene cluster, and their expression is directed by a phase-variable promoter (fimS), which facilitates a switch between piliated and nonpiliated bacterial states (30). UPEC populations are typically heterogeneous in terms of pilus expression, consisting of bacteria that are observed by transmission electron microscopy to be bald or lightly, moderately, or highly piliated (40). The ratio of each piliated fraction shifts depending on the environmental niche. Studies investigating the expression of type 1 pili revealed that UPEC associated with epithelial cells are highly piliated, consistent with the critical role of type 1 pili in colonization of the urothelium (26, 33, 54, 55). In contrast, the fimS promoter in bacteria recovered from urine samples of patients has been shown to be predominantly in the OFF phase (32). Regulation of fimS phase variation is controlled by the FimB, FimE, and FimX recombinases, the expression of which is influenced by numerous regulatory factors (16, 20, 30). We recently identified the QseBC two-component system as one of the factors implicated in the regulation of several CUPs in UPEC, including type 1 pili (19, 31). In a UPEC mutant lacking the sensor kinase QseC, the fim promoter is found primarily in the OFF orientation, resulting in reduced type 1 pilus expression, an effect that stems from uncontrolled activation of the QseB response regulator in this mutant (31). In addition to influencing type 1 pilus expression in the absence of qseC, overactive QseB (31) dysregulates conserved cellular pathways and impacts several other virulence-associated genes, resulting in UPEC attenuation (19, 31).
While the effects of disruption of type 1 pilus production on UPEC pathogenesis are fairly well understood (6, 8, 24, 52) and likely explain at least some of the attenuation of the ΔqseC mutant, the contributions of QseB-misregulated factors in the attenuation of ΔqseC, independent of type 1 pili, are unknown. Therefore, in this study we disengaged type 1 pili from QseBC control to determine the role of other QseB-misregulated factors in acute and chronic stages of bladder infection. By locking the invertible fim promoter element in the ON orientation, we restored type 1 pilus expression in the ΔqseC mutant. The resulting strain, UTI89ΔqseC_LON (LON indicates locked in the ON orientation), was rescued in its ability to cause acute UTI but was rapidly outcompeted by wild-type (wt) UTI89 during acute competitive infection studies and was less efficient at causing chronic cystitis. Thus, despite expression of type 1 pili, the QseB-misregulated processes in the ΔqseC mutant decrease bacterial fitness for acute infection and reduce the ability of UPEC to develop chronic cystitis. We then used small-molecular-weight FimH inhibitors, also known as mannosides, as molecular scalpels to show that transiently blocking FimH function during infection, by oral prophylactic administration, further impeded the ability of ΔqseC to cause chronic cystitis compared to ΔqseC alone. Collectively, our studies extend our understanding of the basis of ΔqseC attenuation, distinguishing the effect of type 1 pili from that of other QseB-misregulated factors during the course of infection, and support that simultaneously targeting QseC and type 1 pili represents a promising avenue for potentiating therapeutics against UPEC.
MATERIALS AND METHODS
Strains, constructs, and growth conditions.
UTI89 is a human cystitis isolate that is highly virulent in a mouse model of UTI (36). UTI89_LON and UTI89ΔqseC_LON were created using λ Red recombinase and primers LON_L (TAAAAAGAGAAGAAGCTTGATTTAACTAATTGATAATAAAGTTAAAAAAGTGTAGGCTGGAGCTGCTTC) and LON_R (CGATGCTTTCCTCTATGAGTCAAAAGAGATCTAATTGTCTTGTATTTATTTGCATATGAATATCCTCCTTAG) so as to mutate 5 out of 9 nucleotides in the left invertible repeat (LIR) of the fim promoter in the chromosome (37) (the wt LIR sequence 5′-TGGCCCCAA-3′ was mutated to 5′-GAGATCTAA-3′ and is underlined in the sequences above). Chromosomally marked LON strains were constructed as shown in the Fig. 2A schematic but without inducing excision of the Kan cassette (the last step of the schematic). Similarly, chromosomally marked UTI89 and UTI89ΔqseC strains were constructed with the Kan resistance cassette in the λ-att or qseC locus, respectively. All strains have previously been tested and behave identically to their nonmarked counterparts. Bacteria were incubated in Luria-Bertani (LB) medium at 37°C for 4 h under shaking conditions, subcultured (1:1,000) in fresh LB medium, and incubated statically at 37°C for 18 h.
Fig 2.

Restoring production of type 1 pili in UTI89ΔqseC does not influence other ΔqseC-mediated defects. (A) Schematic showing the strategy used to lock the phase-variable fim promoter in the ON orientation. LIR, left invertible repeat within the fim promoter; RIR, right invertible repeat within the fim promoter; LIR*, mutated left invertible repeat of the fim promoter. (B and C) Hemagglutination assays and Western blot analyses depicting restoration of type 1 pilus expression in UTI89ΔqseC locked-ON strain (ΔqseC_LON). (D) Locking the fim promoter ON does not interfere with the QseB-mediated dysregulation in the qseC deletion mutant. Relative fold change of qseB and aceB in UTI89_LON and UTI89ΔqseC_LON compared to UTI89 and UTI89ΔqseC, measured by qRT-PCR. Values are normalized to the 16S rrsH gene. (E) Assessment of curli production on YESCA-CR agar verifies that, similar to UTI89ΔqseC, UTI89ΔqseC_LON remains defective for curli expression, exhibiting a white and smooth morphotype. Graphs depict averages of three independent experiments; error bars denote standard errors of the means. Statistical analysis was performed by unpaired two-tailed Student's t test (*, P < 0.05; **, P < 0.0099; ***, P < 0.0001).
