

Abstract
The cytolethal distending toxin (Cdt), produced by some clinically important Gram-negative bacterial species, is related to the family of AB-type toxins. Three heterologous proteins (CdtA, CdtB, and CdtC) and a genotoxin mode of action distinguish the Cdt from others in this toxin class. Crystal structures of several species-specific Cdts have provided a basis for predicting subunit interactions and functions. In addition, empirical studies have yielded significant insights into the in vivo interactions of the Cdt subunits. However, there are still critical gaps in information about the intoxication process. In this study, a novel protein tagging technology was used to localize the subunits in Chinese hamster ovary cells (CHO-K1). A tetracysteine motif was engineered in each subunit, and in subunits with mutations in predicted functional domains, to permit detection with the fluorescein arsenical hairpin binding (FlAsH) dye Lumio green. Live-cell imaging, in conjunction with confocal microscopy, was used to capture the locations of the individual subunits in cells intoxicated, under various conditions, with hybrid heterotrimers. Using this approach, we observed the following. (i) The CdtA subunit remains on the cell surface of CHO cells in association with cholesterol-containing and cholesterol-depleted membrane. (ii) The CdtB subunit is exclusively in the cytosol and, after longer exposure times, localizes to the nucleus. (iii) The CdtC subunit is present on the cell surface and, to a greater extent, in the cytosol. These observations suggest that CdtC, but not CdtA, functions as a chaperone for CdtB entry into cells.
INTRODUCTION
The cytolethal distending toxin (Cdt) is produced by a select number of disease-associated Gram-negative bacterial species, including the oral pathogen Aggregatibacter actinomycetemcomitans (32). This group of toxins has been associated with cell distention, cell cycle arrest in either the G1/S or G2/M transition phase, and/or apoptosis in various mammalian cell types (18).
The Cdt belongs to a general class of AB-type toxins but has several unique features. AB-type toxins typically contain a binding subunit that functions as a dimer in a lectin-like mechanism to attach the toxin to the host cell membrane. A second toxic subunit disrupts an essential internal process or pathway in the infected cell, leading to the inhibition of proliferation or cell death. In contrast, the Cdt contains two heterologous subunits (CdtA and CdtC) that, in theory, form a dimer that attaches the toxin to the cell membrane. The toxic third subunit (CdtB) potentially has multiple activities based on its phylogenetic relationship to a superfamily of enzymes that includes the endonucleases, exonucleases, sphingomyelinases, and inositol polyphosphate 5-phosphatases (15). However, the general consensus is that the primary in vivo action of the Cdt is that of a genotoxin (8, 9, 19, 23, 32). The result of intoxication with the Cdt is cell cycle arrest, leading to the more definitive classification of this group of toxins as cyclomodulins (31, 33).
The specific mechanism by which the toxin enters cells and carries out its genotoxic mission is probably the least understood property, at the molecular level, of the Cdt. CdtA and CdtC appear to function cooperatively in binding of the heterotrimer to target cell membranes (4, 16, 17, 23, 28). Aromatic side chains, clustered on the surface of CdtA, promote cell recognition and, together with a groove formed at the junction of a CdtA and CdtC heterodimer, contribute to binding to a specific receptor (4, 27). A cholesterol recognition/interaction amino acid consensus (CRAC) motif identified in the A. actinomycetemcomitans CdtC may explain an association of the toxin with lipid rafts in certain types of cells (3, 10).
Circumstantial evidence to date implies that CdtC may play dual accessory roles in binding and internalization of the CdtB subunit (1, 23). A theoretical model suggests that the heterotrimer complex is internalized by dynamin-dependent endocytosis associated with outer membrane vesicles, containing lipid rafts, into early and late endosomes (11). The CdtB subunit is subsequently passed to the Golgi complex and then transported, in a retrograde fashion, to the endoplasmic reticulum (ER) lumen, from which it is translocated directly to the nucleoplasm (12, 13). CdtB translocation to the nucleus does not appear to require the ER-associated degradation pathway. Nuclear localization signal (NLS) sequences at either the amino- or carboxy-terminal end of CdtB are thought to provide the mechanism by which the subunit traverses the nuclear membrane (25, 30). Prior translocation studies have relied on the fusion of the CdtB protein to glutathione S-transferase (GST) and detection with fluorescein isothiocyanate (FITC)-conjugated GST antibody or immune-labeling of His6-tagged proteins in standard confocal microscopy.
To further address questions about the fate of the Cdt subunits, especially CdtC, during the intoxication process in a model cell system, we used a fluorescein arsenical hairpin binding (FlAsH) technology. Fluorescent biarsenic dye molecules have distinct advantages over the use of large fusion proteins and allow greater flexibility in designing the labeling site. Using this approach, along with live-cell imaging, we were able to determine the location of each of the wild-type Cdt subunits in toxin-treated cells and to assess the effects of mutations, in predicted functional domains in the subunits, on the intoxication process. Based on the results of these studies, a modified model of Cdt intoxication is proposed.
MATERIALS AND METHODS
Chemicals and reagents.
Except where noted, all reagents were purchased from Invitrogen (Carlsbad, CA). All PCR primer oligonucleotides were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA).
Preparation of wild-type and mutated constructs.
Template DNA for mutagenesis was obtained from the A. actinomycetemcomitans cdtA, cdtB, and cdtC genes individually cloned with a His tag in pET15b (Table 1). A tetracysteine motif, Cys-Cys-Pro-Gly-Cys-Cys (5′-TGTTGTCCTGGCTGTTGC-3′), and spacer sequence, Gly-Ala-Gly-Gly (5′- GGTGCTGGTGGC-3′), were added to each of the subunit genes using the GeneTailor site-directed mutagenesis system. Reactions were carried out as described in the manufacturer's instructions. Two consecutive rounds of mutagenesis were performed to insert the sequences, resulting in CdtALum, CdtBLum, and CdtCLum. DNA from these clones was used as a template to generate subsequent site-specific mutations (Table 1). A binding-deficient subunit, CdtALum, Y214A, was constructed based on the results of a previous study (5). The mutated subunits CdtBLum, CH-1, CdtBLum, CH-2, CdtBLum, A9-3, and CdtBLum, A10-2 were designed to alter catalytic activity, a DNA contact domain, and divalent cation-binding site, respectively (9, 34). CdtBLum, NLS1 and CdtBLum, NLS2 represent mutations in the putative N- and C-terminal NLS reported by Nishikubo et al. (30) and McSweeney and Dreyfus (25), respectively. Amino acids at positions 109 to 112, within the loop insertion between β-sheets 4 and 5, and at positions 141 to 144, within the loop insertion between β-sheets 7 and 8, were altered in mutants CdtCLum, R1 and CdtCLum, R2, respectively (16). CdtCLum, AT was constructed in an attempt to disrupt possible intermolecular β-sheet interactions between CdtC and the hydrophobic C terminus of CdtA (16). All mutations were verified by DNA sequencing (University of Pennsylvania DNA Sequencing Facility).
Table 1.
