

Abstract
Campylobacter jejuni is a leading worldwide bacterial cause of human diarrheal disease. Although the specific molecular mechanisms of C. jejuni pathogenesis have not been characterized in detail, host inflammatory responses are thought to be major contributing factors to the resulting typical acute colitis. The intestinal mucosal chemokine response is particularly important in the initial stages of bacterium-induced gut inflammation. Chemokines attract blood phagocytes and lymphocytes to the site of infection and regulate immune cell maturation and the development of localized lymphoid tissues. The production of chemokines by dendritic cells (DCs) following Campylobacter infection has not yet been analyzed. In the current study, we infected human monocyte-derived DCs with C. jejuni to examine the production of key proinflammatory chemokines and chemokine receptors. The chemokines, including CC families (macrophage inflammatory protein 1α [MIP-1α], MIP-1β, RANTES) and CXC families (growth-related oncogene α [GRO-α], IP-10, and monokine induced by gamma interferon [MIG]), were upregulated in Campylobacter-infected DCs. Chemokine receptors CCR6 and CCR7, with roles in DC trafficking, were also induced in Campylobacter-infected DCs. Further, Campylobacter infection stimulated the phosphorylation of P38, P44/42, and stress-activated protein kinase/Jun N-terminal kinase (SAPK/JNK) mitogen-activated protein kinases (MAPKs) in DCs. NF-κB activation was specifically involved in chemokine induction in DCs infected with C. jejuni. Additionally, STAT3 was significantly increased in Campylobacter-infected DCs compared to that in uninfected DCs. These results suggest that DCs play a significant role in the initiation and modulation of the inflammatory response by enlisting monocytes, neutrophils, and T lymphocytes during human intestinal infection with Campylobacter.
INTRODUCTION
Campylobacter jejuni is a leading cause of human diarrheal disease worldwide (8). The illness caused by C. jejuni is characterized by fever, headache, abdominal cramping, diarrhea, and the presence of erythrocytes, monocytes, and polymorphonuclear leukocytes (PMNs) in the stool (31, 35). C. jejuni is also a leading cause of postinfection irritable bowel syndrome (IBS) and of both Guillain-Barre (GBS) and Reiter's syndromes (24, 32). Although the pathogenic mechanisms of C. jejuni are only now being defined, the host immune and inflammatory responses are thought to be major contributing factors to the resulting acute illness and possibly to the postinfection sequelae, such as IBS, GBS, and Reiter's syndrome.
Campylobacter infection results in illness ranging from a few loose stools to dysentery. The latter is caused by colonic mucosal inflammation (4). The intestinal mucosal chemokine response is particularly important in the initial stages of bacterium-induced inflammation (2, 30, 40). Chemokines attract blood phagocytes and lymphocytes to the site of infection and regulate leukocyte maturation and the development of local lymphoid tissues (e.g., lymphoid follicles in M cells) (1, 23, 26, 29). Chemokine CC and CXC families play important roles in the bacterium-host interaction. Chemokines from the CC family are potent chemoattractant agents for monocytes and T lymphocytes. They include macrophage inflammatory protein 1α (MIP-1α; chemokine ligand CCL3), MIP-1β (CCL4), monocyte chemoattractant protein 1 (MCP-1; CCL2), and RANTES (CCL5). Chemokines from the CXC family include interleukin-8 (IL-8; CXCL8), growth-related oncogene α (GRO-α or CXCL1), gamma interferon (IFN-γ)-inducible protein 10 (IP-10 or CXCL 10), and monokine induced by IFN-γ (MIG) (CXCL9). Interleukin-8 and GRO-α primarily attract neutrophils to the sites of inflammation (7). IFN-γ-inducible protein 10 and MIG have no chemoattractant effect on neutrophils. Rather, they promote the chemotaxis of monocytes and activated T lymphocytes in vivo (19, 33).
The cells within the intestinal mucosa, such as dendritic cells (DCs), phagocytes, and epithelial cells, provide the primary defense against many enteropathogens. In particular, the intestinal DCs assist in determining the nature of the immune response (i.e., tolerance or immune activation). Additionally, they link the innate and adaptive immune responses to microbial pathogens by both producing and responding to inflammatory factors. In general, the immature DCs expressing the chemokine receptor CCR6 are guided by MIP-3α from the blood to intestinal tissues (3). CCR6 is expressed by most B cells and subsets of T cells and DCs found in the gut mucosal immune system. The functional significance of CCR6 was examined by Sakazar-Gonzalez and colleagues (28), who analyzed the role of CCR6+ DCs in the initiation of T cell responses to Salmonella. They found that the expression of the CCR6 receptor on DCs was absolutely required to initiate anti-Salmonella-specific T cell responses. The CCR6+ DCs migrated out of the subepithelial dome of Peyer's patches (PP) after Salmonella infection, whereas CCR6-deficient DCs were unable to respond to bacterial invasion of the PP.
Upon successful recognition of microbial products, immature DCs acquire a mature phenotype, expressing chemokine receptors that drive their migration into secondary lymphoid organs (6, 27, 34). The migration is an important function of DCs as they join T lymphocytes in the lymph nodes, enabling efficient activation of the adaptive immune response. The migratory capacity is controlled by the expression of chemokine receptors, especially CCR7 on the surfaces of DCs (11, 21). CCR7 is activated by two known endogenous ligands, macrophage inflammation protein 3β (MIP-3β; CCL19) and secondary lymphoid tissue chemokine (SLC; CCL 21) (21).
Signal transduction pathways, including mitogen-activated protein kinases (MAPKs) and nuclear factor κB (NF-κB), are important in the bacterium-host interaction. NF-κB family transcription factors regulate the genes involved in acute-phase response, inflammation, and cell growth and differentiation (17). Three major MAPK subfamilies have been characterized: p38, p44/42, and stress-activated protein kinase/Jun N-terminal kinases (SAPK/JNK) (39). The availability of specific inhibitors of NF-κB and antibodies to MAPKs and NF-κB allows evaluation of their involvement in cellular responses. Specifically, the MAPK cascade has been reported to regulate expression of the Campylobacter-stimulated inflammatory response in epithelial cells (15, 36). The signal transduction and activators of transcription proteins (STATs) are also major cell mediators of signaling during immune responses and play roles in cell development and differentiation. It is now well established that many cytokines and STATs are involved in multiple steps of the adaptive immune response.
