

Abstract
The human nasopharynx is the main reservoir for Streptococcus pneumoniae (the pneumococcus) and the source for both horizontal spread and transition to infection. Some clinical evidence indicates that nasopharyngeal carriage is harder to eradicate with antibiotics than is pneumococcal invasive disease, which may suggest that colonizing pneumococci exist in biofilm communities that are more resistant to antibiotics. While pneumococcal biofilms have been observed during symptomatic infection, their role in colonization and the role of host factors in this process have been less studied. Here, we show for the first time that pneumococci form highly structured biofilm communities during colonization of the murine nasopharynx that display increased antibiotic resistance. Furthermore, pneumococcal biofilms grown on respiratory epithelial cells exhibited phenotypes similar to those observed during colonization in vivo, whereas abiotic surfaces produced less ordered and more antibiotic-sensitive biofilms. The importance of bacterial-epithelial cell interactions during biofilm formation was shown using both clinical strains with variable colonization efficacies and pneumococcal mutants with impaired colonization characteristics in vivo. In both cases, the ability of strains to form biofilms on epithelial cells directly correlated with their ability to colonize the nasopharynx in vivo, with colonization-deficient strains forming less structured and more antibiotic-sensitive biofilms on epithelial cells, an association that was lost when grown on abiotic surfaces. Thus, these studies emphasize the importance of host-bacterial interactions in pneumococcal biofilm formation and provide the first experimental data to explain the high resistance of pneumococcal colonization to eradication by antibiotics.
INTRODUCTION
Streptococcus pneumoniae (the pneumococcus) colonizes the mucosal surface of the upper respiratory tract in early childhood, persisting as a symptomless commensal in the nasopharynxes of healthy individuals (27, 37). Although the mechanism(s) whereby pneumococcal colonization progresses to infection of otherwise sterile sites is not fully understood, transition to infection has been associated with preceding virus infections or other compromising assaults that commonly disrupt natural immunological barriers (10, 47, 74). Transition to infection happens often enough to make S. pneumoniae a leading bacterial cause of otitis media, pneumonia, sepsis, and meningitis in children and in elderly or immunocompromised individuals worldwide, causing more than 14.5 million episodes of invasive pneumococcal disease annually and 11% of deaths in children below the age of five (51, 58). Colonizing bacteria are also the source for horizontal spread between individuals, whereas none of the common forms of disease promotes transmission. This suggests that all factors associated with virulence probably evolved to promote persistence and colonization (39).
It has frequently been clinically noted that antimicrobial agents often fail to eradicate bacteria from the nasopharynx, making colonization a more challenging target than treatment of invasive disease (18, 19, 21–23, 26). The reason for this is unknown, but it could suggest that biofilm formation is an important aspect of pneumococcal carriage. Biofilms are organized communities of collaborating bacteria within a self-produced matrix of polymeric substances, attached to an inert or living surface (20, 64). Biofilms are typically found in chronic diseases, where they represent up to 80% of all infections (75). These sessile communities are inherently more resistant to antibacterial agents and are able to resist host immune responses, facilitating persistence and supporting the dissemination of virulent clones (17, 25, 41).
Although it has been speculated that pneumococci colonizing the nasopharynx may form biofilms (50, 52, 59, 67, 73), thus far biofilm formation in vivo has been shown only during disease states and has been found to various degrees in association with adenoids and mucosal epithelia of children with recurrent or chronic middle ear infections and chronic rhinosinusitis (33, 36, 57, 60, 61). Bacterial aggregates have also been documented in the lungs of infected animals (60). These studies provide critical evidence for the role of S. pneumoniae biofilms in disease states, but the role of biofilms during the preceding colonization of the nasopharynx, the main reservoir of the organism, remains unclear.
The studies of pneumococcal biofilm formation in vitro have yielded conflicting information. To date, all S. pneumoniae biofilm work in vitro has been confined to abiotic surfaces, surfaces that this human-exclusive pathogen do not normally colonize (11, 15, 24, 43, 56, 65, 67). Additionally, these studies have been performed mostly for periods of time too short to enable mature biofilm formation. While these studies have made substantial contributions to our understanding of pneumococcal accretion and early biofilm formation, these model systems have failed to produce any associations between the biofilm-producing ability of clinical isolates in vitro and their virulence or isolation site in vivo (43, 65). This leaves a critical need to better understand the processes involved in biofilm formation in vivo that may be used to develop biofilm model systems with the capacity to identify biofilm phenotypes associated with colonization and disease.
The aim of this work was to better understand the basis for the observed increased antibiotic resistance of pneumococcal carriage in patients by investigating bacterial community structure during pneumococcal colonization within the murine nasopharynx and to explore the role of the mucosal epithelium, the niche for pneumococcal carriage, in pneumococcal biofilm formation in order to provide a better framework for studies of host-pneumococcal interactions in the future.
MATERIALS AND METHODS
Reagents.
Cell culture reagents were from Invitrogen, Carlsbad, CA. Bacterial and cell culture media and reagents were from VWR Inc., Radnor, PA. Chemically defined bacterial growth medium (CDM) was from JRH Biosciences, Lexera, KS. Sheep blood was purchased from BioLink, Inc., Liverpool, NY. Remaining reagents were from Sigma-Aldrich, St. Louis, MO.
Bacterial strains.