Plasmids and oligonucleotide primers used in this study
| Plasmid/mutant | Primer sequenceb | Features | Source |
|---|---|---|---|
| pET15bcdtA | NAa | cdtA gene containing 6 His codons at 5′ end | 23 |
| pET15bcdtB | NA | cdtB gene containing 6 His codons at 5′ end | 23 |
| pJDA9 | NA | cdtA gene containing 6 His codons at 3′ end | 5 |
| pJDB7 | NA | cdtB gene containing 6 His codons at 3′ end | 5 |
| pJDC2 | NA | cdtC gene containing 6 His codons at 3′ end | 5 |
| pJDB7 (CdtBH160A) | NA | His at position 160 in active site of CdtB changed to Ala | 8 |
| CdtALum | 5′-aagcaacagcggttaattgttgtcctggctgttgctaagctggatcc-3′ (forward) | Add Lumio binding site CCPGCC at C terminus of CdtA in pET15cdtA | This study |
| 5′-aagcaacagcggttaatggtgctggtggctgttgtcctg-3′ (forward) | Add amino acid linker GAGG at C terminus of CdtA in pET15cdtA | ||
| 5′-attaaccgctgttgcttctaataca-3′ (reverse) | |||
| CdtALum, Y214A | 5′-cctgtaagtccaacatatgcttcaacagttac-3′ (forward) | Change aromatic binding site residue Y214A in CdtALum | This study |
| 5′-atgttggacttacaggattataagtgac-3′ (reverse) | |||
| CdtBLum | 5′-agttttgttcatgatcgctgtcctggctgttgctaaggaggatcc-3′ (forward) | Add Lumio binding site CCPGCC at C terminus of CdtB in pET15cdtB | This study |
| 5′-agtttgttcatgatcgcggtgctggtggctgttgtcctggc-3′ (forward) | Add amino acid linker GAGG at C terminus of CdtB in pET15cdtB | ||
| 5′-gcgatcatgaacaaaactaacaggaaaatg-3′ (reverse) | |||
| CdtBLum, CH-1 | 5′-tgtattttttacagtggctgctttggcc-3′ (forward) | Change catalytic site residue H187A in CdtBLum | This study |
| 5′-cactgtaaaaaatacatcagtaccaatgcgg-3′ (reverse) | |||
| CdtBLum, A9-1 | 5′-gcaacttggaatctggcaggttcttcagct-3′ (forward) | Change predicted DNA contact residue Q62A in CdtBLum | This study |
| 5′-cagattccaagttgctactttgaaatcactca-3′ (reverse) | |||
| CdtBLum, A9-2 | 5′-ttgttggtgatttcgctcgtgcgccggttaat-3′ (forward) | Change predicted DNA contact residue N228A in A9-1 | This study |
| 5′-gaaatcaccaacaaccatccagctatat-3′ (reverse) | |||
| CdtBLum, A9-3 | 5′-tccggtaatattttagatgctgcgattttaca-3′ (forward) | Change predicted DNA contact residue Y266A in A9-2 | This study |
| 5′-atctaaaatattaccggactgatgagtcggtt-3′ (reverse) | |||
| CdtBLum, A10-1 | 5′-tggatggttgttggtgctttcaatcgt-3′ (forward) | Change predicted DNA Mg2+ binding residue D226A in CdtBLum | This study |
| 5′-caccaacaaccatccagctatatcctcgt-3′ (reverse) | |||
| CdtBLum, A10-2 | 5′-tcacaaattacatccgctcattttcct-3′ (forward) | Change predicted DNA Mg2+ binding residue D300A in A10-1 | This study |
| 5′-cggatgtaatttgtgagcgtaactgatt-3′ (reverse) | |||
| CdtBLum, NLS1 | 5′-ttagctatcgtgtcaagtactcaagccgatg-3′ (forward) | Change NLS residues R152S and R153T in CdtBLum | This study |
| 5′-tgacacgatagctaagttcactcggtttg-3′ (reverse) | |||
| CdtBLum, NLS2 | 5′-tcaccatcatcaccggaaagtactggatatagctgg-3′ (forward) | Change NLS residues R216S and R217T in CdtBLum | This study |
| 5′-ttccggtgatgatggtgatgaggtaaaag-3′ (reverse) | |||
| CdtCLum | 5′-taaggagaatactatgtgttgtcctggctgttgcaaaaaatattta-3′ (forward) | Add Lumio binding site CCPGCC at N terminus of CdtC in pJDC2 | This study |
| 5′-ttgtcctggctgttgcggtgctggtggcaaaaaatattta-3′ (forward) | Add amino acid linker GAGG at N terminus of CdtC in pJDC2 | ||
| 5′-catagtattctccttagcgaccatggtatat-3′ (reverse) | |||
| CdtCLum, R1 | 5′-agcaaaatgttttgctgcagcaggagcaggcacaact-3′ (forward) | Change predicted receptor binding residues F109A, P110A, and E112A in CdtCLum | This study |
| 5′-agcaaaacattttgctgcaccgaccactttaa-3′ (reverse) | |||
| CdtCLum, R2 | 5′-ttaatcaaagatgccgcagcaggagcttgtatgacaa-3′ (forward) | Change predicted receptor binding residues L141A, L142A, and F144A in CdtCLum | This study |
| 5′-ggcatctttgattaaaaacgcacctgtattagttgga-3′ (reverse) | |||
| CdtCLum, AT | 5′- gatattgatcatactgcgtttaactctattgcaactaatacagg-3′ (forward) | Change residues V124A, L127S, and P129A in CdtCLum predicted to interact with an α-helix in CdtB | This study |
| 5′-cagtatgatcaatatctttacaatcagttgtgcc-3′ (reverse) |
NA, not applicable.
Underlined bases represent mutated nucleotides.
Expression of recombinant toxin subunits and holotoxin reconstitution.
Recombinant clones Escherichia coli BL21(DE3)(pJDA9), E. coli BL21(DE3)(pJDB7), and E. coli BL21(DE3)(pJDC2) were used to prepare the standard CdtA-His6, CdtB-His6, and CdtC-His6 proteins, respectively (5). All Cdt subunit proteins contain a His6 tag at either the amino- or carboxy-terminal end of the protein. Proteins were isolated from isopropyl-β-d-thiogalactopyranoside (IPTG)-induced cultures by affinity chromatography on nickel-iminodiacetic acid columns (Novagen-EMD Biosciences, San Diego, CA) as described previously (23). The average yields were 40 to 50 μg of protein/ml of culture. The final protein preparations were dialyzed to remove urea while promoting protein refolding, passed through 45-μm filters, and quantified with a Micro BCA protein assay kit (Pierce, Rockford, IL) as described previously (5). Purity was assessed by analysis on 4 to 12% bis-Tris gels stained with Coomassie brilliant blue. Aliquots of the quantified protein samples were stored at −70°C in a buffer containing 10 mM Tris-HCl (pH 7), 100 mM NaCl, 5 mM MgCl2, and 5 mM imidazole.
Wild-type and mutant heterotoxins were reconstituted, in vitro, by mixing equimolar concentrations of the appropriate subunit proteins in a buffer containing 10 mM Tris-HCl (pH 7), 100 mM NaCl, and 5 mM MgCl2. The mixtures were incubated for 1 h at 4°C and used immediately.