Previously, we demonstrated that C. jejuni infection induced human DC maturation and the production of seven cytokines, including IL-8 (13). C. jejuni has also been reported to induce chemokine production in epithelial cells (12, 14, 16). In this study, we employed C. jejuni strain 81-176, which has been shown to cause a dysenteric syndrome in two human feeding studies (4), and two other C. jejuni strains. We investigated whether C. jejuni induces human monocyte-derived DCs to produce CC or CXC chemokines and/or upregulate related chemokine receptors that are essential to recruitment and activation of the host's inflammatory response. Furthermore, we explored the potential signal transduction pathways involved in the interaction between Campylobacter infection and chemokines secreted by human DCs.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Campylobacter jejuni strains 81-176 (obtained following a disease outbreak in Minnesota and shown to cause colitis in two human challenge studies) (4, 33a), and M129 were employed herein. C. jejuni was grown in Mueller-Hinton (M-H) biphasic broth or on M-H agar (Difco) at 37°C under a Campylobacter gas atmosphere of 10% carbon dioxide, 5% oxygen, and 85% nitrogen.
Preparation of human monocyte-derived dendritic cells.
Mononuclear cells were obtained by apheresis of normal volunteer donors as performed by the Blood Services Section of the Department of Transfusion Medicine (DTM) at the NIH Warren G. Magnuson Clinical Center (Bethesda, MD). The mononuclear cells were further enriched for monocytes by centrifugal elutriation as performed by the Cell Processing Section of the DTM. The elutriated monocytes were cultured by previously reported methods (13). The cells were plated in 96-well (5 × 104 cells/well) or 6-well (106 cells/well) plates containing fresh RPMI (Mediatech) with 5% human AB serum Nabi and 800 U/ml granulocyte macrophage colony-stimulating factor (GM-CSF) (Peprotech, Rocky Hill, NJ). This resulted in the generation of immature monocyte-derived DCs that were positive for CD11c but negative for CD14 (data not shown). C. jejuni cells were added to the cultured DCs at different multiplicities of infection (MOIs) without centrifugation and incubated at 37°C in 5% CO2. For chemokine measurements, the infected culture supernatants in 96-well plates were harvested, centrifuged, and frozen at −80°C prior to analysis.
Chemokine measurement of DC supernatants by Luminex assay.
Immature DCs were incubated in 96-well plates (5 × 104 DCs/well) for 4, 24, and 48 h with live C. jejuni 81-176, heat-killed (i.e., treated at 70°C for 30 min) C. jejuni 81-176, lipooligosaccharide (LOS) of C. jejuni 81-176 (extracted by using the hot phenol-water technique), or hot phenol-water-extracted Escherichia coli lipopolysaccharide (LPS; Sigma, St. Louis, MO) (38). Phosphate-buffered saline (PBS; Invitrogen Corporation)-incubated DCs served as unstimulated controls. Chemokine assays were performed by using human chemokine bead kits (Biosource International, Camarillo, CA) and a Luminex 100 analyzer (Luminex Corporation, Austin, TX). Chemokine data analysis was performed using the MasterPlex QT Quantitation Software (MiraiBio, Alameda, CA).
RNA extraction, cDNA synthesis, and PCR analysis.
Human DCs were infected with C. jejuni at an MOI of 10. In an inhibition assay, DCs were incubated with the NF-κB inhibitor caffeic acid phenethyl ester (CAPE; Sigma) at 50 μM for 1 h prior to infection (25). After 4, 8, and 24 h, total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA). Extracted RNA was transcribed into first-strand cDNA by using the ProSTAR first-strand reverse transcriptase PCR (RT-PCR) kit (Stratagene, La Jolla, CA). Negative controls were constructed by omitting RT or RNA. PCR primer pairs that were specific for human GAPDH, chemokines, and chemokine receptor genes were synthesized at our facilities as described previously (13). cDNA was amplified with Taq polymerases (TaKaRa, Madison, WI). GAPDH transcription was included as a constitutive housekeeping gene control. Ten microliters of PCR products were combined with 2 μl of 10× loading buffer, electrophoresed on 2.0% agarose gels, and stained with ethidium bromide. A 0.1-kb DNA ladder was run to provide reference markers. The quantity of RNA was analyzed using GeneTools from Syngene (a division of The Synoptics Group, Frederick, MD).
Western blot assay.
Human DCs were infected with C. jejuni at an MOI of 10 or 100 for 2 or 4 h and lysed in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10 mM NaF, 1 mM Na3VO4, and a protease inhibitor cocktail (Sigma).
After being incubated for 30 min on ice, the lysates were centrifuged and the supernatants were collected. The protein concentration of each sample was quantified by the bicinchoninic acid (BCA) protein assay reagent kit (Pierce, Rockford, IL). The supernatants were separated by 10% SDS-PAGE and then transferred to nitrocellulose membranes, blotted with MAPK, phosphor-MAPK, or phosphor-STAT antibodies according to the manufacturer's instructions (Cell Signaling Technology, Danvers, MA). The quantity of RNA was analyzed using GeneTools from Syngene.
Indirect immunofluorescence analysis.
Dendritic cells were infected with either live or heat-killed C. jejuni 81-176 or reacted with either Campylobacter LOS or E. coli LPS at various concentrations for 2 h, washed 3 times with cold PBS, fixed with 3.7% paraformaldehyde for 15 min, and then blocked with blocking buffer (PBS supplemented with 1% BSA and 0.3% Triton X-100) for 1 h. The PBS-incubated DCs served as unstimulated controls. The cells were labeled with anti-NF-κB p65 (L8F6) mouse monoclonal antibody (Cell Signaling Technology), anti-mouse IgG Fab2 Alexa Flour 488 (green), and Alexa Fluor 594 phalloidin (Invitrogen), which labeled actin filaments red according to the manufacturer's instructions. The stained DCs were mounted with Vectashield mounting medium (Vecta Laboratories, Inc., Burlingame, CA) and viewed with a Zeiss HBO100 fluorescence microscope. The percentage of DCs with green fluorescent nuclei was determined by counting at least 100 cells under each coverslip for 3 different samples.
Statistical analysis.
Results are presented as the means ± standard deviations (SD) of the means from at least three independently conducted assays. P values were calculated using Student's t test between two study groups. Statistical comparisons among three or more different study groups were performed using an analysis of variance (ANOVA) test with a confidence level of 95%.
RESULTS
Chemokine induction in DCs infected with C. jejuni 81-176.
The kinetics and profiles of chemokine secretion from human DCs during C. jejuni infection were first analyzed. Human monocyte-derived DC culture supernatants were collected at 4, 24, and 48 h from uninfected DCs or after infection with C. jejuni at MOIs of 1, 10, and 100 to determine chemokine levels. Campylobacter infection of DCs induced enhanced production of MIP-1α, MIP-1β, RANTES, GRO-α, IP-10, and MIG (Fig. 1). MCP-1 was slightly, but not significantly, increased in C. jejuni-infected cells compared to in uninfected controls (data not shown). The production of MCP-2 or MCP-3 (exotaxin) in Campylobacter-infected DCs was not changed compared to in uninfected DCs (data not shown).