Pneumococcal strains were grown in synthetic medium (CDM) or in Todd-Hewitt medium containing 0.5% yeast extract (THY) as described previously (70). The study used the serotype 19F strains EF3030 and BG8826 (2), the serogroup 6 strain SP670 (31), the serotype 3 strain WU2 (14), the serotype 2 strain D39 (3), and versions of D39 lacking capsule (44), pneumococcal surface protein C (PspC) (4), pneumococcal surface protein A (PspA) (46), pneumolysin (9), and autolysin (LytA) (8). Pneumococci were verified by their sensitivity to optochin, using a optochin-diffusion assay on blood agar, resulting in a clearing zone around the optochin disc (from Fluka Analytical/Sigma-Aldrich) of >15 mm (12).
The MIC for each strain was determined through growth in increasing concentrations of gentamicin and penicillin as described previously (45). In brief, MICs were determined by means of the microdilution broth method, where serial 2-fold dilutions of a starting antibiotic concentration were inoculated into wells of a microtiter plate so that each well contained ∼5 × 105 CFU/ml. The MIC was defined as the lowest antibiotic concentration able to inhibit visible bacterial growth after 18 to 20 h of incubation in ambient air at 37°C. The minimal bactericidal concentration (MBC) was determined in a similar manner but by measuring instead the lowest concentration where no viable bacteria were present.
Imaging of pneumococcal carriage in the mouse nasopharynx.
All experiments performed were approved by the Institutional Animal Care and Use Committee at the University at Buffalo, NY. Six-week-old female BALB/cByJ mice from Jackson Laboratories (Bar Harbor, ME) were maintained in filter-top cages on standard laboratory chow and water ad libitum until use.
Mice were colonized as described previously (70). In short, 20 μl of a bacterial suspension containing 5 × 106 CFU of pneumococci in phosphate-buffered saline (PBS) was pipetted into the nares of nonanesthetized mice. Uninfected control mice received PBS alone. The absence of symptoms associated with pneumonia and invasive disease was confirmed by lack of visual signs such as ruffled fur, inactivity, and labored breathing, as well as lack of fever measured through the abdominal surface temperature, as described previously (6). At 48 h postinoculation, animals were euthanized by CO2 asphyxiation and nasopharyngeal tissue was dissected as described previously (70).
To harvest the mouse nasal septum, the skin was first dissected and completely removed form the skull and nose, and the skull was sectioned in the coronal plane. The remainder of the anterior and posterior skull was then removed and the suture line incised bilaterally to reveal the nasal septum. Scissors were inserted into the posterior nasal cavity and used to separate the nasal septum from the maxillae (laterally) and the ethmoid bone (medially). Tissue present in the nasal conchae was then harvested with forceps. Harvested tissue was either used for analysis by scanning electron microscopy (SEM), as described below, or homogenized and serially diluted on tryptic soy agar supplemented with 5% sheep blood (blood agar) to measure the CFU per mouse nasopharyngeal tissue as a measure for colonization load. In some experiments, injecting PBS retrograde through the trachea preceded harvesting of tissue and collecting it from the nares (nasopharyngeal lavage), and bacterial load was measured by determining viable plate counts.
SEM.
Tissue samples and biofilms grown in vitro (see below) were fixed using 2.5% glutaraldehyde, 0.075% ruthenium red, and 0.075 M lysine acetate in 0.1 M sodium cacodylate buffer (pH 7.2) for 1 h at room temperature. This procedure has been shown to retain carbohydrate structures and improve preservation of biofilm morphology (35). Samples were washed three times without shaking for 15 min at room temperature in 0.075% ruthenium red in 0.2 M sodium cacodylate buffer and were then dehydrated with a graded series of ethanol solutions (10, 30, 50, 75, 95, and 100%) at room temperature, with 15 min used for each step. Samples were exchanged into 100% hexamethyldisilazane and allowed to air dry before being mounted onto stubs, carbon coated, and analyzed using an SU70 scanning electron microscope at an acceleration voltage of 5.0 kV (available through the South Campus Instrumentation Center, University at Buffalo, NY).
Antibiotic resistance of bacterial colonization.
Mice colonized for 48 h were treated with 20 μl gentamicin (0 to 1,000 μg/ml) in the nares for 6 h. The colonization burden was then assessed after euthanizing the animals by enumerating viable bacteria both in a nasopharyngeal lavage specimen and from harvested nasopharyngeal tissue after the nasal wash, as described above.
Cells.
NCI-H292 mucoepidermoid bronchial carcinoma cells (ATCC CCL-1848) were grown on various surfaces as described previously (30). Primary human bronchial epithelial cells were obtained by bronchoscopic biopsy from a nonsmoking, healthy volunteer in association with a prior study investigating epithelial cell function during chronic obstructive pulmonary disease, according to protocols approved by the Institutional Review Board for Human Studies at the University at Buffalo, Buffalo, NY. Cells were cultured on collagen-coated inserts (Millicell; Millipore) in 24-well plates as described previously (40). Once confluent, cultures were shifted to an air-liquid interface by removal of apical medium. Three-week-old cultures with transepithelial resistance of >600 Ω/cm2 were used in experiments and showed airway cells with cilia and mucin production by scanning electron microscopy and gel analysis of surface secretions.
Static biofilm model.