Heterotoxin assembly.
The ability of combinations of wild-type and mutated subunit proteins to form a stable heterotoxin complex was determined by differential dialysis as described and validated previously (5). Each mutant protein was substituted for wild-type CdtA, CdtB, or CdtC in the reconstitution preparation. Following dialysis, each dialyzed sample was analyzed by Western blotting. Retained proteins were detected with anti-His tag monoclonal antibody or subunit-specific rabbit polyclonal antibodies (4). These experiments were performed twice.
Cell cycle.
For cell cycle analysis by flow cytometry (6), Chinese hamster ovary (CHO-K1) cells were grown to 75% confluence in Ham's F-12 medium supplemented with 7.5% fetal bovine serum and 1% antibiotic-antimycotic solution at 37°C in 5% CO2. Reconstituted heterotoxin preparations, containing various combinations of wild-type and mutated subunits, were then added to cultures at a concentration of 10 μg (120 nM) of total protein/ml of medium. Controls consisted of no toxin and toxin made with CdtBH180A (a biologically inactive subunit) and wild-type CdtA and CdtC. After 36 h of exposure, nuclei from 1 × 106 cells were stained with propidium iodide and analyzed on a FACSCalibur flow cytometer at the University of Pennsylvania Cancer Center Flow Cytometry and Cell Sorting Shared Resource Facility. The data collected from 30,000 events, from two independent Cdt-treated cultures, were analyzed using ModFit 3.0 (Verity Software House, NH).
Cell proliferation.
A standard colony formation assay (6) was performed in triplicate to assess the ability of the various reconstituted heterotoxin preparations to inhibit cell proliferation. CHO-K1 cells were plated at a density of 350 cells per well in 6-well plates containing Ham's F-12 medium supplemented with 7.5% fetal bovine serum and 1% antibiotic-antimycotic solution at 37°C in 5% CO2. Separated cultures were treated with increasing concentrations of heterotrimer complex reconstituted with various combinations of wild-type and mutated subunits. Control wells contained either no toxin or toxin made with CdtBH160A and wild-type CdtA and CdtC. Following 5 to 6 days of incubation, colonies were fixed and stained with crystal violet. Cell counts were expressed as numbers of CFU.
Kinetics of CdtA binding to cells.
Wild-type and mutated CdtALum subunits were assessed for their ability to bind to cells in an enzyme-linked immunosorbent assay (ELISA) essentially as described previously (5). CHO-K1 cells were seeded in standard culture medium at a density of 1.5 × 104 per well in 96-well plates. After 48 h of growth, the confluent cultures were washed, fixed, and exposed, in triplicate wells, to various concentrations of purified CdtA, CdtALum, or CdtALum, Y214A in phosphate-buffered saline (PBS) containing 3% bovine serum albumin. One set of triplicate wells received no toxin and served as a negative control. After 1 h of incubation at room temperature, the plates were washed with PBS containing 0.1% Tween 20 and probed with a 1:10,000 dilution of rabbit anti-CdtA polyclonal antibody (4), followed by a 1:3,000 dilution of anti-rabbit IgG-horseradish peroxidase conjugate (GE Healthcare, Piscataway, NJ) and 100 μl/well of the horseradish peroxidase substrate ABTS-100 (Rockland, Gilbertsville, PA). The absorbance was measured at 405 nm in a Synergy 2 microplate reader (BioTek Instruments, Inc., Winooski, VT).
Fluorescent detection of subunit proteins using the biarsenical compound Lumio green.
CHO-K1 cells were plated at a density of 1.5 × 105 cells in Delta TPG 0.17-mm (black) dishes (Thermo Scientific) containing F-12 medium supplemented with 7.5% fetal calf serum and 1% antibiotic-antimycotic solution. Cells were allowed to attach to the dishes overnight at 37°C in 5% CO2. Subconfluent cultures were treated for various amounts of time with 0, 2.5, 5, 10, and 20 μg of reconstituted toxin (total protein) per ml of medium. For certain experiments, cholesterol associated with the CHO-K1 cell membrane was depleted by treatment of the cells with 10 mM methyl-β-cyclodextrin (MβCD) (20) for 1 h prior to the addition of toxin. The concentration of MβCD was maintained at 1 mM for the duration of the experiment. Following incubation, the cells were washed with Hanks' balanced salt solution (HBSS) and the Lumio green labeling reagent was added at a concentration of 2.5 μM in HBSS for 30 min at 37°C in the dark. In some cases, the cells were colabeled with a 1:1,000 dilution of wheat germ agglutinin (WGA)-Alexa Fluor 555 conjugate. After being washed with HBSS, the cells were covered with Disperse blue 3 solution prepared according to the manufacturer's protocol. Cell images were taken on a Bio-Rad Radiance 2100 confocal microscope (Bio-Rad, Hercules, CA) using a 60× objective and the same laser settings for each sample. High-resolution, high-contrast live-cell images were obtained with a Nikon A1R laser scanning confocal microscope using a 60× objective. Representative images were analyzed using Nikon Elements advanced research software, package 3.2 (Nikon Instruments Inc., Melville, NY).
To quantify the amount of cholesterol depleted from the CHO-K1 cell membrane following MβCD treatment, additional cell cultures were grown to subconfluence in T75 flasks and exposed to 10 mM MβCD for 1 h. MβCD-treated and untreated control cells were harvested, and cell membrane-associated lipid rafts were isolated according to a published protocol (2). The cholesterol concentration was determined using the Amplex red cholesterol assay kit according to the manufacturer's instructions. Cholesterol levels were expressed as μg of cholesterol per 1 μg of total protein as determined using the Micro BCA protein assay kit (Thermo Scientific, Rockford, IL).
Isolation of nuclei.
CHO-K1 cells (1 × 106) were plated per T75 flask and allowed to attach overnight. The cells were then treated for 48 h with 10 μg/ml of reconstituted heterotrimer containing various combinations of wild-type and mutated Cdt subunits. One set of cells received only the reconstitution buffer. Following incubation, the cells were washed and collected by centrifugation. Cell pellets were suspended in 5 ml of ice-cold hypotonic buffer (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM dithiothreitol [DTT]) and kept on ice for 10 min. The cells were then broken open to release nuclei using 10 strokes with a tight pestle in a 7-ml Dounce homogenizer. The preparations were centrifuged at 228 × g (Sorvall centrifuge, H-1000B rotor) for 5 min at 4°C. Nuclear pellets were suspended in 3 ml of 0.25 M sucrose containing 10 mM MgCl2 and layered over a 3-ml cushion of 0.88 M sucrose containing 0.5 mM MgCl2. The samples were centrifuged at 2,135 × g for 10 min at 4°C. Gradient fractions containing the nuclei were thoroughly washed and labeled with a 1:1,000 dilution of Lumio green labeling reagent for 30 min at 37°C in the dark. The labeled nuclei were washed and suspended in Disperse blue 3. A drop of each sample was placed on a glass slide and mounted using Pro-Long Gold with 4′,6-diamidino-2-phenylindole (DAPI). The nuclei were visualized immediately using a Bio-Rad 2000 confocal microscope and a 60× objective.