Fig 1.

Chemokine induction in human DCs infected with C. jejuni 81-176. Culture supernatants were tested at 4, 24, and 48 h postinfection with C. jejuni 81-176 at various MOIs of 1, 10, or 100. Phosphate-buffered saline-incubated DCs served as uninfected controls. Chemokine assays were performed by using human chemokine bead kits and a Luminex 100 analyzer. Data analysis was performed using MasterPlex QT quantitation software (MiraiBio, Alameda, CA). The levels of MIP-1α, MIP-1β, RANTES, GRO-α, IP-10, and MIG are shown in A, B, C, D, E, and F, respectively. Data are presented as the means ± SD of duplicate wells from at least 3 independent assays, and ANOVA was used for statistical comparisons among three groups.
At 4 h postinfection with C. jejuni 81-176, the production of MIP-1α, MIP-1β, and IP-10 was increased at virtually all MOIs, except for IP-10 at an MOI of 1. For example, the titers of MIP-1α, MIP-1β, and IP-10 were increased, respectively, 160.6-fold (22,322.86 ± 5,673.72 pg/ml), 14.7-fold (22,979.94 ± 7,426.06 pg/ml), and 10.7-fold (269.80 ± 68.87 pg/ml), at an MOI of 10 after 4 h compared to uninfected control levels (i.e., 138.96 ± 73.38 pg/ml, 1,563.62 ± 785.68 pg/ml, and 25.20 ± 16.38 pg/ml, respectively) (Fig. 1A, B, E) (P < 0.001, 0.01, 0.01). Although the titers of RANTES, GRO-α, and MIG were not changed relative to those of the uninfected control at the earliest time point tested, production of these chemokines was significantly increased to 55.5-fold (4,749.45 ± 1,564.31 pg/ml), 5.5-fold (157.10 ± 43.35 pg/ml), and 7.9-fold (448.33 ± 149.50 pg/ml), respectively, at an MOI of 10 at 24 h postinfection compared to uninfected control levels (i.e., 85.64 ± 31.93 pg/ml, 28.61 ± 10.00 pg/ml, 56.62 ± 30.52 pg/ml, respectively) (Fig. 1C, D, F; P < 0.001, 0.05, 0.01). The increase in Campylobacter MOI from 1 to 100 did not have a significant impact on overall chemokine production (P > 0.05). Moreover, the data showed that chemokine production generally increased over time with peak amounts at 24 or 48 h, corresponding with the timing of typical disease symptoms and with the timing of maximal DC maturation and cytokine production as demonstrated in our previous study (13).
Chemokine induction in DCs infected with live versus heat-killed C. jejuni.
Clinical isolates of wild-type, invasive C. jejuni (strains 81-176, 11168, and M129) and heat-killed 81-176 (i.e., heated at 70°C for 30 min) were compared for their ability to trigger human DC secretion of selected chemokines at an MOI of 10 at 24 h postinfection (Fig. 2). C. jejuni NCTC11168 and M129 induced similar amounts of the targeted chemokines as strain 81-176, compared to PBS-incubated DCs (P < 0.01 for all). Heat-killed bacteria were employed to determine if perhaps live Campylobacter's ability to invade host cells contributed to the induction of the tested chemokines. Live bacteria did induce slightly higher levels of chemokines than heat-killed bacteria, but these differences were not statistically significant (P > 0.05). Thus, both metabolically inactive, heat-killed bacteria and live, active bacteria stimulated similar chemokine responses.
Fig 2.

Comparison of chemokine levels induced in DCs treated with live C. jejuni strains 81-176, NCTC 11168, or M129 or heat-killed strain 81-176. Culture supernatants were tested at 24 h postinfection with C. jejuni at an MOI of 10 and compared to an uninfected control (un). The heat-killed C. jejuni cells were treated at 70°C for 30 min and subsequently confirmed by plate count to be nonviable. The titers of MIP-1α, MIP-1β, RANTES, GRO-α, IP-10, and MIG are shown in A, B, C, D, E, and F, respectively. Data are presented as the means ± SD of duplicate wells from 3 independent assays.
Are bacterial polysaccharides responsible for inducing chemokine production in DCs?
Lipooligosaccharide was prepared from C. jejuni 81-176 by the hot phenol-water extraction method (38) and identified as LOS on silver-stained SDS-PAGE gels (data not shown). An equal amount of Westphal-purified LPS from E. coli, known to induce cytokine production in DCs, was used as a positive control. Both LOS and LPS at 0.01, 0.1, or 1.0 μg/ml induced chemokine production by DCs after 24 h of incubation. MIP-1α, RANTES, GRO-α, and IP-10 showed maximally increased levels when DCs were treated with either 0.1 or 1 μg/ml LOS (Fig. 3A, C to E). MIG was stimulated to slightly higher levels with 1 μg/ml LOS than with 0.1 μg/ml LOS (Fig. 3F). However, the ANOVA test revealed no significant difference among the doses of LOS in terms of MIG induction. The levels of chemokines produced were comparable to that observed after infection with whole bacteria. Although it is difficult to equate DC responses to live bacteria versus pure LOS, these data suggest that C. jejuni LOS may be a key contributor to the induction of chemokines by DCs.
Fig 3.

Chemokine levels induced in DCs by C. jejuni 81-176 LOS or control E. coli LPS, compared to uninfected DCs (un). LOS (1, 10, 100 ng) from C. jejuni 81-176 or LPS (1, 10, 100 ng) from E. coli was added to DCs per well (100 μl medium/well) for 24 h. The titers of MIP-1α, MIP-1β, RANTES, GRO-α, IP-10, and MIG are shown relative to the values observed in uninfected DCs. Data are presented as means ± SD from duplicate wells from 3 independent assays.
RT-PCR, using the specific LPS inhibitor, polymyxin B (50 μM), was utilized to confirm the role of Campylobacter LOS in stimulating human DCs. Polymyxin B, which binds to LPS/LOS, clearly blocked chemokine expression induced by LOS and LPS of RANTES, GRO-α, IP-10, and MIG (P < 0.01). The most heavily induced chemokines, MIP-1α and MIP-1β, which are induced to >20,000 pg/ml following addition of LOS/LPS, were markedly but not totally inhibited by polymyxin addition to either the LPS or LOS (P < 0.05). It is possible that these chemokine genes are very sensitive to NF-κΒ activation and the polymyxin B levels added did not totally bind all available LOS/LPS sites under the conditions/times employed. These results are included in Fig. S1 in the supplemental material.