Pneumococci were grown in CDM to mid-logarithmic phase (optical density at 600 nm [OD600] = 0.5), washed, and resuspended in fresh prewarmed CDM to a density of 2 × 104 CFU per 500 μl, and suspensions were used to seed sterile, round glass coverslips in the bottom of polystyrene 24-well plates with or without a substratum of confluent H292 cells or inserts with differentiated primary epithelial cells. Biofilms were cultured at 34°C in 5% CO2 for the indicated times with change of culture medium every 12 h and used for SEM studies or to assess biomass and antibiotic resistance as described below. To measure bacterial viability in these biofilms, LIVE/DEAD stain (Invitrogen) was added to 48-h biofilms according to the manufacturer's descriptions, and the biofilm layers were inspected by confocal microscopy using a Zeiss LSM5 laser scanning confocal microscope (Zeiss Inc., Thornwood, NY).
Biomass and antibiotic resistance of static biofilms.
Biofilms washed with PBS to eliminate planktonic bacteria were exposed to 500 μg/ml gentamicin for 3 h at 34°C in 5% CO2 or with PBS alone as a control to determine total initial biomass. Biofilms were then washed with PBS, sealed, floated on a sonicator water bath, and sonicated for 2 s to disperse the bacteria (verified by light microscopy) as described previously (66). Cells were then collected and vortexed twice for 20 s at high speed to ensure a homogenous solution, and the dispersed biofilm cells were used to determine viable CFU per ml by viable plate counts. Results are reported as the total number of CFU per biofilm.
RNA isolation and quantitative RT-PCR (qRT-PCR).
Briefly, frozen stocks of D39 were used to inoculate a 10-ml culture in CDM for planktonic growth. When the bacteria reached an OD600 of ∼0.6, 1 ml was removed and bacteria were pelleted by centrifugation at 9,000 × g for 2 min at 4°C. For isolation of RNA from biofilms, biofilms were seeded over a prefixed epithelial substratum and maintained for 48 h as described above. After 48 h, biofilms were sonicated, resuspended in 1 ml of PBS, and pelleted by centrifugation at 9,000 × g for 2 min at 4°C. Pellets were resuspended in 0.5 ml of 0.9% NaCl, 1 ml of RNAprotect (Qiagen, Valencia, CA) was added, and the mixture was incubated at room temperature for 5 min. Cells were then pelleted at 9,000 × g for 2 min at room temperature, and RNA was purified using Qiashreeder columns and the RNeasy minikit as described previously (70).
Statistical analysis.
Column comparisons were analyzed for statistical significance using a two-tailed Student t test for unpaired data. Multivariance analysis was done using one-way analysis of variance (ANOVA) that was corrected for variance using the Bartlett variance test and for multiple comparisons using the Bonferroni multiple-comparison test. For both tests a P value of <0.05 was considered significant. Statistical analysis was performed using the GraphPad Prism software (version 5.0d; GraphPad Software Inc., La Jolla, CA).
RESULTS
S. pneumoniae forms biofilms during carriage of the mouse nasopharynx.
Although a role of biofilm formation during pneumococcal disease has been shown, the growth phenotype of bacteria colonizing the human nasopharynx, its main biological niche, has not been visually documented. To obtain the best chance of observing biofilm formation in vivo and to produce the most clinically relevant colonization possible, we inoculated BALB/cByJ mice intranasally for 48 h with a clinical strain of pneumococci, EF3030, that has been well documented, by us and others, to effectively establish noninvasive colonization of high bacterial density (4, 55, 62). This procedure produced a mean carriage of 5 × 106 pneumococci per nasopharyngeal tissue after 48 h, without any evidence of disease. These numbers are in agreement with other studies using the EF3030 strain (13, 55, 62). Pneumococcal growth in the tissues was verified by morphology of the bacteria on blood agar plates in combination with testing several isolated clones for optochin sensitivity. No other bacterial species were identified from the nasal tissues of infected mice, and nasal wash or tissue from uninfected mice showed no bacterial growth.
Tissues from infected mice were analyzed by scanning electron microscopy (SEM), and all tissues showed the same colonization pattern, with an increasing bacterial burden along the anterior-to-posterior length of the nasopharynx (Fig. 1A shows a low-magnification view). In the anterior region of the nasal septum, pneumococci could be observed scattered over the ciliated epithelium primarily as single cells or diplococci (Fig. 1B). More posterior, larger aggregates of interconnected pneumococci forming tower structures organized in filamentous structures embedded in extracellular matrix were observed covering the ciliated epithelium (Fig. 1C and D). In the most posterior region of the nasopharyngeal tissue, large mature biofilm structures could be observed, where networks of bacteria were embedded within structured matrices (Fig. 1D and E). In contrast, mice that were mock infected with sterile PBS alone showed a generally uniform ciliated epithelial surface devoid of any bacteria or bacterial material (Fig. 1F).
Fig 1.

Biofilm formation during asymptomatic pneumococcal carriage in the mouse nasopharynx. (A to F) Scanning electron microscopy images of murine nasopharyngeal tissue colonized with S. pneumoniae EF3030 for 48 h. (A) Low-magnification view of the nasal septum. The figure is labeled to indicate the anterior and posterior views, with the anterior view being the area closer to the nare openings of the mouse. (B) Image from the anterior area of the nasopharynx showing individual pneumococci lying on top of the ciliated epithelium. The inset shows a 2.5-fold-increased magnification of part of the field. Arrows point out pneumococci as well as the ciliated epithelium. (C) A midposterior view with more organized and mature biofilm structures where pneumococci can be observed enclosed in extracellular matrix lying on top of the ciliated epithelium. The inset shows a 2.5-fold-increased magnification of part of the field showing biofilm on top of ciliated epithelium. Arrows indicate the presence of cilia and bacteria. (D) A posterior view of the colonized nasopharynx, showing tower formations and biofilm architecture (left) as well as filaments observed to connect the bacteria (right). (E) Posterior view at higher magnification showing bacteria covered by extracellular matrix and fibrous interbacterial connections in a thick biofilm. The inset shows bacteria and fibrous connections at a 2.5-fold-increased magnification. The experiment was repeated on 5 animals, and images originate from two of those animals. (F) High-magnification view of the uninfected epithelium.