Statistical methods.
Mean values and standard deviations were plotted where appropriate. The t test was used to predict if experimental values were significantly different from the control values.
RESULTS
The addition of tetracysteine and spacer motifs to Cdt subunits does not alter the cytotoxicity of reconstituted holotoxin.
To track the fate of each of the three Cdt subunits during the intoxication process, a tetracysteine motif that binds the Lumio green reagent and a spacer sequence that enhances binding were engineered into either the amino- or carboxy-terminal end of each protein (Fig. 1A). Each of the modified subunits formed a heterotrimer when combined with the corresponding wild-type subunits as verified in a differential dialysis assay. An increase in the number of CHO cells having a 4N DNA content and the extent of inhibition of CHO cell proliferation in response to the toxin preparations were determined using independent assays. The results confirmed that heterotrimers reconstituted using combinations of wild-type proteins and each modified subunit protein had activity comparable to that of the wild-type heterotoxin (Table 2).
Fig 1.

Summary of genetic modifications. (A) Positions of amino acid additions and substitutions (boxed residues) in recombinant A. actinomycetemcomitans CdtA-His6, CdtB-His6, and CdtC-His6. The locations of the histidine and tetracysteine tags and active site in CdtB are underlined. The spacer sequence to improve Lumio green labeling is in italics. Predicted NLSs (25, 30) are in labeled boxes. The location where the wild-type amino acid sequence begins is marked with an arrow. (B) Ribbon backbone structure of the A. actinomycetemcomitans Cdt showing the locations of the mutated sites designated in panel A and in Table 1. The structure was modeled in Chimera 1.6 using Protein Data Bank file accession number 2F2F (37). Side chains are shown only for the amino acids targeted for mutation in sites AD1 in CdtA and AT, R1, and R2 in CdtC. Amino acids designated for mutation in sites A9, A10, and CH-1 in CdtB cluster around a catalytic center marked by a thin orange polygon. Amino acids targeted for mutation in the two NLS sites align along the back side of the structure as designated by the thin green line. (C) The same structure as shown in panel B except that CdtA and CdtC are depicted as a surface model and the heterotrimer is rotated approximately 90° vertically. Only the surface-exposed sites AD1, R1, and R2 are labeled.
Table 2.
Biological activity of toxin reconstituted with the mutated Cdt subunits
| Subunit composition of the Cdt | % of CHO cells that were diploid in: |
CHO cell proliferationa (% of surviving CFU) | |||
|---|---|---|---|---|---|
| G1 (2N DNA content) | G2 (4N DNA content) | S | G2/G1 | ||
| None | 65.48 | 14.84 | 19.68 | 0.23 | 100 ± 5 |
| CdtABC | 30.36 | 53.10 | 16.46 | 1.75 | 0 |
| CdtABH160AC | 78.40 | 0.02 | 21.58 | 0 | 99 ± 2 |
| CdtALumBC | 27.01 | 48.34 | 24.65 | 1.79 | 8 ± 14 |
| CdtABLumC | 19.51 | 63.96 | 16.53 | 3.27 | 10 ± 10 |
| CdtABCLum | 17.45 | 66.85 | 15.70 | 3.83 | 5 ± 6 |
| CdtALum, Y214ABC | 48.90 | 26.37 | 24.73 | 0.54 | 70 ± 9 |
| CdtABLum, CH-1 | 77.71 | 3.77 | 18.53 | 0.05 | 98 ± 1 |
| CdtABLum, A9-3C | 55.76 | 22.41 | 21.83 | 0.41 | 96 ± 3 |
| CdtABLum, A10-2C | 60.62 | 19.72 | 19.67 | 0.33 | 100 ± 6 |
| CdtABLum, NLS1C | 42.11 | 34.97 | 22.92 | 0.83 | 6 ± 5 |
| CdtABLum, NLS2C | 45.99 | 35.44 | 18.57 | 0.77 | 83 ± 5 |
| CdtABCLum, R1 | 46.12 | 28.48 | 25.40 | 0.62 | 0 |
| CdtABCLum, R2 | 42.46 | 31.82 | 25.72 | 0.75 | 5 ± 27 |
| CdtABCLum, AT | 75.68 | 1.84 | 22.49 | 0.02 | 95 ± 2 |
| None + MβCD | 48.24 | 17.72 | 34.04 | 0.37 | ND |
| CdtABC + MβCD | 57.36 | 0 | 42.64 | 0 | ND |
CHO cells (350 cells/well) exposed to 5 μg/ml of total Cdt protein. Cells were counted as CFU. ND, not determined.
Kinetics of Cdt subunit translocation and effects of the depletion of membrane cholesterol.
CHO cells were treated with 10 μg/ml of CdtALumBC, CdtABLumC and CdtABCLum/ml for 5 min to 18 h and colabeled with Lumio green reagent and WGA. Examination by fluorescence microscopy revealed a limited number of Lumio green-labeled (green fluorescence) cells per field by 5 min of exposure to CdtABLumC and CdtABCLum (Fig. 2A). Relatively less CdtALum was detected in association with cells compared to CdtBLum and CdtCLum. All of the cells labeled with WGA-Alexa Fluor 555 (red fluorescence). By 18 h there was a significant increase in the number of Lumio green-labeled cells per field for each of the reconstituted heterotrimers. There was also clear evidence of cell distention at this time point. For this reason, fluorescence detection of subunits in association with cells exposed to the toxin for longer than 18 h is not shown. Lumio green fluorescence was not detected when cells were treated with CdtABC that lacked the tetracysteine sequence. Background fluorescence due to CHO cell proteins possibly containing native tetracysteines was not observed. Dose response to a heterotrimer containing CdtABLumC showed that 10 μg of total Cdt protein was optimal for detection of Lumio green-stained cells (Fig. 2B).
Fig 2.

Localization of the Cdt subunits in intoxicated cells. (A) Cultures of CHO-K1 were treated with 10 μg/ml (120 nM) of heterotrimer reconstituted with each of the subunits, containing tetracysteine and spacer sequences. Cells were labeled with Lumio green (green fluorescence) and WGA-Alexa Fluor 555 (red fluorescence) at 5 min, 4 h, and 18 h postintoxication. Cells were incubated with either no toxin (panel labeled Untreated) or wild-type heterotrimer (panel labeled CdtABC) as controls. In a subset of experiments, CHO-K1 cells were treated with MβCD, immediately prior to intoxication for 18 h, as described in Materials and Methods. Cells were colabeled as described above. The merged images are shown for all experiments. Scale bar = 50 μm. (B) To examine dose response, CHO-K1 cultures were exposed to 2.5, 5, and 10 μg/ml of heterotrimer reconstituted with CdtABLumC for 18 h. The cells were then labeled with Lumio green. Scale bar = 50 μm. (C) Quantification of cholesterol in isolated membrane rafts and total cell lysate before and after treatment with MβCD for 18 h. The results were expressed as μg of cholesterol per 1 μg of total protein in the sample. Statistically significant differences are marked by asterisks (*, P = 0.001; **, P = 0.0002). The cholesterol standard curve is shown in the inset.