C. jejuni induced the expression of CCR6 and CCR7 by DCs.
We used RT-PCR to examine the expression of CCR6 and CCR7 in C. jejuni-infected DCs at an MOI of 10. After 4, 8, and 24 h of infection, total DC RNA was extracted with TRIzol reagent. Extracted RNA was assessed via RT-PCR; GAPDH served as a constitutive control. The mRNA of CCR6 and CCR7 was upregulated in infected DCs at 4, 8, and 24 h postinfection (Fig. 4A; lanes 2, 4, 6) compared to the uninfected cells (lanes 1, 3, 5). The fold changes of CCR6 receptor mRNA were 5.9 (4 h, lane 2), 4.8 (8 h, lane 4), and 3.6 (24 h, lane 6), and those of CCR7 were 7.1 (4 h), 15.3 (8 h), and 30 (24 h), compared to the uninfected controls at the same time points (P < 0.01 for all time points). The changes in gene expression of these receptors were verified by semiquantitative analysis (Fig. 4B). The densitometry analysis of the agarose gel of CCR6 and CCR7 mRNA was normalized against GAPDH, which served as an internal control. The results demonstrate that the expression of both chemokine receptors CCR6 and CCR7 is significantly upregulated in Campylobacter-infected human DCs.
Fig 4.

Levels of chemokine receptor CCR6 or CCR7 mRNA produced by human DCs during Campylobacter infection. DCs were infected with C. jejuni 81-176 at an MOI of 10. At 4, 8, and 24 h postinfection, total RNA was extracted with TRIzol reagent and analyzed following RT-PCR. (A) Lanes 1, 3, and 5 represent mRNA from PBS-incubated cells for 4, 8, and 24 h, respectively, while lanes 2, 4, and 6 represent mRNA from DCs infected with C. jejuni for 4, 8, and 24 h, respectively. The fold change was analyzed using GeneTools from Syngene. (B) The changes in gene expression of CCR6 and CCR7 were verified by semiquantitative analysis. The densitometry analysis of the agarose gel was normalized against constitutively expressed GAPDH. The relative mRNA levels of CCR6 and CCR7 versus GAPDH are presented as means ± SD from three samples from independent experiments.
The chemokines secreted by Campylobacter-infected DCs were accompanied by the activation of MAPKs.
To explore if MAPK signal transduction pathways were involved in chemokine production, uninfected DCs were compared to cells treated with live or heat-killed Campylobacter, as well as LOS for 2 h (Fig. 5). After DC lysis, the supernatant proteins were separated by SDS-PAGE, blotted to nitrocellulose membranes, and analyzed via Western blotting with phospho-MAPK or MAPK antibodies. C. jejuni infection was observed to induce phosphorylation of all MAPK proteins tested. p38 showed a 354.5-fold increase at an MOI of 10 and a 535.7-fold increase at an MOI of 100 (Fig. 5A, lanes 2 and 3). p44/42 and SAPK/JNK (p54/46), respectively, increased 2.2/2.0-fold and 2.2/1.9-fold at an MOI of 10, and 10/1-fold and 154/92-fold at an MOI of 100 (Fig. 5B and C, lanes 2 and 3), compared to respective PBS-incubated DCs (Fig. 5, lane 1) (P < 0.05, for all except p46 at an MOI of 10). The same blots were then stripped and reprobed with specific MAPK antibodies to examine total MAPK protein expression. Total p38 MAPK protein expression was significantly upregulated in infected DCs (P < 0.01). However, total p44/42 and SAPK/JNK proteins did not show significant upregulation between the treated and untreated cells.
Fig 5.
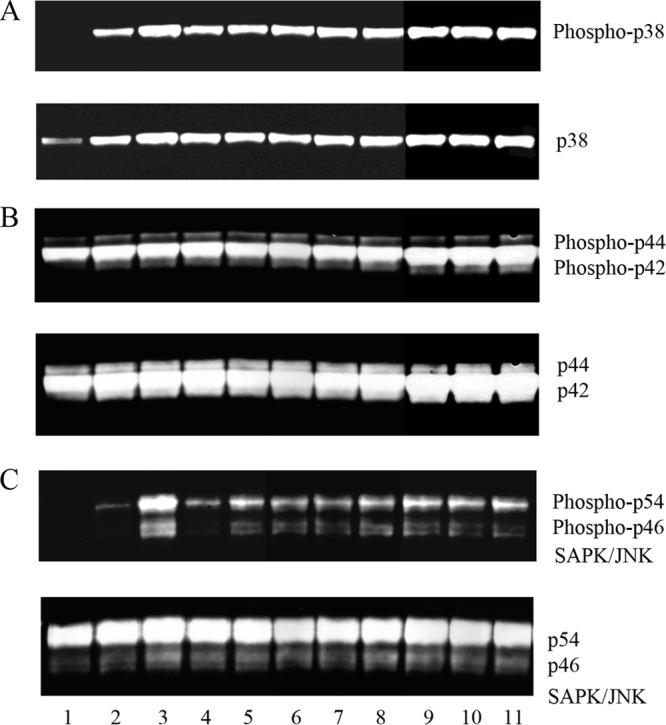
Live or heat-killed C. jejuni strain 81-176, Campylobacter LOS, or E. coli LPS activate MAP kinases in human DCs. The activation and expression of phospho-MAPKs were assessed by Western blotting. One representative experiment (n = 3) is shown. Lane 1 contains the uninfected control. Lanes 2 and 3 represent DCs infected with live 81-176 at MOIs of 10 and 100, and lanes 4 and 5 represent DCs infected with heat-killed 81-176 at MOIs of 10 and 100. Lanes 6 to 8 are DCs treated with 0.01, 0.1, and 1.0 μg/ml LOS, respectively, and lanes 9 to 11 are DCs treated with 0.01, 0.1, and 1.0 μg/ml LPS, respectively. (A) Phospho-p38 activation after 2-h treatments compared to total p38 in the same blot; (B) phospho-p44/42 activation after 2-h treatments compared to total p44/42 in the same blot; (C) phospho-SAPK/JNK (p54/p46) activation after 2-h treatments compared to total SAPK/JNK in the same blot.