Tissue-associated pneumococci in the mouse nasopharynx are more resistant to antibiotic treatment than loosely associated bacteria.
Antimicrobial resistance is a general characteristic of biofilms (17, 25, 41, 59). To further characterize the phenotype of S. pneumoniae during nasopharyngeal carriage in the mouse, we evaluated the susceptibility of colonizing pneumococci to both the bactericidal antibiotic gentamicin and the more commonly used antibiotic penicillin and compared that to their sensitivity in vitro. We selected the antibiotic gentamicin because while aminoglycosides are rapidly bactericidal against planktonic cells, they do not generally penetrate biofilms very well (1, 5, 16), making it an excellent discriminator of biofilm organization. In addition, we also evaluated the biofilm-specific resistance to penicillin to make our results more clinically relevant. S. pneumoniae EF3030 had an MIC of 16 μg/ml and a minimal bactericidal concentration (MBC) of 32 μg/ml to gentamicin in vitro. However, completely eradicating a bacterial inoculum of 107 CFU/ml of EF3030 pneumococci in 6 h required 500 μg/ml of gentamicin, correlating well with the increased concentrations needed for eradicating pneumococci in time-kill assays with other antibiotics (38, 63). The MIC of EF3030 for penicillin G was 0.01 μg/ml; however, consistent with the bacteriostatic nature of penicillin G, eradication of a bacterial inoculum of 107 CFU/ml could not be achieved in 6 h even at concentrations exceeding 100 μg/ml. Therefore, we increased the incubation time to 12 h, after which the entire inoculum could be eradicated by 1 μg/ml.
Mice colonized with EF3030 pneumococci for 48 h were treated intranasally with increasing doses of gentamicin for 6 h or penicillin G for 12 h. The survival of pneumococci was then assessed both in a nasal wash and the nasopharyngeal tissue after the nasal wash by viable counts. Untreated mice showed a total colonization burden of 5 × 106 CFU per mouse. The majority of the bacteria were associated with the tissue, with a minor portion of the bacterial carriage being loosely enough associated with the nasopharyngeal tissue to be washed out in a nasal lavage (mean of 7 × 104 CFU per nasal wash).
After treatment with gentamicin for 6 h or penicillin G for 12 h, the colonizing population recovered from the nasal wash, and more loosely associated with the tissue, was eradicated at a significantly lower concentration of gentamicin or penicillin G than the more adherent population (Fig. 2). Although specific tissue concentrations of gentamicin or penicillin could not be measured, the in vivo data suggest that the bacteria recovered in the nasal washes had gentamicin and penicillin G sensitivities similar to those of their in vitro broth-grown counterparts, whereas the tissue-associated bacteria were much more resistant to antibiotic treatment. The majority (≥90%) of the nasal wash population was eradicated at doses of 3 μg of gentamicin and 1 μg penicillin G, representing concentrations of 150 μg/ml gentamicin and 50 μg/ml penicillin G. The population was completely eradicated at 10 μg gentamicin, or 500 μg/ml (Fig. 2, squares), similar to the concentration needed to eradicate planktonic cells. However, bacteria associated with the tissue were highly resistant to both gentamicin and penicillin. For gentamicin, although >98% of bacteria were eradicated at 30 μg of gentamicin (1,500 μg/ml) (Fig. 2, circles), a bacterial carriage of 103 organisms was still detected even after treatment with a dose of 100 μg (5,000 μg/ml), 10 times the dose required to eradicate a similar inoculum in vitro. For penicillin G, no significant eradication compared with the control was observed even at a dose of 100 μg penicillin (5,000 μg/ml).
Fig 2.

Gentamicin and penicillin G resistance of tissue-associated pneumococci of colonized mice. Mice were colonized with S. pneumoniae EF3030 for 48 h, treated with various doses of gentamicin intranasally for 6 h, and sacrificed; a nasal wash and nasopharyngeal tissue were then collected, and the bacterial burden was determined by viable counts. (A and D) Plots of the bacterial survival in nasal washes (squares) and nasopharyngeal tissue (circles) after gentamicin or penicillin G treatment as percentage of mock-treated animals (treated with PBS alone). Statistical analysis was performed using one-way ANOVA (P < 0.001 and P = 0.84 for tissue association for penicillin G and P < 0.001 for nasal wash and P = 0.31 for tissue association for gentamicin). (B, C, E, and F) Bacterial burden in tissue (B and E) or nasal wash (C and F). Six animals were used per experiment, and 2 experiments were performed. Statistical analysis was performed using the unpaired Student t test. Significance is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
The increased resistance to both gentamicin and penicillin G seen for pneumococci colonizing the nasopharyngeal tissue was not associated with an acquired gentamicin or penicillin resistance, as both bacteria from colonized, untreated animals and bacteria surviving treatment with gentamicin retained an MIC of 16 μg/ml and an MBC of 32 μg/ml for gentamicin and 0.01 μg/ml for penicillin G. The increased resistance of tissue-associated bacteria to antimicrobial treatment further supports the hypothesis that pneumococci make biofilms during colonization of the nasopharyngeal mucosa.