Cdt intoxication of some, but not all, Cdt-sensitive types of cells has been correlated with the formation of lipid rafts and reported to be dependent on the concentration of membrane cholesterol in the rafts (2, 10, 13, 21). To determine if membrane cholesterol was essential for intoxication of CHO cells, cells were treated with MβCD prior to the addition of the hybrid heterotrimers (Fig. 2C). This treatment resulted in a 69% ± 8% reduction in total cholesterol content in Triton X-100-insoluble membrane raft preparations and a 44% ± 4% decrease in cholesterol in the total cell lysate, compared to controls, when normalized to 1 μg of total protein in the samples. Each of the three subunits was detected in association with MβCD-treated cells exposed to toxin for 18 h after addition of MβCD (Fig. 2A). In addition, the cells were drastically distended, which was a clear indication of intoxication. Thus, Cdt intoxication of CHO cells can occur following a significant reduction in the concentration of membrane cholesterol.
Localization of Cdt subunits in live cells.
Standard confocal fluorescence microscopy cannot easily distinguish between cell surface and intracellular localization of the Cdt subunits. Therefore, high-resolution, high-contrast live-cell imaging was used to track each subunit, in separate cultures, exposed to CdtALumBC, CdtABLumC, or CdtABCLum. A Z stack series was obtained for CHO cells treated for 18 h with each of the hybrid toxins (Fig. 3). There were indications that CdtALum was distributed on the cell surface. Images colabeled with Lumio green and WGA-Alexa Fluor 555 show that the merged fluorescence (yellow) is concentrated at the edges of the cell (see arrowheads). The more pronounced merged Lumio green and WGA-Alexa Fluor 555 fluorescence in the largest cell in the field is most likely at the bottom edge of the cell where it contacts the plastic dish. Significantly less CdtALum, Y214A, a binding-deficient subunit, was detected in association with the cell membrane relative to CdtALum.
Fig 3.

Live-cell imaging of CHO-K1 cells exposed to 10 μg/ml of the heterotrimer CdtALumBC, CdtABLumC, or CdtABCLum. Successive Z sections from representative fields were taken from top to bottom. Cells were colabeled with Lumio green (green fluorescence) and WGA-Alexa Fluor 555 (red fluorescence) at 18 h postintoxication. Cells treated with a heterotrimer containing a binding-deficient CdtA subunit (CdtALum, Y214ABC) were used as a control. The insets in panel CdtABLumC show a representative field of cells containing fragmented nuclei (FN). S, cell surface; P, polar end. Scale bar = 50 μm.
The distribution of Lumio green fluorescence in Z stack sections of live cells treated with CdtABLumC was throughout the cytosol and not associated with the cell surface. In some fields CdtBLum was detected in cells having fragmented nuclei (FN; see inset). Nuclear fragmentation is often a consequence of Cdt activity. CdtCLum also appeared to be widely distributed throughout the cells. Merged Lumio green and WGA-Alexa Fluor 555 fluorescence was concentrated at the polar ends of the cells (see arrowheads), indicating that a portion of the CdtABCLum was concentrated at the cell surface. Cells intoxicated with heterotrimer containing either CdtBLum or CdtCLum, and labeled with Lumio green, consistently colabeled poorly with WGA-Alexa Fluor 555.
To confirm the location of the Cdt subunits, XY projections were constructed from the Z stack sections shown in the fourth column in Fig. 3. CdtALum was detected only on the surface of the CHO cells in association with WGA-Alexa Fluor 555-labeled membrane, as indicated by yellow in the merged images (Fig. 4). In contrast, CdtBLum was found exclusively in the cytosol (green fluorescence). This subunit was detected in relatively large clusters of fluorescence, suggesting that the protein was concentrated possibly within the ER. An XY projection of a cell with fragmented nuclei is shown in the inset and provides evidence of the presence of CdtBLum in the nucleus. CdtCLum was distributed both on the cell surface and in the cytosol.
Fig 4.

Localization of the Cdt subunits in X-Y projection views of live CHO-K1 cells exposed to 10 μg/ml of the heterotrimer CdtALumBC, CdtABLumC, or CdtABCLum. XY projections were reconstructed from assembled single z sections from the live-cell experiment shown in Fig. 3. The inset in panel CdtABLumC shows a representative field of cells containing fragmented nuclei (FN). S, cell surface; C, cytosol. Scale bar = 50 μm.
Since our data showed that the CdtCLum subunit was present in the cytosol of intoxicated cells, predicted functional sites in CdtCLum were mutated to attempt to validate the localization of this subunit. Mutations in predicted receptor binding sites in CdtCLum, R1 and CdtCLum, R2 had no effect on cytotoxicity (Table 2) and subunit localization (Fig. 5). A mutation in the β-sheet region of CdtCLum, predicted to interact with the hydrophobic C-terminal tail of CdtA and an α-helix in CdtB, disrupted holotoxin activity (Table 2) even though assembly of the holotoxin was not affected (data not shown). The intracellular localization of this CdtCLum, AT subunit, as determined by the fluorescent confocal microscopy, was significantly altered (Fig. 5). The fact that only the two CdtCLum mutations predicted to affect receptor binding failed to affect internalization of the subunit indicated that CdtC has a relatively minor role in receptor recognition in live cells.
Fig 5.
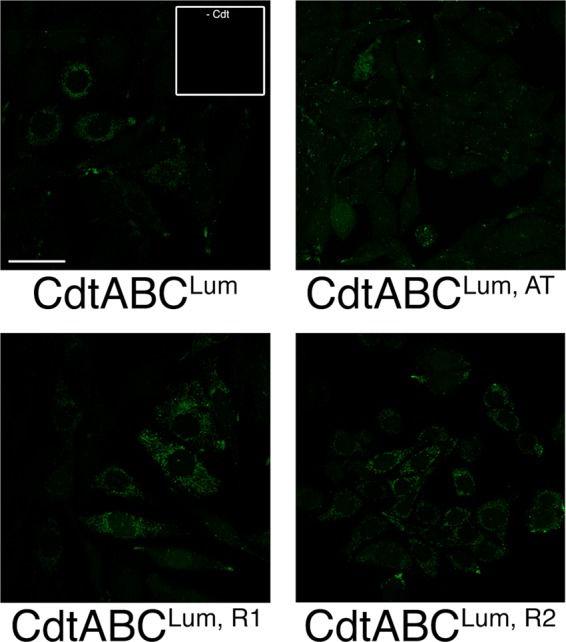
Localization of mutated CdtC subunits in live cells. Cells were treated with 10 μg/ml of toxin reconstituted with either CdtCLum, AT and CdtCLum, R1 or CdtCLum, R2. Cells were labeled with Lumio green 18 h postintoxication. Lumio green-labeled cells not exposed to toxin are shown in the inset. Scale bar = 50 μm.
CdtB is detected in association with the nucleus following intoxication of live cells.