Mouse infections.
All procedures involving mice were performed in compliance with current federal guidelines and institutional policies at Washington University in Saint Louis and ensured the proper and humane treatment of animals. Female C3H/HeN mice (Harlan), 7 to 9 weeks old, were used for all studies described below, and in each case mice were infected with 107 bacteria.
Short-term infections.
Mice were transurethrally infected with bacteria carrying the green fluorescent protein (GFP)-expressing plasmid pANT4 (9) as previously described (31), and they were sacrificed at 1 and 3 h postinfection (hpi). Bladders were aseptically removed, homogenized, and plated for total bacterial enumeration or gentamicin treated to determine intracellular bacterial titers. The experiment was repeated 3 times, and statistical analysis was performed using two-tailed Mann-Whitney tests (P < 0.05 was considered significant).
Ex vivo gentamicin assay.
Bladders were bisected and washed 3 times in 500 μl phosphate-buffered saline (PBS). The washes were collected and plated for CFU enumeration to determine luminal bacteria. Washed bladders were incubated for 90 min at 37°C with 100 μg/ml gentamicin to kill adherent extracellular bacteria. Following washes (3×) with PBS, bladders were homogenized in 1 ml PBS and plated to determine intracellular bacterial titers. The two-tailed Mann-Whitney test (P < 0.05) was used for statistical analysis.
Acute infection studies.
Mice were transurethrally infected with 107 bacteria carrying the plasmid pANT4 as previously described (31). Experiments were repeated three times and statistically analyzed using the two-tailed Mann-Whitney test (P < 0.05 was considered significant).
Long-term infection studies.
Mice were infected with chromosomally marked strains of wt UTI89, UTI89_LON, UTI89ΔqseC, or UTI89ΔqseC_LON. Mice were sacrificed at 2 weeks postinfection (wpi), and organs were processed for CFU enumeration on LB-kanamycin agar plates. The experiment was repeated 2 times. Cumulative data from all experiments are presented.
Competition assays.
Cohorts of 10 mice were used per time point. Bacterial inocula were prepared such that they comprised 50% UTI89_LON and 50% UTI89ΔqseC_LON (Kanr) in a total of 3 × 107 to 4 × 107 bacteria per 50 μl, and the inocula were transurethrally introduced in each mouse. Mice were sacrificed at 6, 16, 30, 48, and 72 hpi, and organs were processed for CFU enumeration on LB and LB-Kan agar plates. To account for possible variation due to antibiotic resistance, experiments were repeated using UTI89_LON (Kanr) and UTI89ΔqseC_LON. The experiment was repeated 3 times. Cumulative data from all experiments are presented. Statistical analysis was performed using the Wilcoxon matched-pairs signed-rank test.
Mannoside studies.
One hundred μl of a 10 mg/ml (50 mg/kg of body weight) solution of ZFH-02056 {IUPAC name: N1,N3-dimethyl-5-[4-](2R,3S,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl[oxyphenyl]benzene-1,3-dicarboxamide} was administered to each mouse by oral gavage 30 min prior to bacterial inoculation. Control mice were gavaged with 100 μl of PBS 30 min prior to bacterial inoculation. Mice were euthanized at 2 and 4 wpi, and organs were processed for CFU enumeration.
IBC enumeration by confocal microscopy.
For IBC enumeration, mice were infected with fluorescent bacteria carrying the GFP-plasmid pANT4 as described above for acute infections. Bladders excised from infected animals were treated for confocal microscopy as previously described (53). Briefly, bladders were bisected, splayed, and fixed in 4% paraformaldehyde overnight at 4°C. Fixed bladders were washed and counterstained for 20 min with the nuclear ToPro3 (Molecular Probes) stain and rhodamine-conjugated wheat germ agglutinin to outline the facet cells (1:700 dilution for each). For IBC enumeration, the entire bladder was scanned using a Zeiss LSM 510 Meta laser scanning inverted confocal microscope (Thornwood).
Immunoblotting, HA, and phase assays.
Bacteria were grown statically in LB for 18 h at 37°C. Immunoblotting (using anti-type 1 pilus antibody), hemagglutination (HA), and phase assays were performed on normalized cells (optical density at 600 nm [OD600] of 1) as previously described (25, 31).
qPCR analyses.
RNA extraction, DNase treatment, and reverse transcription were performed using reagents and methods as reported by Kostakioti et al. (31). Relative transcript abundance was determined by quantitative PCR (qPCR) as previously described (31) using aceB-specific (19) or qseB-specific (31) primers.
RESULTS
Deletion of QseC impairs the ability of UPEC to colonize and invade the bladder.