Pneumococcal biofilms formed on epithelial cells in vitro show similarities with in vivo biofilms.
As S. pneumoniae does not colonize any other niche than the mucosal surface of the nasopharynx by forming biofilms, we were interested in evaluating the role of bacterial-host cell interactions during biofilm formation in vitro.
We first tested the ability of pneumococci to form biofilms on primary ciliated tracheobronchial epithelial cells from healthy volunteers. S. pneumoniae EF3030 was inoculated at a multiplicity of infection (MOI) of 1 bacterium per cell into the upper compartment of a Transwell with live primary ciliated tracheobronchial epithelial cells grown to confluence in an air-liquid interface. The cell culture medium in the lower chamber was changed every 12 h. At 24 h, S. pneumoniae had formed early-stage biofilms, while the ciliated tracheobronchial epithelial cells remained largely intact (Fig. 3). Bacterial aggregates in more than one layer were observed on top of the cells, and extracellular matrix formation and bacterial interconnections were visible at this stage. However, at later times points, the epithelium disintegrated, causing transmigration of bacteria, and biofilm formation was difficult to evaluate.
Fig 3.

S. pneumoniae forms biofilms over live primary ciliated tracheobronchial epithelial cells in vitro. S. pneumoniae EF3030 was inoculated on top of differentiated tracheobronchial epithelial cells growing in an air-liquid interphase for 24 h and prepared for SEM analysis. (A) Low-magnification view showing S. pneumoniae EF3030 attachment and early biofilm formation in aggregates on the epithelial cell surface, with some detectable matrix formation. (B) High-magnification view showing pneumococcal attachment to cells and other pneumococci through fibrous structures as well as early aggregation of bacteria, with some extracellular material being produced (arrows). This figure shows a representative of one infection culture. However, the experiment has been reproduced more than three times with similar results.
The NCI-H292 mucoepidermoid bronchial carcinoma cells were not useful for live biofilm assays, as the cells were rapidly killed by the bacterial inoculum (within 12 h). These results confirm our earlier observation that pneumococci are toxic to epithelial cells in the absence of a host response (29). However, we were positively surprised to see that primary differentiated cells were much more resistant to pneumococcal toxicity than cell lines, suggesting that these cells are a useful tool for investigating early pneumococcal-host cell interactions.
To determine the usefulness of a respiratory epithelial substratum for pneumococcal biofilm formation over longer periods of time, NCI-H292 or primary differentiated tracheobronchial epithelial cells were grown to confluence and fixed in 4% paraformaldehyde. To evaluate the role of fixation on bacterial association, pneumococci were added at a multiplicity of infection (MOI) of 10 bacteria per cell for 1 h and total cell-associated bacteria were measured. There was no significant difference in adherence between live and fixed cells, suggesting that the adhesive ability of the cells remained intact after fixation (Fig. 4).
Fig 4.
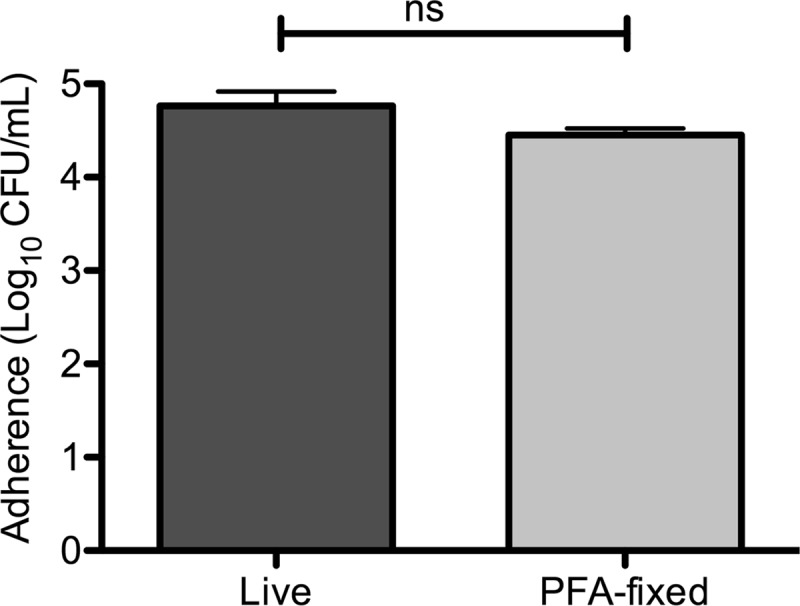
Adherence of pneumococci to live and prefixed bronchial epithelial cells. Bronchial carcinoma NCI-H292 cells were grown to confluence and washed in PBS or fixed with 4% paraformaldehyde. Live and prefixed cells were inoculated with 107 S. pneumoniae EF3030 bacteria for 1 h, and cell association was measured. The results represent three separate experiments with duplicate samples. Statistical analysis was performed using the unpaired Student t test. ns, not significant.