To further determine if both CdtB and CdtC target the nucleus, nuclear fractions were isolated from cells intoxicated with CdtABLumC or CdtABCLum and labeled with Lumio green. Fluorescence microscopy of the nucleus preparations clearly showed CdtBLum, but not CdtCLum, associated with the nuclei (Fig. 6A). CdtBLum was detected in the nuclei by 4.5 h postintoxication. There was a reduction in the number of Lumio green-labeled nuclei if the cells were treated with MβCD prior to intoxication. Growth of cells treated with MβCD and Cdt was arrested. However, the block in cell cycle progression occurred at the S phase of growth (Table 2).
Fig 6.

Nuclear localization of the Cdt subunits in live CHO-K1 cells treated with 10 μg/ml of CdtABLumC and CdtABCLum. (A) Nuclei were isolated from cells exposed to the hybrid toxins for 48 h as described in Materials and Methods. The nuclei were costained with DAPI and Lumio green. In some experiments, cells were treated with MβCD immediately prior to intoxication. The insets show merged DAPI and Lumio green images. The arrowheads mark nuclei positively labeled with Lumio green (green fluorescence) and their corresponding positions in the merged images. The circles show background staining (most likely membrane fragments that do not colocalize with DAPI-stained nuclei). Scale bar = 50 μm. (B) Cells were treated with Cdt containing the various mutated CdtB and CdtC subunits. Nuclei were isolated and labeled as in panel A. Only the merged images are shown.
The biological activity of the CdtB subunit was altered by changing, in separate mutation events, one of the conserved histidines in the catalytic site (CdtBLum, CH-1) as well as predicted DNA contact (CdtBLum, A9-3) and Mg2+ binding (CdtBLum, A10-2) residues. These mutations significantly reduced the biological activity of the toxin (Table 2) but had no effect on the translocation of the subunit to the nucleus (Fig. 6B). A heterotrimer made with CdtBLum, NLS2, a mutated subunit that contained alterations in one of two predicted NLSs, had significantly reduced cytotoxicity and was not detected in association with nuclei up to 48 h postintoxication.
DISCUSSION
Our ongoing studies of the Cdt have been focused on the interactive roles of the individual subunits in toxin assembly and activity (4, 5, 6, 23). We have specifically studied the Cdt produced by A. actinomycetemcomitans because this bacterium is a model pathogen in oral infectious diseases and is the only indigenous oral bacterium identified to date that carries and expresses the cdt genes. The structural and biological complexity and uniqueness of the Cdt heterotrimer have presented a significant challenge to determining the specific functions of the individual subunits in the both the assembly of the holotoxin and intoxication of cells. Significant insight into the structure and function of the Cdt subunits has been obtained by compiling data from studies of the various species-specific members of the Cdt family as well as using information from the crystal structures of the Cdt from Haemophilus ducreyi (29) and A. actinomycetemcomitans (37). However, questions remain regarding the mechanism(s) by which the Cdt interacts with target cells and the fate of the individual subunits during the intoxication process.
In previous studies, immunodetection of His6-tagged Cdt subunits in fixed CHO cells (23) and HeLa cells exposed to the toxin reconstituted with a GST-CdtB subunit (13) resulted in low-intensity immunofluorescence. Since cells are three dimensional, the localization of immune-labeled subunits cannot be precisely determined by standard two-dimensional confocal microscopy (1). Other investigations that used biochemical and flow cytometry analyses of immunolabeled cells suggested that CdtA and CdtC mediate toxin binding to target cell membranes and facilitate internalization of the active CdtB subunit (16, 17, 28). However, the precise localization of GST-CdtB, as well as the other subunits, was not evident in those studies.
In the current study, we employed an innovative FlAsH technology to track and localize each of the cytolethal distending toxin subunits during consecutive stages of intoxication of live cells with active heterotrimers. This technology has been successfully used to examine virus entry and trafficking in cells (26, 27, 36) and secretion of the cytosolic and periplasmic Xcp polar protein in Pseudomonas aeruginosa (35). Optimal biarsenical compound binding increased resistance to compound release by dithiol reducing agents, and increased fluorescence quantum yield was achieved by engineering the tetracysteine motif, Cys-Cys-Pro-Gly-Cys-Cys, and a four-amino-acid spacer (Gly-Ala-Gly-Gly) into each subunit protein (24) (Invitrogen). Even though the biarsenical dye molecule could potentially label a range of endogenous tetracysteine-containing proteins in the live cells, the technology is optimized for binding to proteins containing the CCXXCC motif (14). In our experiments, the addition of the extra amino acids at either the amino or carboxy termini of the subunit proteins did not significantly affect heterotoxin assembly or function.
Fluorescent biarsenic dye tagging of proteins and live-cell imaging showed that the CdtALum subunit remained on the cell surface of CHO cells from 5 min to 48 h postexposure. This observation confirmed our earlier interpretation of the results of CdtA localization experiments (23). We also found that CdtA remained in association with both cholesterol-containing and cholesterol-depleted membranes. Our experimental conditions for cholesterol depletion by MβCD resulted in the maximum achievable decrease in cholesterol in membrane rafts and total cell lysate. No appreciable loss of cell viability was detected at this level of cholesterol depletion. These values are in accordance with data published by others indicating that more than 50% depletion of total cellular cholesterol leads to cell death (22). Our experiments also showed that a substantial reduction in the amount of cholesterol in the CHO-K1 cell membrane did not abolish the internalization of CdtBLum and CdtCLum. Earlier studies, using Jurkat cells and model membranes isolated from these cells, indicated that raft-associated cholesterol is important for A. actinomycetemcomitans Cdt binding to cell membranes (2, 3). Using an alternative strategy, it was found that intoxication of CHO-K1 cells with the Cdt from A. actinomycetemcomitans was dependent on membrane cholesterol loading of cells (10). The concentration of Cdt used in that study was 2-fold higher than that used in our experiments. Other notable differences among species-specific Cdts have been reported. Intoxication of CHO-K1 cells with the Campylobacter jejuni Cdt was not dependent on cholesterol membrane loading (10). However, in a separate study it was found that CHO-K1 membranes, depleted of cholesterol by treating cells with MβCD, resulted in reduced intoxication with the C. jejuni Cdt (21). It is apparent that the divergent results obtained in independent studies of cholesterol requirements for intoxication support the hypothesis that the mechanism of entry of the Cdt into the cells depends on (i) the specific cell type or line, (ii) species-specific origin of the toxin, (iii) toxin concentration, and (iv) reorganization of the putative receptor for toxin binding within protein components of the cell membrane. Evidence for the existence of a protein receptor is supported by recent findings that implicate TMEM181, a presumed G protein-coupled receptor in KBM7 cells, in the recognition of the Cdt from E. coli (7). It seems reasonable to speculate that a similar protein receptor for the binding of Cdt from A. actinomycetemcomitans may also exist in the CHO-K1 cell membrane and may be relatively insensitive to cholesterol levels.