Heat-killed C. jejuni induced strong phosphorylation of p38 (i.e., increased 360.2-fold over uninfected control), p44/42 (increased 2.1/1.8-fold), and p54 (increased 33.5-fold) at an MOI of 10 (Fig. 5A, B, and C, lane 4). Heat-killed bacteria at an MOI of 100 enhanced the phosphorylation of the MAPKs even more than that observed at an MOI of 10 (lane 5). Finally, treatment with 0.01 μg/ml of Campylobacter LOS significantly increased phosphorylation of p38 (541-fold), p44/42 (2.4/1.8-fold), and p54/46 (59.2/15.8-fold) (lane 6). Higher concentrations of LOS (0.1 and 1.0 μg/ml; lanes 7 and 8) did not enhance phosphorylation of MAPKs over that seen with 0.01 μg/ml LOS (lane 6). E. coli LPS served as a positive control, and all three concentrations of LPS (0.01, 0.1, and 1.0 μg/ml) induced dramatic phosphorylation of the three MAP kinases (lanes 9 to 11). These data show that MAPK phosphorylation is stimulated in DCs treated with live or heat-killed C. jejuni or with only LOS.
Expression of chemokine mRNAs was upregulated in Campylobacter-induced DCs, and the upregulation was NF-κB dependent.
The expressions of mRNA for MIP-1α, MIP-1β, RANTES, GRO-α, IP-10, and MIG were assessed via RT-PCR in human DCs infected with strain C. jejuni 81-176 for 4, 8, and 24 h. These chemokine mRNAs were upregulated in infected DCs (Fig. 6A, lanes 2, 5, 8) compared to uninfected controls (lanes 1, 4, 7). mRNA expressions for MIP-1α, MIP-1β, and RANTES were upregulated 112.6-fold, 12.3-fold, and 11.7-fold, respectively, at 4 h (Fig. 6A, lane 2; P <0.01) compared to uninfected DCs at the same time points (lane 1). The mRNA levels for GRO-α, IP-10, and MIG were slightly upregulated at 4 h but were increased 3.2-fold, 48.9-fold, and 59.5-fold, respectively, by 8 h (lane 5), compared to uninfected DCs after 8 h of incubation (lane 4).
Fig 6.

Levels of proinflammatory chemokine mRNAs produced by human DCs infected with C. jejuni. DCs were infected with C. jejuni 81-176 at an MOI of 10 for 4, 8, and 24 h. Extracted RNA was transcribed into cDNA. Negative controls were constructed by omitting either the RT or RNA (not shown). cDNA was amplified with Taq polymerase. GAPDH transcription was included as a constitutive housekeeping gene. (A) Lanes 1, 4, and 7 show uninfected controls after incubation with PBS for 4, 8, or 24 h, respectively. Lanes 2, 5, and 8 show cells infected with C. jejuni for 4, 8, and 24 h, respectively. Lanes 3, 6, and 9 show the chemokine mRNAs detected in human DCs treated with the NF-κB inhibitor CAPE at 50 μM for 1 h prior to adding bacteria. These data are representative of three independent experiments. The fold change was analyzed using GeneTools from Syngene. (B) The changes in gene expression were verified by semiquantitative analysis. The densitometry analysis of the agarose gel was normalized against GAPDH.
Caffeic acid phenethyl ester (CAPE) is a specific inhibitor of NF-κB activation (25). DCs treated with 50 μM CAPE for 1 h prior to inoculation with strain 81-176 showed markedly reduced transcription at 4, 8, and 24 h (Fig. 6A, lanes 3, 6, and 9). Inhibition of NF-κB activation greatly reduced the mRNA expression of MIP-1α, MIP-1β, RANTES, IP-10, and MIG (P < 0.05). GRO-α mRNA was not affected by CAPE treatment. The changes in gene expression of the six chemokines were verified by semiquantitative analysis (Fig. 6B). The densitometry analysis of the agarose gel for each chemokine mRNA was normalized against GAPDH.
The results show that Campylobacter infection upregulates the transcription of MIP-1α, MIP-1β, RANTES, GRO-α, IP-10, and MIG. Further, the transcription of these chemokine genes, with the exception of GRO-α, was NF-κB dependent in C. jejuni-infected DCs.
Indirect immunofluorescence analysis of NF-κB in untreated or treated DCs.
To understand the mechanisms of NF-κB activation in human DCs infected with Campylobacter or treated with LOS, the indirect immunofluorescence technique was employed. Dendritic cells were infected with C. jejuni 81-176 or treated with Campylobacter LOS or control E. coli LPS for 2 h. PBS-incubated DCs were used as a control. NF-κB p65 mouse monoclonal antibodies were utilized to recognize the endogenous levels of total NF-κB p65 proteins. The DCs that reacted with the anti-NF-κB p65 antibody presented green fluorescence, while the actin filaments of the DCs were labeled with red phalloidin for cell shape recognition. The percentage of DCs with green nuclei was estimated by counting at least 100 DCs under each coverslip. The averages from 3 separate experiments are presented in Fig. 7A. About 37% of the DCs were activated with C. jejuni at an MOI of 10 for 2 h; the P value was <0.01 compared to PBS-treated cells. The higher MOI of 100 increased the activated cell percentage to ∼59%. Incubation of DCs with LOS and LPS at 0.1 μg/ml resulted in 57% of the nuclei reacting with the anti-NF-κB antibody.
Fig 7.