S. pneumoniae EF3030 was then inoculated on top of prefixed epithelial substrata for 48 h, and the biofilms were inspected by SEM and compared with the biofilms observed in murine nasopharyngeal tissues (Fig. 5). Low-magnification images show extensive 3-dimensional structures with deep layers of bacteria embedded and surrounded by an amorphous matrix in most areas. In the higher-magnification images we chose areas where bacterial density and structural interactions could be more clearly visualized and matrix formation was minimal. Biofilms formed both on prefixed carcinoma cells and on prefixed primary ciliated tracheobronchial epithelial cells shared morphology and general architecture with biofilms formed in vivo, with regard to both the general architecture and matrix formation (Fig. 5A to C) and the bacterial organization (Fig. 5D to F). This suggests that biofilms formed on both live and fixed epithelial substrata provide suitable in vitro models for pneumococcal biofilm formation in vivo.
Fig 5.

Comparison of S. pneumoniae biofilms formed on epithelial cells in vitro with biofilms formed in vivo in the mouse nasopharynx. Scanning electron microscopy images show the morphology of S. pneumoniae EF3030 biofilms formed on paraformaldehyde-fixed NCI-H292 carcinoma cells (A and D), on primary, differentiated, tracheobronchial epithelial cells (B and E), or in the murine nasopharynx in vivo (C and F) at low (A to C) or high (D to F) magnification. S. pneumoniae biofilms formed in vitro over respiratory epithelial cells under static conditions for 48 h display phenotypes similar to those of biofilms formed in vivo within the mouse nasopharynx after 48 h.
Abiotic surfaces provide less ample support for biofilm formation than epithelial cells.
To evaluate how biofilms produced on epithelial substrata compare with those produced on abiotic surfaces, S. pneumoniae EF3030 cells were seeded on glass surfaces or prefixed NCI-H292 cells and their morphology was evaluated by SEM (Fig. 6A), whereas accumulation of biomass (Fig. 6B) and resistance to gentamicin (Fig. 6C) or penicillin G (Fig. 6D and E) were evaluated by viable counts over time. Biofilm formation on epithelial cells was initiated faster than that on glass, with clear aggregate formation seen already after 6 h, and resulted in a more advanced and specialized architecture of the biofilm over time, with water channels and tower structures in a general “honeycomb”-like structure with extracellular matrix formation and a higher degree of biomass (Fig. 6A; Fig. 7 shows more structural detail). The biofilm formation on glass was delayed, with mere accretion after 6 h, and lacking in the phenotypic structures seen both for the biofilms on epithelial cells and the biofilms observed in vivo during nasopharyngeal colonization, suggesting that biofilm formation on abiotic surfaces failed to mimic the in vivo situation equally well.
Fig 6.

Impact of epithelial substratum on in vitro biofilm formation by EF3030 pneumococci. S. pneumoniae EF3030 was seeded on glass or on paraformaldehyde-fixed NCI-H292 cells. (A) Biofilm morphology was detected at 6, 24, and 48 h on both surfaces. A major difference was observed in the density, matrix formation, and organization of the biofilms, with epithelial cells showing increased support for biofilm formation. Insets show 2.5-fold increased magnifications of part of the field. (B) Biomass was measured by viable counts over time. (C to E) Biofilm sensitivity to gentamicin at 500 μg/ml (C), penicillin G at 1 μg/ml (D), and penicillin G at 10 μg/ml. All data in panels B to E represent six individual data points from three separate experiments. Statistical analysis was performed using the unpaired Student t test. Significance is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Fig 7.

Characteristics of pneumococci growing as biofilms on epithelial cells. S. pneumoniae EF3030 was seeded on ciliated primary bronchial epithelial cells or on paraformaldehyde-fixed NCI-H292 cells, and biofilm structure was investigated. (A) Matrix production in a 48-hour biofilm of S. pneumoniae D39 formed over a ciliated primary bronchial epithelium. Also visible in this image is the “honeycomb” structure that the pneumococci form within the larger biofilm. (B) Tower formations and “honeycomb” structure formations in a 48-h biofilm of S. pneumoniae D39 formed over an NCI-H292 epithelial substratum. (C) Low-magnification view demonstrating architectural complexity of a 48-h D39 biofilm formed over a ciliated primary bronchial epithelium, with towers and water channels and extracellular matrix formation covering the bacteria. (D) High-magnification view of a water channel within a mature D39 biofilm formed over a ciliated primary bronchial epithelium.
This was confirmed by measuring the biomass and antibiotic resistance of the respective biofilms (Fig. 6B and Fig. 6C to E, respectively). Biofilm biomass initially peaked at between 6 and 8 h after inoculation on both surfaces, being generally unchanged over the next 12 to 18 h, when bacteria were seen to organize into three-dimensional structures, and then increasing again to reach a steady state at 48 h, which remained stable over the next 48 h, with a significantly (>3 log10 CFU/biofilm) higher biomass of biofilms formed on the epithelial substratum (P < 0.01) (Fig. 6B). The difference in organization and maturation of the biofilms on glass and epithelial cells was mirrored by an increased resistance to gentamicin of biofilms formed on epithelial cells compared with those formed on glass (Fig. 6C). On both substrata, 6-hour biofilms were susceptible to gentamicin, but whereas the glass-derived biofilms remained somewhat sensitive to gentamicin over the next 48 h, epithelial cell-derived biofilms became increasingly and significantly (P < 0.05) more resistant to gentamicin than those on glass. Similar results were obtained when performing the experiments on plastic (not shown), suggesting that the type of abiotic surface did not play a role. Similar increases in resistance were observed also when penicillin was used at different concentrations (Fig. 6D and E) and with erythromycin (not shown). At the lower concentration of penicillin (1 μg/ml) (Fig. 6D), differences in antibiotic sensitivity were observed mainly at the earlier time points when biofilm maturation was ongoing, whereas later on, when the biofilms had completely matured, little antibacterial activity was observed regardless of the substratum. Using an increased dose of penicillin (10 μg/ml), early-stage biofilms were completely eradicated, whereas there was a significant difference in sensitivity to penicillin G after 48 h, when the biofilms had matured (Fig. 6E).