We have previously shown that deletion of qseC in UTI89 leads to reduced bladder bacterial titers during the acute stages of infection (6 and 16 h postinfection) in a cystitis murine model (31). Given that type 1 pili are critical for bladder adherence and invasion (33, 35) and are significantly reduced in the absence of QseC, we rationalized that the in vivo attenuation of UTI89ΔqseC could be, at least partly, attributed to reduced type 1 pilus-mediated colonization and invasion of the bladder. Thus, we assessed the ability of UTI89ΔqseC to adhere to and invade the bladder epithelium compared to the parent strain. Female C3H/HeN mice were transurethrally inoculated with 107 wild-type (wt) UTI89 or UTI89ΔqseC, and bacterial colonization and invasion were assessed at 1 and 3 h postinfection (hpi), time points previously shown to be suitable for studying the invasion events in vivo (29, 33, 35). Enumeration of CFU recovered from infected bladders indicated that compared to wt UTI89, UTI89ΔqseC had significantly lower overall bacterial titers (which include both intracellular and luminal populations) at both time points (Fig. 1A). Treatment of infected bladders with gentamicin to eradicate the luminal bacterial population while leaving the intracellular population unharmed revealed decreased intracellular numbers for UTI89ΔqseC (Fig. 1B). Despite the observed reduced internalization of UTI89ΔqseC, the number of luminal UTI89ΔqseC bacteria was not higher but rather was comparable to wt UTI89 luminal populations (Fig. 1B); given the reduced expression of type 1 pili by UTI89ΔqseC, these data suggest that reduced adherence of UTI89ΔqseC leads to faster clearance by micturition. Thus, our data indicate that deletion of qseC affects the ability of UTI89 to initiate infection in part by compromising its ability to colonize and invade the host bladder.
Fig 1.
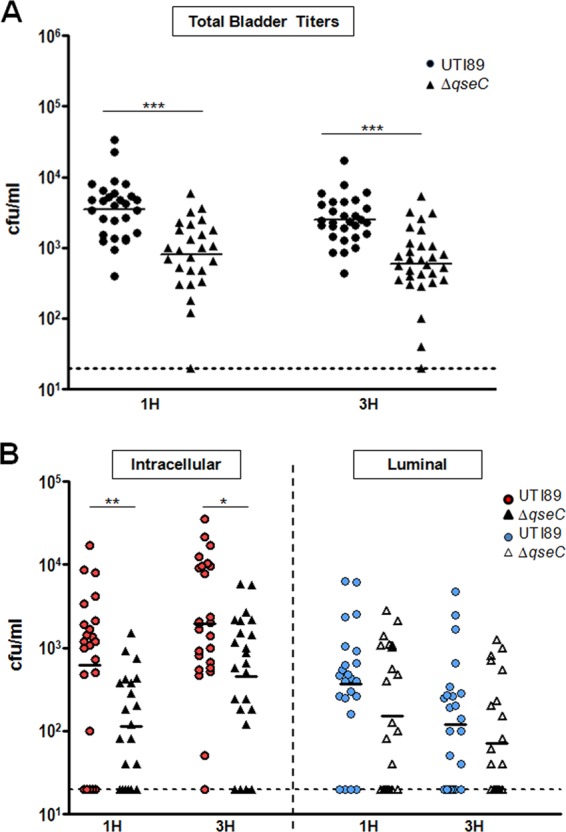
UTI89ΔqseC has a defect in bladder invasion. (A) Bladder titers at 1 and 3 hpi showing bacterial numbers recovered from bladders infected with UTI89ΔqseC or wt UTI89. (B) Bacterial titers recovered from gentamicin-treated bladders (intracellular population) after washing 3 times with PBS to remove the extracellular, adherent bacteria (luminal population). Data from 3 independent experiments are shown (*, P < 0.02; **, P < 0.003; ***, P < 0.0001; all by two-tailed Mann-Whitney).
Expression of type 1 pili rescues the adherence properties of UTI89ΔqseC in vitro but does not influence other ΔqseC-related defects.
Our findings demonstrate that attenuation of UTI89ΔqseC is linked to reduced bacterial internalization due to decreased type 1 pilus production. We therefore investigated whether restoration of type 1 pilus expression alone would be sufficient to overcome the in vivo defects of UTI89ΔqseC. We have previously reported that in the absence of QseC, the phase-variable fim promoter driving type 1 pilus expression, fimS, is primarily switched to the OFF orientation (31). We thus locked fimS in the ON orientation in the chromosome of wt UTI89 and UTI89ΔqseC to attain expression of type 1 pili independently of QseC (Fig. 2A). UTI89_LON and UTI89ΔqseC_LON exhibited comparable mannose-sensitive hemagglutination (HA) properties, which were higher than that of the wt UTI89 because the inversion of the promoter to the OFF state can no longer occur (Fig. 2B). Consistent with this, Western blot analyses probing for FimA, the major type 1 pilus subunit, verified that the level of type 1 pilus production in UTI89ΔqseC_LON is comparable to that of UTI89_LON, but again it is higher than that of wt UTI89 (Fig. 2C). Electron microscopy revealed that the resulting strains, UTI89_LON and UTI89ΔqseC_LON, were not hyperpiliated, rather a larger population of piliated versus nonpiliated bacteria was observed compared to wt UTI89 (data not shown).