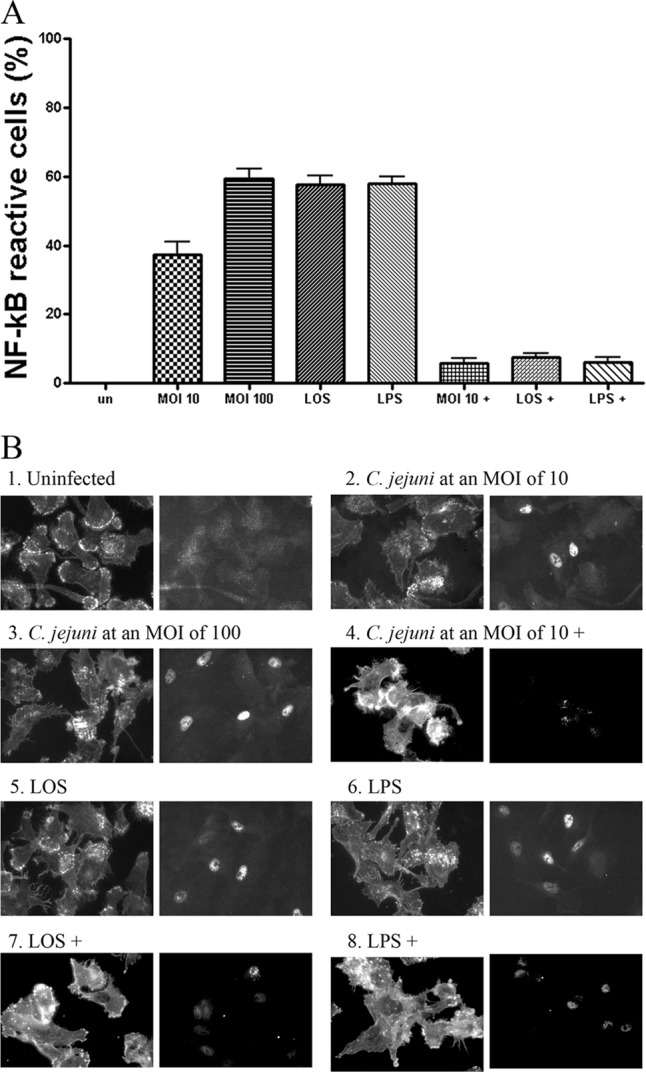
Indirect immunofluorescence analysis of NF-κB activation in DCs. Dendritic cells were infected with C. jejuni 81-176 or treated with 0.1 μg/ml LOS or LPS for 2 h. Treated DCs were then reacted with NF-κB p65 (L8F6) mouse monoclonal antibody, anti-mouse IgG Fab2 Alexa Flour 488 (green), and Alexa Fluor 594 phalloidin (red). (A) The percentage of the NF-κB reactive DCs with green nuclei is presented. Approximately 100 DCs under each coverslip for each experiment were counted. Data are presented as the means ± SD from 3 independent assays. (B) The fluorescent images labeled by phalloidin (left) and NF-κB antibody (right) are presented: 1, uninfected controls; 2 and 3, DCs infected with C. jejuni 81-176 at MOIs of 10 or 100, respectively; 4, DCs were preincubated with 50 μM CAPE (designated with a plus sign) for 1 h and infected with C. jejuni 81-176 at an MOI of 10; 5, DCs treated with LOS; 6, DCs treated with LPS; 7 and 8, DCs preincubated with 50 μM CAPE for 1 h and treated with LOS or LPS, respectively.
The immunofluorescent images from the DCs infected with C. jejuni 81-176 or treated with LOS or LPS are presented in Fig. 7B. More intense fluorescent nuclei were observed in the DCs infected with C. jejuni than those treated with LOS or LPS (Fig. 7B). Although bacteria at an MOI of 10 resulted in less nuclei reacting with the anti-NF-κB antibody (Fig. 7A), these nuclei presented stronger fluorescence than those from the LOS- or LPS-treated DCs (Fig. 7B). Therefore, it is possible that live C. jejuni causes stronger NF-κb activation within individual cells than engendered by other treatments. The specific inhibitor, CAPE, strongly inhibited NF-κB activation caused by treatment with Campylobacter, LOS, or LPS, as evidenced by comparison to untreated DCs (P < 0.05; Fig. 7A and B).
Chemokine secretion by Campylobacter-infected DCs was accompanied by the activation of STAT3.
After DCs were infected with C. jejuni 81-176 for 2 and 4 h at an MOI of 10, the cells were lysed, supernatants were separated by SDS-PAGE, transferred onto a nitrocellulose membrane, and then blotted with anti-phospho-STAT3-1 (Tyr705) or anti-phospho-STAT3-2 (Ser727) antibodies. Figure 8 shows that Campylobacter infection induced the phosphorylation of STAT3 in human DCs, and the fold changes were 33.8 at 2 h postinfection and 48.0 at 4 h postinfection (Fig. 8A, lanes 2 and 3) compared to the uninfected control (lane 1), as detected with phospho-STAT3-2 antibody. C. jejuni induced slight phosphorylation at 4 h postinfection (1.6-fold) as detected with the phospho-STAT3-1 antibody. These data demonstrate that STAT3 was significantly phosphorylated at Ser727 (P < 0.001) but only marginally at Tyr705 in C. jejuni-infected DCs.
Fig 8.
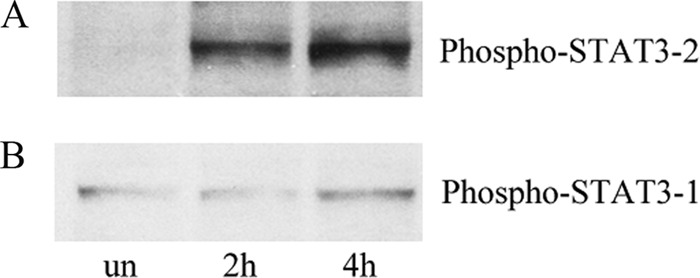
C. jejuni-induced STAT3 phosphorylation in human DCs. After human DCs were infected with C. jejuni 81-176 for 2 or 4 h, whole-cells extracts were prepared, and phospho-STAT3 was detected by Western blotting. PBS-incubated DCs served as an uninfected control (un). (A) Phospho-STAT3-2 (Ser727) activation at 2 or 4 h postinfection; (B) phospho-STAT3-1 (Tyr705) activation at 2 or 4 h postinfection.
DISCUSSION
The current study was conducted to enhance our understanding of the early immune responses induced by C. jejuni during infection. The data now available suggest that Campylobacter induces different chemokine profiles when infecting different cell types in humans or animals. In previous studies employing human intestinal epithelial cell lines, Campylobacter infection upregulated several chemokines, including IL-8, GRO-α, MCP-1, and MIP-1α (12, 14). In the current study, MIP-1α, MIP-1β, and RANTES were induced dramatically, and MCP-1 was induced only slightly in human DCs. Further, the titers of MIP-1α and MIP-1β induced in DCs were much higher than the levels observed previously in an infected epithelial cell line (14). The receptors for MIP-1α, MIP-1β, and RANTES are CCR1 and CCR5, which are present on monocytes and T cells (16). Therefore, these data suggest that DCs play important roles in attracting monocytes/macrophages, NK cells, and particularly T cells (via secretion of MIP-1α, MIP-1β, and RANTES) during the early stages of C. jejuni infection, perhaps allowing them to initiate a Th1-directed immune response, as suggested earlier (13). Similarly, IP-10 and MIG also favor the Th1 immune response by interacting with the receptor CXCR3, found primarily on Th1 cytokine-producing effector T cells (10), thereby establishing a positive-feedback regulatory loop to maintain the residence of the effector T-cell population at the site of infection (16). Interleukin-8 upregulation by DCs, shown previously (13), likely enlists PMNs to the site of infection. GRO-α also presumably attracts neutrophils to the site of infection. These results correlate with the clinical observation of infiltration of the lamina propria with monocytes and neutrophils during Campylobacter infection (31).