Based on fluorescence detection with Lumio green, we found that the CdtBLum subunit is initially exclusively in the cytosol and subsequently (within 4.5 h postintoxication) also in the nucleus. These results were in good agreement with those of several studies that employed less direct methods (reviewed in reference 11). In another study, CdtB was detected in the nuclei of COS-1 and REF52 cells 4 h after microinjection of purified C. jejuni protein (19). Using fluorescent biarsenic dye technology, we also found that site-directed mutagenesis of two consecutive arginine residues (CdtBLum, NLS2) in a putative nuclear localization motif reported by McSweeney and Dreyfus (25) not only substantially decreased the biological activity of reconstituted toxin but also abolished nuclear localization. Interestingly, site-directed substitutions of two arginine residues (CdtBLum, NLS1) in the N-terminal nuclear localization sequence described by Nishikubo et al. (30) did not have any effect on the biological activity or nuclear localization. This discrepancy may be due to the possibility that monopartite or bipartite arginine and/or lysine residues necessary for the classical NLS function may not necessarily be required by this atypical NLS. Mutating sites in CdtBLum that resided in the catalytic center of the protein (CdtBLum, CH-1, CdtBLum, A9-3, and CdtBLum, A10-2) resulted in altered biological activity of hybrid heterotrimers but did not affect translocation of the subunit to the nucleus.
It has been more difficult to confirm the fate of the CdtC subunit during intoxication. By using a direct labeling method in combination with live-cell imaging, the CdtCLum subunit was found on the cell surface but to a greater extent in the cytosol. These results are contrary to predicted functions for this subunit based on Cdt structure and comparisons to toxins having the standard A-B arrangement. However, the Lumio green labeling experiments supported the results of our earlier fluorescence experiments and those from standard confocal microscopy studies (1), suggesting that CdtC enters the cell (23). The A. actinomycetemcomitans CdtC may have a relatively minor or accessory role in binding of the toxin to the cell surface, as supported by the presence of a cholesterol recognition amino acid consensus sequence (CRAC) region (3). Our experiments, based on adhesion assays (4, 5) and specific labeling in live cells, suggest that the CdtA subunit plays a more active role in Cdt-cell recognition. Additional evidence that CdtC may be more active in the internalization of CdtB than in attaching the toxin to the cell surface was obtained from analysis of the effects of specific mutations on the localization of the subunit. Mutations altering two solvent-accessible molecular surface-exposed loop regions predicted to be involved in cell surface binding (CdtCLum, R1 and CdtCLum, R2) had a moderate effect on Cdt activity and did not affect holotoxin assembly or intracellular localization of the subunit. However, toxin reconstituted with CdtCLum, AT, which has a mutation in a site predicted to interact with both CdtA and CdtB, did not induce cell cycle arrest or inhibit CHO cell proliferation. The localization of CdtCLum, AT, as determined by fluorescence microscopy, was significantly altered.
In summary, a fluorescent biarsenic dye tagging technology and live-cell imaging was used, for the first time, to examine the cellular location of the Cdt subunits following intoxication of CHO cells with hybrid heterotrimers made with combinations of wild-type and mutated subunit proteins. Taken together, the results of this study, and those of earlier investigations (1, 12, 13), allow us to propose a modified model of intoxication with particular emphasis on the role of CdtC (Fig. 7). This model is based on results obtained with the Lumio green system, a single species-specific Cdt, and CHO cells. Therefore, the model should be viewed in relation to these restrictions. In this model, CdtA is primarily responsible for anchoring the toxin to a specific receptor on the cell surface through interactions of the aromatic patch region and a groove formed between CdtA and CdtC in the heterotrimer complex. In this regard, CdtC plays an accessory role in toxin binding. However, the main function of CdtC is to facilitate the endocytosis of CdtB by interacting with cholesterol-enriched lipid rafts through a CRAC motif. The association with cholesterol-rich regions of the membrane is not required for uptake in CHO cells but enhances the process. CdtA remains on the cell surface, due to relatively tight binding to receptor, while a CdtB-CdtC dimer or CdtB monomer is delivered to the ER possibly through the Golgi. CdtC may be eliminated by the ER-associated degradation (ERAD) pathway. CdtB is then processed through the ER and delivered to the nucleus. A significant role for CdtC in the internalization of CdtB may provide an alternative target for therapeutic intervention. Strategies designed to inhibit CdtC could potentially both block binding of the toxin to cells and impede the uptake of the CdtB subunit.
Fig 7.
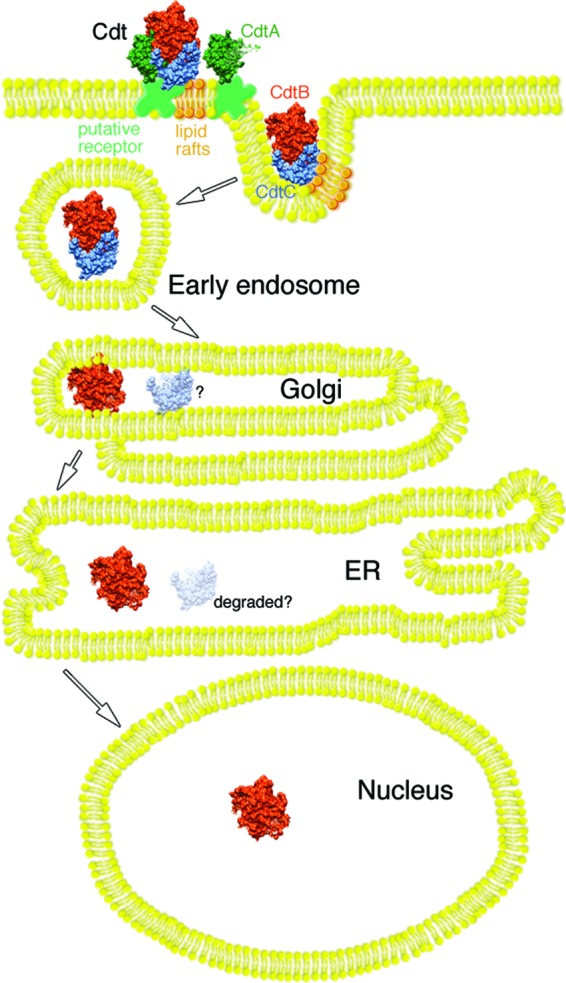
Model of Cdt intoxication of CHO-K1 cells. The CdtA subunit anchors the heterotrimer to a putative specific receptor in the cell membrane, while CdtC plays an accessory role in the binding process. Interaction of the CdtC with lipid rafts, through a cholesterol recognition amino acid consensus (CRAC) domain, enhances the binding and may facilitate enclosure of a CdtB-CdtC dimer in early endosomes. A CdtB-CdtC dimer retrogradely passes to the endoplasmic reticulum from the Golgi apparatus. During this process, CdtC most likely dissociates from the CdtB subunit and is degraded by the ER-associated degradation (ERAD) pathway (depicted by the faded CdtC subunit and the question mark). Evidence suggests that CdtB is subsequently processed through the ER and delivered to the nucleus bypassing the ERAD pathway (12).
ACKNOWLEDGMENTS
We thank Sunday Akintoye, Department of Oral Medicine, for the use of a fluorescence microscope and Kathleen Boesze-Battaglia and the University of Pennsylvania School of Dental Medicine Live-Cell Imaging Facility for use of the Nikon A1R laser scanning confocal microscope.
This work was supported by Public Health Service grant DE012593 from the National Institute of Dental and Craniofacial Research.