In addition to defects in type 1 pilus expression, deletion of qseC results in perturbation of central cellular processes due to aberrant upregulation and activation of the QseB response regulator (19, 31). This prompted us to verify that restoration of type 1 pilus expression does not affect the QseB-mediated dysregulation. We first performed quantitative reverse transcription-PCR (qRT-PCR) to probe the expression of qseB in the fim-locked-ON (LON) strains compared to wt UTI89 and UTI89ΔqseC. We found that the LON strains behaved similarly to their non-LON counterparts, with UTI89ΔqseC and UTI89ΔqseC_LON exhibiting increased qseB levels compared to both wt UTI89 and UTI89_LON (Fig. 2D). These data suggest that the massive pleiotropy imparted by QseB-mediated misregulation in the absence of QseC remains unaffected upon restoration of type 1 pilus expression. To test this, we examined the expression of AceB, a metabolic factor, and curli, a bacterial amyloid, both known to be misregulated in the absence of QseC (19). We confirmed by qRT-PCR that aceB, which is upregulated in UTI89ΔqseC (19), displays elevated transcript levels in UTI89ΔqseC_LON compared to both UTI89 and UTI89_LON (Fig. 2D), while curli expression was abolished, which is the previously reported defect of UTI89ΔqseC (Fig. 2E). Curli formation can be assessed by growth on yeast extract/Casamino Acids agar supplemented with the Congo red dye (YESCA-CR), since curli bind CR, giving rise to red, dry, and rough colonies (3). When grown under curli-inducing conditions, UTI89ΔqseC_LON had a white and smooth morphotype on YESCA-CR medium similar to UTI89ΔqseC and in contrast to the red, dry, and rough curli-positive phenotype of UTI89 and UTI89_LON (Fig. 2E). As expected, based on these results, production of the CsgG outer membrane pore, critical for secretion of the curli components, was diminished in UTI89ΔqseC_LON but remained unaffected in UTI89_LON (data not shown). These findings show that locking the fim promoter in the ON orientation does not interfere with qseB overexpression and the associated downstream defects imparted by the qseC deletion.
UTI89ΔqseC_LON is restored for virulence in the acute infection stages.
We investigated whether restoration of type 1 pilus expression would rescue the qseC phenotype during the acute stages of infection. Our previous studies demonstrated that deletion of qseC results in reduced bladder titers and diminished IBC formation at 6 and 16 hpi (31). Since type 1 pili are essential for attachment, invasion, and IBC formation (52), we tested the ability of UTI89ΔqseC_LON to survive within the urinary tract and form IBCs during these times. Mice were infected with wt UTI89, UTI89ΔqseC, UTI89_LON, or UTI89ΔqseC_LON, and bacterial invasion was evaluated at 1 hpi by gentamicin protection assays. UTI89_LON and UTI89ΔqseC_LON had comparable amounts of internalized bacteria, in contrast to what was observed for UTI89ΔqseC (Fig. 3A). In contrast to UTI89ΔqseC, the bacterial titers recovered from UTI89ΔqseC_LON-infected bladders were comparable to those recovered for wt UTI89 and UTI89_LON at both 6 and 16 hpi (Fig. 3B). Similarly, UTI89ΔqseC_LON formed IBCs as efficiently as wt UTI89 and UTI89_LON (Fig. 3C). Thus, restoring expression of type 1 pili in the absence of QseC is sufficient for rescuing IBC formation and survival within the bladder at the acute infection stages.
Fig 3.

UTI89ΔqseC_LON is restored for virulence at the early and acute infection stages. (A) Bacterial numbers recovered from infected bladders at 1 hpi showing that UTI89ΔqseC_LON invades the bladder as efficiently as the parent strain. (B and C) Bacterial titers and IBC numbers at 6 and 16 hpi reveal that UTI89ΔqseC_LON can survive inside the host at levels comparable to wt UTI89 during the acute stages of infection. Data from 3 independent experiments are shown (*, P < 0.04; **, P < 0.009; ***, P < 0.0003; all by two-tailed Mann-Whitney test).
UTI89ΔqseC_LON is outcompeted by UTI89_LON within the host.
To investigate whether QseB-mediated misregulation in the UTI89ΔqseC_LON strain imparted a fitness disadvantage independent of the conferred type 1 piliation defect in UPEC acute pathogenesis, we monitored the infection outcome in mice coinfected with equal bacterial loads of differentially marked UTI89_LON and UTI89ΔqseC_LON strains during a 72-h period, marking the acute and subacute stages. Our data revealed that UTI89ΔqseC_LON began exhibiting a fitness disadvantage as early as 6 hpi in the presence of UTI89_LON, as indicated by a median competitive index (CI) of 0.1860 (CIs ranged from 0.0496 to 6.951) (Fig. 4A). The fitness disadvantage was more prominent at the later stages of infection, with the CI median dropping 16-fold, from 0.1860 to a value of 0.0115 at 16 hpi, with a range of 0.000074 to 0.2368 (Fig. 4A). These data suggest that restoration of type 1 pili does not suffice for complete restoration of in vivo fitness.
Fig 4.

Restored type 1 pilus expression cannot sustain wild-type persistence of the qseC deletion mutant inside the host. (A) Competition assays showing that UTI89ΔqseC_LON has a survival disadvantage compared to the parent strain and is rapidly cleared from the host bladder. Competitive index (CI) was calculated by dividing the ratio of UTI89ΔqseC_LON versus UTI89_LON titers by the ratio of the corresponding inocula. A CI of <1 indicates that UTI89ΔqseC_LON has a fitness disadvantage compared to UTI89_LON. Data from 3 independent experiments are shown (*, P < 0.03; **, P < 0.005; ***, P < 0.0009; all by Wilcoxon matched-pairs signed-rank test). (B) Graph depicting the bladder bacterial titers recovered at 2 wpi. Data from 3 independent experiments are shown (*, P < 0.03; **, P < 0.005; ***, P < 0.0009; all by two-tailed Mann-Whitney test).