Heat-killed 81-176 cells induced only slightly lower chemokine levels than live 81-176 cells, and this difference was not statistically significant. Live Campylobacter isolates 11168 and M129 induced statistically similar levels of chemokine production as strain 81-176. Additionally, in our previous study, the noninvasive C. jejuni 2× CheY mutant, RY213, induced slightly lower levels of IL-8 and other cytokines than the wild type, but these differences were not significant (13). These data, together with our previous studies (13), suggest that there are no large differences in the quantities of chemokines and cytokines induced by heat-killed versus live C. jejuni. In fact, LOS may be largely responsible for the chemokine induction observed in the current study.
To understand the signal transduction pathways activated during Campylobacter infection of DCs, we examined MAP kinases, NF-κB, and STAT3 activation by Western blotting, RT-PCR with specific inhibitors, and indirect immunofluorescence assays. We demonstrated here the increased expression of mRNA for MIP-1α, MIP-1β, RANTES, GRO-α, IP-10, and MIG in DCs infected with Campylobacter. Further, we showed that chemokine upregulation was correlated with phosphorylation of MAP kinases and activation of NF-κB and STAT3.
Three MAP kinases were observed to be increased in phosphorylation in DCs treated with either live or heat-killed C. jejuni or with LOS. The phosphorylation of p38 and SAPK/JNK was more significantly increased after infection with live C. jejuni 81-176 at an MOI of 100, in comparison to other treatments. These results might indicate that live Campylobacter cells induce the phosphorylation of p38 and SAPK/JNK not only through LOS but also by the abilities of live bacteria to secrete protein or to adhere to and/or to invade DCs.
Expression of mRNA for MIP-1α, MIP-1β, RANTES, IP-10, and MIG was significantly reduced in infected DCs pretreated with an NF-κB inhibitor compared to that in nontreated cells. Similar to previous studies in human epithelial cell lines (14, 16), these data suggest that transcription of these chemokine genes proceed largely through the activation of NF-κB. The incomplete inhibition for some of these chemokines at particular time points, however, might be due to the activity of other compensatory transcription factors (14).
The indirect immunofluorescence assay showed that both Campylobacter cells (at an MOI of 100) and LOS induced a similar percentage of DCs showing NF-κB activation. However, more intense fluorescence was observed in DCs infected with live Campylobacter at both MOIs of 10 and 100, compared with those treated with LOS or LPS. The data suggest that live bacterial activities (such as protein secretion and adherence to and invasion into human DCs) might play additional roles in NF-κB activation in DCs. Interestingly, higher titers (0.1 and 1.0 μg/ml) of either Campylobacter LOS or E. coli LPS induced similar levels of NF-κB activation in DCs as seen with the 0.01-μg/ml treatment dose (data not shown). These results might reflect the dynamics of the 2 h of incubation time (i.e., NF-κB activation may not have reached its maximum or may have peaked and declined at higher LOS doses) or that induction is saturated at all doses.
Among seven STATs, STAT3 has emerged as an important regulator of T cell differentiation, as well as of survival versus apoptosis (9, 18). Activated STAT3 in DCs results in a “tolerogenic” outcome that may be important in limiting the host inflammatory response in the gut. STAT3 can be activated by several cytokines, including IL-6 and IL-11. Increased activation of STAT3 is observed at early time points in mouse macrophages following infection with Salmonella (20). In another study, Helicobacter pylori activated the STAT3 signaling pathway in human epithelial cells, and this activation was independent of IL-6 or IL-11 (5). Finally, during E. coli-induced meningitis, phosphorylated STAT3 interacts with the C-terminal domain of EC-gp96 to initiate bacterial invasion of human brain microvascular endothelial cells (22). Rapid nuclear translocation and phosphorylation of STAT3 at Ser727 via MAPK or mammalian target of rapamycin (mTOR) pathways are required for optimal transcriptional activation in response to growth factors and cytokines, including IFN-γ (37). STAT3 can be activated by IL-6 and other chemokines, followed by activation of the gp130 receptor on single tyrosine residues (Tyr705). Our STAT3 phosphorylation results suggest that STAT3 activation in DCs following Campylobacter infection is IL-6 independent and might occur via the MAPK pathway.
In summary, we have demonstrated significant upregulation of MIP-1α, MIP-1β, RANTES, GRO-α, IP-10, and MIG from DCs, via MAPK, NF-κB, and STAT3 activation during Campylobacter infection of human DCs. These results together with our previous report of increased proinflammatory cytokines by Campylobacter-infected DCs suggest that DCs play significant roles in the initiation and modulation of the inflammatory response by enlisting monocytes, neutrophils, and T lymphocytes during Campylobacter infection. It would appear that Campylobacter invasion of the colonic epithelium and stimulation of expression of proinflammatory chemokines/cytokines and chemokine receptors in epithelial cells and DCs contribute to the pathogenesis and clinical symptoms of Campylobacter infection, as well as represent the early innate/adaptive immune responses to infection.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge T. T. Wai for help in preparing bacterial cultures and Manuel Osorio for help with the Luminex 100 analyzer. We thank Mustafa Akkoyunlu and Kensuke Shima for critical review of the manuscript.