Footnotes
Published ahead of print 29 May 2012
REFERENCES
- 1. Akifusa S, Heywood W, Nair SP, Stenbeck G, Henderson B. 2005. Mechanism of internalization of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Microbiology 151:1395–1402 [DOI] [PubMed] [Google Scholar]
- 2. Boesze-Battaglia K, et al. 2006. Cholesterol-rich membrane microdomains mediate cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Cell. Microbiol. 8:823–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boesze-Battaglia K, et al. 2009. Cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J. Biol. Chem. 284:10650–10658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cao L, Bandelac G, Volgina A, Korostoff J, DiRienzo JM. 2008. Role of aromatic amino acids in receptor binding activity and subunit assembly of the cytolethal distending toxin of Aggregatibacter actinomycetemcomitans. Infect. Immun. 76:2812–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao L, Volgina A, Huang CM, Korostoff J, DiRienzo JM. 2005. Characterization of point mutations in the cdtA gene of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Mol. Microbiol. 58:1303–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao L, Volgina A, Korostoff J, DiRienzo JM. 2006. Role of intrachain disulfides in the activities of the CdtA and CdtC subunits of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Infect. Immun. 74:4990–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carette JE, et al. 2009. Haploid genetic screens in human cells identify host factors used by pathogens. Science 326:1231–1235 [DOI] [PubMed] [Google Scholar]
- 8. DiRienzo JM, Cao L, Volgina A, Bandelac G, Korostoff J. 2009. Functional and structural characterization of chimeras of a bacterial genotoxin and human type I DNAse. FEMS Microbiol. Lett. 291:222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elwell CA, Dreyfus LA. 2000. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 37:952–963 [DOI] [PubMed] [Google Scholar]
- 10. Eshraghi A, et al. 2010. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol. J. Biol. Chem. 285:18199–18207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guerra L, Cortes-Bratti X, Guidi R, Frisan T. 2011. The biology of the cytolethal distending toxins. Toxins 3:172–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guerra L, et al. 2009. A novel mode of translocation for cytolethal distending toxin. Biochim. Biophys. Acta 1793:489–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guerra L, et al. 2005. Cellular internalization of cytolethal distending toxin: a new end to a known pathway. Cell. Microbiol. 7:921–934 [DOI] [PubMed] [Google Scholar]
- 14. Hearps AC, et al. 2007. The biarsenical dye Lumio exhibits a reduced ability to specifically detect tetracysteine-containing proteins within live cells. J. Fluoresc. 17:593–597 [DOI] [PubMed] [Google Scholar]
- 15. Hofmann K, Tomiuk S, Wolff G, Stoffel W. 2000. Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc. Natl. Acad. Sci. U. S. A. 97:5895–5900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu X, Nešić D, Stebbins CE. 2006. Comparative structure-function analysis of cytolethal distending toxins. Proteins 62:421–434 [DOI] [PubMed] [Google Scholar]
- 17. Hu X, Stebbins CE. 2006. Dynamics and assembly of the cytolethal distending toxin. Proteins 65:843–855 [DOI] [PubMed] [Google Scholar]
- 18. Jinadasa RN, Bloom SE, Weiss RS, Duhamel GE. 2011. Cytolethal distending toxin: a conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 157:1851–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lara-Tejero M, Galán JE. 2000. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 290:354–357 [DOI] [PubMed] [Google Scholar]
- 20. Leitinger B, Hogg N. 2002. The involvement of lipid rafts in the regulation of integrin function. J. Cell Sci. 115(Pt 5):963–972 [DOI] [PubMed] [Google Scholar]
- 21. Lin CD, et al. 2011. Cholesterol depletion reduces entry of Campylobacter jejuni cytolethal distending toxin and attenuates intoxication of host cells. Infect. Immun. 79:3563–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mahammad S, Parmryd I. 2008. Cholesterol homeostasis in T cells. Methyl-beta-cyclodextrin treatment results in equal loss of cholesterol from Triton X-100 soluble and insoluble fractions. Biochim. Biophys. Acta 1778:1251–1258 [DOI] [PubMed] [Google Scholar]
- 23. Mao X, DiRienzo JM. 2002. Functional studies of the recombinant subunits of a cytolethal distending holotoxin. Cell. Microbiol. 4:245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin BR, Giepmans BNG, Adams SR, Tsien RY. 2005. Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity. Nat. Biotechnol. 23:1308–1314 [DOI] [PubMed] [Google Scholar]
- 25. McSweeney LA, Dreyfus LA. 2004. Nuclear localization of the Escherichia coli cytolethal distending toxin CdtB subunit. Cell. Microbiol. 6:447–458 [DOI] [PubMed] [Google Scholar]
- 26. Mire CE, Dube D, Delos SE, White JM, Whitt MA. 2009. Glycoprotein-dependent acidification of vesicular stomatitis virus enhances release of matrix protein. J. Virol. 83:12139–12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mire CE, White JM, Whitt MA. 2010. A spatio-temporal analysis of matrix protein and nucleocapsid trafficking during vesicular stomatitis virus uncoating. PLoS Pathog. 6:e1000994 doi:10.1371/journal.ppat.1000994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mise K, et al. 2005. Involvement of ganglioside GM3 in G2/M cell cycle arrest of human monocytic cells induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 73:4846–4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nešić D, Hsu Y, Stebbins CE. 2004. Assembly and function of a bacterial genotoxin. Nature 429:429–433 [DOI] [PubMed] [Google Scholar]
- 30. Nishikubo S, et al. 2003. An N-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J. Biol. Chem. 278:50671–50681 [DOI] [PubMed] [Google Scholar]
- 31. Nougayrède JP, Taieb F, De Rycke J, Oswald E. 2005. Cyclomodulins: bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 13:103–110 [DOI] [PubMed] [Google Scholar]
- 32. Ohara M, Oswald E, Sugai M. 2004. Cytolethal distending toxin: a bacterial bullet targeted to nucleus. J. Biochem. 136:409–413 [DOI] [PubMed] [Google Scholar]
- 33. Oswald E, Nougayrede JP, Taieb F, Sugai M. 2005. Bacterial toxins that modulate host cell-cycle progression. Curr. Opin. Microbiol. 8:83–91 [DOI] [PubMed] [Google Scholar]
- 34. Pan CQ, Ulmer JS, Herzka A, Lazarus RA. 1998. Mutational analysis of human DNase I at the DNA binding interface: implications for DNA recognition, catalysis, and metal ion dependence. Protein Sci. 7:628–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Senf F, Tommassen J, Koster M. 2008. Polar secretion of proteins via the Xcp type II secretion system in Pseudomonas aeruginosa. Microbiology 154:3025–3032 [DOI] [PubMed] [Google Scholar]
- 36. Whitt MA, Mire CE. 2011. Utilization of fluorescently-labeled tetracysteine-tagged proteins to study virus entry by live cell microscopy. Methods 55:127–136 [DOI] [PubMed] [Google Scholar]
- 37. Yamada T, Komoto J, Saiki K, Konishi K, Takusagawa F. 2006. Variation of loop sequence alters stability of cytolethal distending toxin (CDT): crystal structure of CDT from Actinobacillus actinomycetemcomitans. Protein Sci. 15:362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]