UTI89ΔqseC_LON is less efficient in establishing chronic cystitis.
We used enumeration of bladder titers at 2 weeks postinfection (wpi) as a surrogate marker of chronic cystitis to determine whether QseB-mediated misregulation reduces the ability of UPEC to establish chronic cystitis, even in the presence of type 1 pilus expression. Cohorts of female C3H/HeN mice were inoculated with wt UTI89, UTI89ΔqseC, UTI89_LON, or UTI89ΔqseC_LON, and CFU were enumerated at 2 wpi. Our findings demonstrated that, compared to wt UTI89, mice infected with UTI89ΔqseC had significantly lower titers by 2 wpi, with only 2 out of 20 mice (10%) displaying high bladder titers (>104) indicative of chronic cystitis (Fig. 4B). These results are consistent with the lower bacterial burden observed for UTI89ΔqseC at the acute infection stages (Fig. 3). The LON strains were significantly more competent in establishing chronic cystitis, due to their inability to revert fimS in the OFF orientation, than their non-LON counterparts. UTI89ΔqseC_LON was as efficient as wt UTI89 in developing chronic cystitis. However, significantly fewer mice infected with UTI89ΔqseC_LON had high bacterial titers indicative of chronic cystitis (51.7%) compared to the mice infected with UTI89_LON (70.0% developed chronic infection) (Fig. 4B). These findings argue that QseB-mediated misregulation in the absence of QseC imparts a fitness disadvantage independent of the conferred type 1 piliation regulatory defects.
The effect of FimH inhibitors that blunt type 1 pilus function is potentiated by deletion of qseC.
Deletion of qseC reduces but does not abolish expression of type 1 pili (31). We rationalized that if we temporarily blunt type 1 pilus function in the ΔqseC mutant during the acute infection stage, we will exacerbate the inability of this mutant to establish chronic infection. Compounds ZFH-02056 and ZFH-04269 (mannosides) bind with high affinity to the mannose-binding pocket of the FimH adhesin and thus are potent inhibitors of type 1 pilus function (11). Specifically, oral administration of ZFH-02056 and ZFH-04269 significantly reduces bacterial titers in chronically infected mice within 6 h (11). We used compound ZFH-02056 as a molecular scalpel to temporarily blunt FimH function in wt UTI89 and UTI89ΔqseC early in infection and assess long-term infection outcome. Mice were pretreated with one dose of 50 mg/kg ZFH-02056 and infected 30 min after treatment with wt UTI89 or UTI89ΔqseC. Compound ZFH-02056 becomes bioavailable in the bladder within 30 min of oral delivery and at this dosage is sustained for 6 h above the MIC (11). Mice treated with PBS prior to infection were included as controls. Bladder bacterial titers were enumerated at 2 wpi to estimate the propensity for chronic infection. Even though ZFH-02056 is bioavailable at the dose delivered in the bladder for only 6 h (11), we observed that this single dosing significantly affected the course of infection 2 wpi. Compared to nontreated mice, at 2 wpi there was a 2-log reduction in CFU in mannoside-treated mice infected with wt UTI89 (Fig. 5, UTI89 versus UTI89_MAN). Thus, this single-dose mannoside treatment of wt UTI89 resulted in the same degree of attenuation observed with UTI89ΔqseC; both had a similar reduction in CFU (Fig. 5, UTI89_MAN versus UTI89ΔqseC). Notably, in mannoside-treated animals infected with UTI89ΔqseC, bacterial reduction was further enhanced, as indicated by an additional 1.5-log drop in CFU, compared to mannoside-treated animals infected with wt UTI89 (Fig. 5, UTI89_MAN versus UTI89ΔqseC_MAN). Our findings strongly indicate that dual targeting of QseC and type 1 pili could have an additive effect, thus potentiating bacterial clearance from the bladder.
Fig 5.
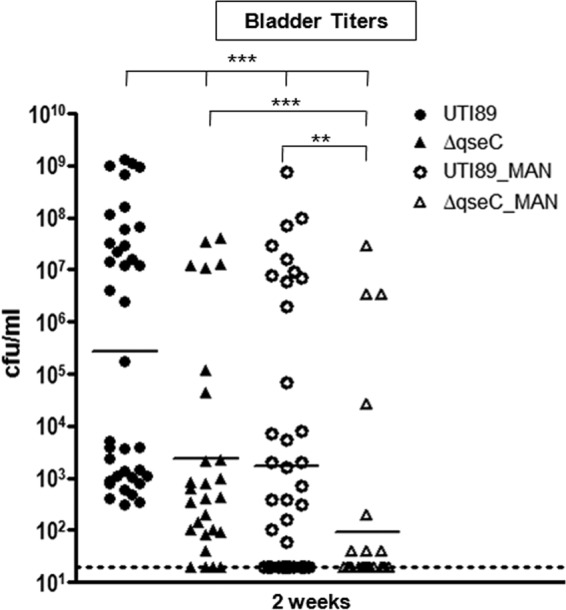
Coinhibition of type 1 pili and QseC as a prophylactic measure for UTIs. Chart depicting the bladder CFU obtained at 2 wpi from mice pretreated with mannoside and subsequently infected with either wt UTI89 or UTI89ΔqseC (used as a proxy for a QseC inhibitor). Significantly fewer CFU (1.5-log reduction) were obtained from pretreated mice infected with UTI89ΔqseC. The averages from 3 independent experiments are shown (**, P < 0.01; ***, P < 0.001; each by two-tailed Mann-Whitney). UTI89_MAN, mice pretreated with mannoside and challenged with wt UTI89; ΔqseC_MAN, mice pretreated with mannoside and challenged with UTI89ΔqseC; UTI89, mice pretreated with PBS and challenged with wt UTI89; and ΔqseC, mice pretreated with PBS and challenged with UTI89ΔqseC.