Footnotes
Published ahead of print 11 June 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Baggiolini M. 1998. Chemokines and leukocyte traffic. Nature 392:565–568 [DOI] [PubMed] [Google Scholar]
- 2. Baggiolini M, Dewald B, Moser B. 1997. Human chemokines: an update. Annu. Rev. Immunol. 15:675–705 [DOI] [PubMed] [Google Scholar]
- 3. Bilsborough J, Viney JL. 2004. Gastrointestinal dendritic cells play a role in immunity, tolerance, and disease. Gastroenterology 127:300–309 [DOI] [PubMed] [Google Scholar]
- 4. Black RE, Levine MM, Clements ML, Hughes TP, Blaser MJ. 1988. Experimental Campylobacter jejuni infection in humans. J. Infect. Dis. 157:472–479 [DOI] [PubMed] [Google Scholar]
- 5. Bronte-Tinkew DM, et al. 2009. Helicobacter pylori cytotoxin-associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo. Cancer Res. 69:632–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cella M, et al. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nature Med. 5:919–923 [DOI] [PubMed] [Google Scholar]
- 7. Charo IF, Ransohoff RM. 2006. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354:610–621 [DOI] [PubMed] [Google Scholar]
- 8. Coker AO, Isokpehi RD, Thomas BN, Amisu KO, Obi CL. 2002. Human campylobacteriosis in developing countries. Emerg. Infect. Dis. 8:237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Darnell JE., Jr 1997. STATs and gene regulation. Science 277:1630–1635 [DOI] [PubMed] [Google Scholar]
- 10. Dwinell MB, Lügering N, Eckmann L, Kagnoff MF. 2001. Regulated production of interferon-inducible T-cell chemoattractants by human intestinal epithelial cells. Gastroenterology 120:49–59 [DOI] [PubMed] [Google Scholar]
- 11. Förster R, et al. 1999. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99:23–33 [DOI] [PubMed] [Google Scholar]
- 12. Hickey TE, Baqar S, Bourgeois AL, Ewing CP, Guerry P. 1999. Campylobacter jejuni-stimulated secretion of interleukin-8 by INT407 cells. Infect. Immun. 67:88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu L, Bray MD, Osorio M, Kopecko DJ. 2006. Campylobacter jejuni induces maturation and cytokine production in human dendritic cells. Infect. Immun. 74:2697–2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu L, Hickey TE. 2005. Campylobacter jejuni induces secretion of proinflammatory chemokines from human intestinal epithelial cells. Infect. Immun. 73:4437–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu L, McDaniel JP, Kopecko DJ. 2006. Signal transduction events involved in human epithelial cell invasion by Campylobacter jejuni 81-176. Micro. Pathogen. 40:91–100 [DOI] [PubMed] [Google Scholar]
- 16. Johanesen PA, Dwinell MB. 2006. Flagellin-independent regulation of chemokine host defense in Campylobacter jejuni-infected intestinal epithelium. Infect. Immun. 74:3437–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kraft S, Novak N, Katoh N, Bieber T, Rupec RA. 2002. Aggregation of the high-affinity IgE receptor FcεRI on human monocytes and dendritic cells induces NF-κB activation. Soc. Invest. Derm. 118:830–837 [DOI] [PubMed] [Google Scholar]
- 18. Leonard WJ, O'Shea JJ. 1998. Jaks and stats: biological implications. Annu. Rev. Immunol. 16:293–322 [DOI] [PubMed] [Google Scholar]
- 19. Liao F, Rabin R, Farber JM. 1995. Human mig chemokine: biochemical and functional characterization. J. Exp. Med. 182:1301–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin T, Bost KL. 2004. Stat3 activation in macrophages following infection with Salmonella. Biochem. Biophy. Res. Commun. 321:828–834 [DOI] [PubMed] [Google Scholar]
- 21. Martin-Fontecha A, et al. 2003. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J. Exp. Med. 198:615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maruvada R, Argon Y, Prasadarao NV. 2008. Escherichia coli interaction with human brain microvascular endothelial cells induces signal transducer and activator of transcription 3 association with the C-terminal domain of Ec-gp96, the outer membrane protein A receptor for invasion. Cell. Microbiol. 10:2326–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maurer M, von Stebut E. 2004. Macrophage inflammatory protein-1. Intern. J. Biochem. Cell Bio. 36:1882–1886 [DOI] [PubMed] [Google Scholar]
- 24. Mishu B, et al. 1993. Serologic evidence of previous Campylobacter jejuni infection in patients with the Guillain-Barré syndrome. Ann. Intern. Med. 118:947–953 [DOI] [PubMed] [Google Scholar]
- 25. Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. 1996. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. U. S. A. 93:9090–9095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nelson PJ, Krensky AM. 1998. Chemokines, lymphocytes and viruses: what goes around, comes around. Curr. Opin. Immunol. 10:265–270 [DOI] [PubMed] [Google Scholar]
- 27. Rissoan MC, et al. 1999. Reciprocal control of T helper cell and dendritic cell differentiation. Science 283:1183–1186 [DOI] [PubMed] [Google Scholar]
- 28. Sakazar-Gonzalez RM, et al. 2006. CCR6-mediated dendritic cell activation of pathogen-specific T cells in Peyer's patches. Immun. 24:623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schall TJ, Bacon K, Toy KJ, Goeddel DV. 1990. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 347:669–671 [DOI] [PubMed] [Google Scholar]
- 30. Schluger NW, Rom WN. 1997. Early responses to infection: chemokines as mediators of inflammation. Curr. Opin. Immunol. 9:504–508 [DOI] [PubMed] [Google Scholar]
- 31. Skirrow MB, Blaser MJ. 2002. Campylobacter jejuni, p 719–739 In Blaser MJ, Smith PD, Raving JI, Greenberg HB, Guerrant RL. (ed), Infections of the gastrointestinal tract, 2nd ed Lippincott Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 32. Tam CC, et al. 2006. Incidence of Guillain-Barré syndrome among patients with Campylobacter infection: a general practice research database study. J. Infect. Dis. 194:95–97 [DOI] [PubMed] [Google Scholar]
- 33. Taub DD, et al. 1993. Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes adhesion to endothelial cells. J. Exp. Med. 177:1809–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33a. Tribble, et al. 2010. Assessment of the duration of protection in Campylobacter jejuni experimental infection in humans. Infect. immun. 78:1750–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vieira PL, de Jong EC, Wierenga EA, Kapsenberg ML, Kaliński P. 2000. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J. Immunol. 164:4507–4512 [DOI] [PubMed] [Google Scholar]
- 35. Wassenaar TM, Blaser MJ. 1999. Pathophysiology of Campylobacter jejuni infections of humans. Microbes Infect. 1:1023–1033 [DOI] [PubMed] [Google Scholar]
- 36. Watson RO, Galán JE. 2005. Signal transduction in Campylobacter jejuni-induced cytokine production. Cell. Microbiol. 7:655–665 [DOI] [PubMed] [Google Scholar]
- 37. Wen Z, Zhong Z, Darnell JE., Jr 1995. Maximal activation of transcription by stat1 and stat3 requires both tyrosine and serine phophorylation. Cell 82:241–250 [DOI] [PubMed] [Google Scholar]
- 38. Westphal O, Jann K. 1965. Methods in carbohydrate chemistry, vol 5, p 83 Academic Press, New York, NY [Google Scholar]
- 39. Wuyts WA, Vanaudenaerde BM, Dupont LJ, Demedts MG, Verleden GM. 2003. Involvement of p38 MAPK, JNK, p42/p44 ERK and NF-kappaB in IL-1beta-induced chemokine release in human airway smooth muscle cells. Respir. Med. 97:811–817 [DOI] [PubMed] [Google Scholar]
- 40. Yang S-K, Eckmann L, Panja A, Kagnoff MF. 1997. Differential and regulated expression of C-X-C, C-C, and C-chemokines by human colon epithelial cells. Gastroenterology 113:1214–1223 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.