Similar phenotypes were detected for another encapsulated S. pneumoniae strain, D39, indicating that the effects are not strain specific. This strain also showed increased bacterial adhesion, accumulation, and structural maturity during biofilm formation on the epithelial surface compared with glass and showed the same differences in biomass accumulation and antibiotic resistance as those seen for S. pneumoniae EF3030 (Fig. 8). Overall, these results indicate that an epithelial substratum potentiates biofilm formation in a way similar to the morphology and antibiotic resistance patterns observed in vivo and that biofilms formed on glass or plastic lack features that may be important in vivo.
Fig 8.

Impact of epithelial substratum on in vitro biofilm formation by D39 pneumococci. S. pneumoniae D39 was seeded on glass or on paraformaldehyde-fixed NCI-H292 cells. (A) Biofilm morphology was detected at 6, 24, and 48 h on both surfaces. (B) Biomass was measured by viable counts over time. (C to E) Biofilm sensitivity to gentamicin at 500 μg/ml (C), penicillin G at 1 μg/ml (D), and penicillin G at 10 μg/ml (E). All data in panels B to E represent six individual data points from three separate experiments. Statistical analysis was performed using the unpaired Student t test. Significance is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Correlation between in vivo colonization ability and biofilm formation on epithelial cells.
Although EF3030 and D39 pneumococci both showed an increased ability to form biofilms on epithelial cells compared with abiotic surfaces, the strains were not equally good at forming biofilms on epithelial cells. Compared with that by strain EF3030, biofilm formation by D39 was slower and demonstrated less structural maturity as evidenced by the development of decreased biofilm-specific antimicrobial resistance (Fig. 9A). The superior biofilm formation and resistance to insult (chemotherapeutic or potentially host derived) correlate well with the propensity for EF3030 (a serotype 19F clinical otitis isolate) to form stable colonization within the murine nasopharynx for extended periods of time. In contrast, D39 colonizes poorly and is more prone to disseminate to the bloodstream (Fig. 9B). To verify the correlation between biofilm formation in vitro on epithelial cells and colonization in vivo, three additional clinical strains, BG8826 (a serotype 19F strain isolated from the nasopharynx of a healthy child), SP670 (a serogroup 6, penicillin-resistant otitis isolate), and WU2 (a serotype 3, mouse-passaged clinical isolate) were included as comparisons. The nasopharyngeal isolate BG8826 produced well-developed and mature biofilms with high resistance to antibiotics, whereas SP670 formed poor biofilms with low resistance to antibiotics (Fig. 9A). The strains' ability to form structured biofilms on epithelial cells and their resistance to antibiotic treatment correlated well with each strain's ability to colonize the nasopharynxes of mice (Fig. 9B). Importantly, when producing biofilms on abiotic surfaces, no correlation between biofilm formation and antibiotic resistance on one hand and colonization on the other could be detected (not shown). The WU2 strain failed to form mature biofilms on epithelial cells, resulting in accretion in the range of 103 CFU/well that was completely eradicated by gentamicin. This correlates well with this strain's inability to colonize the nasopharynx at high levels (with a mean of 4 × 104 CFU/tissue measured after 24 h, after which it declined) and its ability to quickly disseminate to the bloodstream, causing sepsis in mice.
Fig 9.

Correlation between nasopharyngeal colonization and biofilm formation on epithelial surfaces. (A) Gentamicin sensitivity presented as log10 death of 48-h biofilms formed over prefixed NCI-H292 cells inoculated with S. pneumoniae BG8826, EF3030, D39, or SP670. This experiment was repeated three times. (B) Mice were colonized with the same strains for 48 h, and nasopharyngeal tissue was harvested and bacterial burden enumerated from viable counts of tissue homogenate. The results are based on one group of six mice for each strain. Statistical analysis was performed using the unpaired Student t test. Significance is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Correlation between in vivo colonization ability of mutants lacking virulence determinants and biofilm formation.
To further validate the important role for epithelial-bacterial cell interactions in biofilm formation and the usefulness of biofilm formation on epithelial surfaces to predict colonization efficiency, we used the wild-type strain D39 and deletion mutants lacking each of five virulence-associated molecules (autolysin, pneumolysin, PspC, PspA, and capsule). The D39 strain background rather than the EF3030 strain background was chosen both because these strains have been used previously in biofilm experiments on abiotic surfaces, which would allow comparisons with previous literature in the field, and because they are well studied for their ability to interact with host structures and impact colonization or invasive disease (4, 8, 9, 28, 53). The strains were grown on glass or prefixed NCI-H292 epithelial cells for various times and evaluated by SEM, as well as for biomass and resistance to gentamicin. To ensure that these strains were equally sensitive to gentamicin as the wild-type strain, the MIC and MBC for each strain were determined and were found to be 16 μg/ml and 32 μg/ml, respectively, for all stains.