DISCUSSION
UPEC is the primary cause of UTI and is one of the most common bacterial infections afflicting the human population due to its high rate of occurrence and recurrence (14, 18). Among the patients experiencing acute UTI symptoms, approximately one-third will present with a recurrent infection within 6 to 12 months, while others may experience other debilitating sequelae, such as chronic infections or, in more severe cases, pyelonephritis, systemic infection, and death (14, 18, 28). The ability of UPEC to persist in multiple niches in the urinary tract is responsible, at least in part, for these disease outcomes (21, 28, 38, 43). Studies supporting this have shown that UPEC can establish chronic infection (21) or latently persist within the underlying epithelium, forming quiescent intracellular reservoirs (QIRs) that can reinitiate infection as the epithelium layers mature (38). These disease outcomes seem to be at least partly dependent on the number of IBCs formed (43). Given the frequency of recurrent UTIs, the rapid emergence and dissemination of antibiotic-resistant strains, and the potentially harmful effects of suppressive antibiotic therapy to prevent recurrences (12, 47), the identification of new strategies to combat or prevent UTIs becomes imperative. Here, we focus on distinguishing the effects of two factors, type 1 pili and QseC, which not only have been individually proposed to be ideal drug targets due to their effects on UPEC virulence, but also have been shown to be linked via a regulatory cascade that leads to fim misregulation upon qseC deletion.
Type 1 pili and QseC have been extensively implicated in UPEC virulence. Type 1 pili physically participate in the interactions that lead to host colonization and establishment of infection, while the QseC sensor kinase influences numerous cellular processes that in turn affect virulence-associated factors, including type 1 pili (19, 31, 33, 35, 51, 52). In this study, we show that restoring type 1 pilus expression in the absence of qseC rescues the adherence and invasion defects of this mutant and restores its ability to properly form IBCs inside the host during the acute infection stages. Thus, attenuation of UTI89ΔqseC is largely linked to reduced expression of type 1 pili, which decreases the population of internalized bacteria. Of note, locking the fim promoter in the ON orientation shifted the overall population toward piliated bacteria without yielding more pili per cell, which indicates that the levels of pili on the bacterial surface are not exclusively a function of phase variation but also are a function of transcriptional and/or posttranscriptional regulation. Our studies show that by shifting the population toward piliated bacteria, the LON strains are more efficient at colonizing and invading the host than their non-LON counterparts, leading to more bacteria being internalized, which is likely perpetuated with every IBC cycle. This explains the higher levels of colonization in the LON bacteria at 2 wpi compared to the non-LON counterparts, as a previous study demonstrated an association between the number of IBCs formed during acute infection and the development of chronic cystitis (21, 43). Interestingly, although UTI89ΔqseC_LON is enhanced in its ability to cause chronic infection compared to UTI89ΔqseC, it is still significantly less fit for establishing chronic cystitis than UTI89_LON. We have found that the establishment of chronic cystitis is dependent on host-pathogen interactions within the first 24 h of acute infection (21, 43). Indeed, although UTI89ΔqseC_LON alone does not have a defect during acute infection in single-infection experiments, it is rapidly outcompeted by UTI89_LON in the bladder when coinoculated with this strain. Thus, QseB-mediated misregulation in the absence of QseC results in a significant defect in sustaining infection and establishing chronic cystitis, even when type 1 piliation is restored. These data suggest that simultaneous inactivation of these factors have an additive effect on interfering with the onset of chronic UTI.
Research aimed at the development of therapeutics for UTIs has shown that compound ZFH-02056, an inhibitor of type 1 pilus function, significantly reduces UPEC titers in the host bladder when used as a treatment for chronic cystitis (11). The qseC deletion leads to reduced but not abolished expression of type 1 pili (31). We showed that pretreatment with one oral dose of ZFH-02056 significantly reduced the ability of UTI89ΔqseC to establish chronic cystitis. Thus, drugs targeting QseC may potentiate mannosides in the treatment of UTI. Previous studies identified LED209 as an inhibitor of the kinase activity of QseC (41). However, our studies have established that the defects of a qseC deletion mutant are associated with the phosphatase function of QseC, the absence of which results in an overactive QseB response regulator that cannot be dephosphorylated and deactivated (31). In vitro analyses demonstrated that LED209 had no inhibitory effect on the phosphatase activity of UPEC QseC and did not impact virulence-associated gene expression in UPEC (see Fig. S1 in the supplemental material). These data further indicate that it is the phosphatase activity of QseC that needs to be targeted to attenuate virulence, an area that we are currently exploring.
Collectively, our work extends our understanding of the molecular basis of attenuation of UPEC ΔqseC and shows that drugs targeting QseC hold promise to further potentiate mannoside efficacy for the treatment and prevention of UTIs. Patients who suffer from chronic or recurrent UTIs currently require suppressive antibiotics which disrupt the normal flora, causing unwanted side effects (14). The development of antivirulence agents would provide an alternative prevention means with the potential to eliminate UPEC colonization by effectively targeting both bacterial attachment and invasion, but most importantly persistence within the host, thereby greatly reducing the possibility of chronic and nonretractable infections.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Zhenfu Han for synthesizing compound ZFH-02056. We thank Eduardo A. Groisman for provision of protocols and facilities for the radioactivity experiments described in the supplemental material.
This work was supported by NIH grants P50 DK64540-06, R01 AI048689-08, and R37 AI02549-18 (to S.J.H.).
Footnotes
Published ahead of print 4 June 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Anderson GG, Goller CC, Justice S, Hultgren SJ, Seed PC. 2010. Polysaccharide capsule and sialic acid-mediated regulation promote biofilm-like intracellular bacterial communities during cystitis. Infect. Immun. 78:963–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson GG, et al. 2003. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 301:105–107 [DOI] [PubMed] [Google Scholar]
- 3. Barnhart MM, Chapman MR. 2006. Curli biogenesis and function. Annu. Rev. Microbiol. 60:131–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bishop BL, et al. 2007. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat. Med. 13:625–630 [DOI] [PubMed] [Google Scholar]
- 5. Bouckaert J, et al. 2005. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesin. Mol. Microbiol. 55:441–455 [DOI] [PubMed] [Google Scholar]
- 6. Chen SL, et al. 2009. Positive selection identifies an in vivo role for FimH during urinary tract infection in addition to mannose binding. Proc. Natl. Acad. Sci. U. S. A. 106:22439–22444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen SL, et al. 2006. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc. Natl. Acad. Sci. U. S. A. 103:5977–5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Connell I, et al. 1996. Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proc. Natl. Acad. Sci. U. S. A. 93:9827–9832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cormack BP, Valdivia RH, Falkow S. 1996. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173:33–38 [DOI] [PubMed] [Google Scholar]
- 10. Cusumano CK, Hung CS, Chen SL, Hultgren SJ. 2010. Virulence plasmid harbored by uropathogenic Escherichia coli functions in acute stages of pathogenesis. Infect. Immun. 78:1457–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cusumano CK, et al. 2011. Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci. Transl Med. 3:109ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dethlefsen L, Huse S, Sogin ML, Relman DA. 2008. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6:e280 doi:10.1371/journal.pbio.0060280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eto DS, Jones TA, Sundsbak JL, Mulvey MA. 2007. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PLoS Pathog. 3:e100 doi:10.1371/journal.ppat.0030100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Foxman B. 2010. The epidemiology of urinary tract infection. Nat. Rev. Urol. 7:653–660 [DOI] [PubMed] [Google Scholar]
- 15. Foxman B. 2002. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am. J. Med. 113(Suppl. 1A):5S–13S [DOI] [PubMed] [Google Scholar]
- 16. Gally DL, Leathart J, Blomfield IC. 1996. Interaction of FimB and FimE with the fim switch that controls the phase variation of type 1 fimbriae in Escherichia coli K-12. Mol. Microbiol. 21:725–738 [DOI] [PubMed] [Google Scholar]
- 17. Garofalo CK, et al. 2007. Escherichia coli from urine of female patients with urinary tract infections is competent for intracellular bacterial community formation. Infect. Immun. 75:52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Griebling TL. 2007. Urologic diseases in America, vol 07–5512. NIH, Washington, DC [Google Scholar]
- 19. Hadjifrangiskou M, et al. 2011. A central metabolic circuit controlled by QseC in pathogenic Escherichia coli. Mol. Microbiol. 80:1516–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hannan TJ, et al. 2008. LeuX tRNA-dependent and -independent mechanisms of Escherichia coli pathogenesis in acute cystitis. Mol. Microbiol. 67:116–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hannan TJ, Mysorekar IU, Hung CS, Isaacson-Schmid ML, Hultgren SJ. 2010. Early severe inflammatory responses to uropathogenic E. coli predispose to chronic and recurrent urinary tract infection. PLoS Pathog. 6:e1001042 doi:10.1371/journal.ppat.1001042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Henderson JP, et al. 2009. Quantitative metabolomics reveals an epigenetic blueprint for iron acquisition in uropathogenic Escherichia coli. PLoS Pathog. 5:e1000305 doi:10.1371/journal.ppat.1000305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hochhut B, et al. 2006. Role of pathogenicity island-associated integrases in the genome plasticity of uropathogenic Escherichia coli strain 536. Mol. Microbiol. 61:584–595 [DOI] [PubMed] [Google Scholar]
- 24. Hultgren SJ, Porter TN, Schaeffer AJ, Duncan JL. 1985. Role of type 1 pili and effects of phase variation on lower urinary tract infections produced by Escherichia coli. Infect. Immun. 50:370–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hultgren SJ, Schwan WR, Schaeffer AJ, Duncan JL. 1986. Regulation of production of type 1 pili among urinary tract isolates of Escherichia coli. Infect. Immun. 54:613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hung CS, et al. 2002. Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol. Microbiol. 44:903–915 [DOI] [PubMed] [Google Scholar]
- 27. Hunstad DA, Justice SS. 2010. Intracellular lifestyles and immune evasion strategies of uropathogenic Escherichia coli. Annu. Rev. Microbiol. 64:203–221 [DOI] [PubMed] [Google Scholar]
- 28. Ikaheimo R, et al. 1996. Recurrence of urinary tract infection in a primary care setting: analysis of a 1-year follow-up of 179 women. Clin. Infect. Dis. 22:91–99 [DOI] [PubMed] [Google Scholar]
- 29. Justice SS, et al. 2004. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 101:1333–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klemm P. 1986. Two regulatory fim genes, fimB and fimE, control the phase variation of type 1 fimbriae in Escherichia coli. EMBO J. 5:1389–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kostakioti M, Hadjifrangiskou M, Pinkner JS, Hultgren SJ. 2009. QseC-mediated dephosphorylation of QseB is required for expression of genes associated with virulence in uropathogenic Escherichia coli. Mol. Microbiol. 73:1020–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lim JK, et al. 1998. In vivo phase variation of Escherichia coli type 1 fimbrial genes in women with urinary tract infection. Infect. Immun. 66:3303–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, Hultgren SJ. 2000. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 19:2803–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morschhauser J, Vetter V, Korhonen T, Uhlin BE, Hacker J. 1993. Regulation and binding properties of S fimbriae cloned from E. coli strains causing urinary tract infection and meningitis. Zentralbl. Bakteriol. 278:165–176 [DOI] [PubMed] [Google Scholar]
- 35. Mulvey MA, et al. 1998. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 282:1494–1497 [DOI] [PubMed] [Google Scholar]
- 36. Mulvey MA, Schilling JD, Hultgren SJ. 2001. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 69:4572–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murphy KC, Campellone KG. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 4:11 doi:10.1186/1471-2199-4-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mysorekar IU, Hultgren SJ. 2006. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. U. S. A. 103:14170–14175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pallett A, Hand K. 2010. Complicated urinary tract infections: practical solutions for the treatment of multiresistant Gram-negative bacteria. J. Antimicrob. Chemother. 65(Suppl. 3):iii25–iii33 [DOI] [PubMed] [Google Scholar]
- 40. Pinkner JS, et al. 2006. Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc. Natl. Acad. Sci. U. S. A. 103:17897–17902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rasko DA, et al. 2008. Targeting QseC signaling and virulence for antibiotic development. Science 321:1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rosen DA, Hooton TM, Stamm WE, Humphrey PA, Hultgren SJ. 2007. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 4:e329 doi:10.1371/journal.pmed.0040329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schwartz DJ, Chen SL, Hultgren SJ, Seed PC. 2011. Population dynamics and niche distribution of uropathogenic Escherichia coli during acute and chronic urinary tract infection. Infect. Immun. 79:4250–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Snyder JA, Lloyd AL, Lockatell CV, Johnson DE, Mobley HL. 2006. Role of phase variation of type 1 fimbriae in a uropathogenic Escherichia coli cystitis isolate during urinary tract infection. Infect. Immun. 74:1387–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thankavel K, et al. 1997. Localization of a domain in the FimH adhesin of Escherichia coli type 1 fimbriae capable of receptor recognition and use of a domain-specific antibody to confer protection against experimental urinary tract infection. J. Clin. Investig. 100:1123–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thumbikat P, et al. 2009. Bacteria-induced uroplakin signaling mediates bladder response to infection. PLoS Pathog. 5:e1000415 doi:10.1371/journal.ppat.1000415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ubeda C, et al. 2010. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J. Clin. Investig. 120:4332–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Waksman G, Hultgren SJ. 2009. Structural biology of the chaperone-usher pathway of pilus biogenesis. Nat. Rev. Microbiol. 7:765–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Welch RA, et al. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 99:17020–17024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wiles TJ, Kulesus RR, Mulvey MA. 2008. Origins and virulence mechanisms of uropathogenic Escherichia coli. Exp. Mol. Pathol. 85:11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wright KJ, Hultgren SJ. 2006. Sticky fibers and uropathogenesis: bacterial adhesins in the urinary tract. Future Microbiol. 1:75–87 [DOI] [PubMed] [Google Scholar]
- 52. Wright KJ, Seed PC, Hultgren SJ. 2007. Development of intracellular bacterial communities of uropathogenic Escherichia coli depends on type 1 pili. Cell Microbiol. 9:2230–2241 [DOI] [PubMed] [Google Scholar]
- 53. Wright KJ, Seed PC, Hultgren SJ. 2005. Uropathogenic Escherichia coli flagella aid in efficient urinary tract colonization. Infect. Immun. 73:7657–7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xie B, et al. 2006. Distinct glycan structures of uroplakins Ia and Ib: structural basis for the selective binding of FimH adhesin to uroplakin Ia. J. Biol. Chem. 281:14644–14653 [DOI] [PubMed] [Google Scholar]
- 55. Zhou G, et al. 2001. Uroplakin Ia is the urothelial receptor for uropathogenic Escherichia coli: evidence from in vitro FimH binding. J. Cell Sci. 114:4095–4103 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.