All mutants, with the exception of the unencapsulated strain, showed decreased biomass on plastic, as measured by crystal violet staining after 6 h (Fig. 10A), when only accretion of bacteria was visible (Fig. 11A). This difference disappeared after 48 h (Fig. 10B and C). In accordance, these strains were all equally sensitive to gentamicin treatment as the wild-type strain at this time point (Fig. 10E), except for the autolysin-negative strain, which showed a somewhat increased sensitivity.
Fig 10.

Biofilm-associated properties of pneumococci lacking colonization-associated virulence determinants on epithelial cells and abiotic surfaces. Biofilms were formed on glass or paraformaldehyde-fixed NCI-H292 cells by S. pneumoniae D39 and D39 mutant strains lacking PspA, PspC, autolysin (LytA), or pneumolysin (Ply). (A) Crystal violet staining after 6 h of growth on glass. One-way ANOVA to investigate variance of the means showed a P value of <0.05. (B) Crystal violet staining after 48 h of growth on glass (P = 0.83 using one-way ANOVA). (C) Biomass after 48 h of growth on glass, determined by viable counts (P = 0.36 using one-way ANOVA). (D) Biomass after 48 h of growth on epithelial cells, determined by viable counts (P < 0.01 using one-way ANOVA). (E) Gentamicin (500 μg/ml) sensitivity of biofilms developed on glass for 48 h (P < 0.05 using one-way ANOVA). (F) Gentamicin (500 μg/ml) sensitivity of biofilms developed on epithelial cells for 48 h (P < 0.001 using one-way ANOVA). All results are based on three to five separate experiments with duplicate samples. Statistics displayed on the graph represent column comparisons using the unpaired Student t test. Significance is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Fig 11.


Correlation between colonization-associated pneumococcal virulence determinants and biofilm formation on epithelial cells. Biofilms were formed on glass or paraformaldehyde-fixed NCI-H292 cells by S. pneumoniae D39 and D39 mutant strains lacking PspA, PspC, autolysin (LytA), or pneumolysin (Ply). Biofilm architecture was visualized by scanning electron microscopy after 48 h on glass and epithelial cells. (A) Forty-eight-hour biofilm development on abiotic surfaces. Insets show 20-fold-increased magnifications of parts of the field. (B) Forty-eight-hour biofilm formation over a prefixed epithelial cell substratum. Insets show 20-fold-increased magnifications of parts of the field.
The situation was very different for biofilms formed on epithelial cells. Autolysin, pneumolysin, and PspC were all required for optimal formation of S. pneumoniae biofilms on epithelial cells, and biofilms of pneumococci lacking these proteins were less visually robust (Fig. 11B), showed less biomass (Fig. 10D), and were significantly more sensitive to gentamicin than wild-type D39 pneumococci (Fig. 10F). Morphologically, strains lacking autolysin showed the most deficient biofilm phenotype (Fig. 11B). These pneumococci produced no visual matrix material and appeared as featureless bacterial chains rather than as the architecturally complex biofilms formed by the wild-type strain. Pneumolysin-negative pneumococci maintained a similar matrix production and formation of “honeycomb” patterns, but these were organized in a different pattern and with less density than those of the wild-type strain (Fig. 11B). The strain lacking PspC created biofilms on epithelial cells with a different architecture, showing more prominent cell clusters and less matrix material or fibrous connections between the bacteria (Fig. 11B).
Deletion of PspA had no effect on biofilm formation over 48 h as assessed either by measuring biomass (Fig. 10D) or visually by SEM (Fig. 11B) and caused no increased sensitivity to gentamicin (Fig. 10F).
Role of capsule in pneumococcal biofilm formation.
Interestingly, despite the absolute capsular requirement for colonization (30), the acapsular strain AM1000 (D39-Cps) showed a significant increase in bacterial binding to plastic after 6 h compared with D39 pneumococci (Fig. 12A). Although this initial increase was less pronounced after 48 h of incubation (Fig. 12A), SEM images still provided evidence for increased biofilm formation, with more pronounced maturation and organization than for any of the encapsulated strains (Fig. 12B), and was associated with an increased resistance to gentamicin (Fig. 12C and D).
Fig 12.

Impact of epithelial substratum on in vitro biofilm formation by unencapsulated pneumococci. S. pneumoniae D39-Cps (AM1000) was seeded on glass or on paraformaldehyde-fixed NCI-H292 cells. (A) D39 and D39-Cps were seeded on glass for 6 and 48 h, and biomass was measured with uptake of crystal violet. The results represent three individual experiments with duplicate samples. (B) Biofilm morphology inspected by SEM at 6 and 48 h on both surfaces. (C) D39 and D39-Cps were seeded on glass for 48 h and tested for their sensitivity to gentamicin (500 μg/ml). The results represent three individual experiments with duplicate samples. (D) Biomass was measured by viable counts over time. The results represent six individual data points from three separate experiments. (E) D39-Cps (AM1000) sensitivity to gentamicin (500 μg/ml) was observed throughout the time course of the experiment. The data represent six individual data points from three separate experiments. Statistical analysis was performed using the unpaired Student t test. Significance is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
The same was seen on epithelial surfaces, where D39-Cps showed an increased initial adherence about 10-fold higher than that of the encapsulated wild-type strain after 6 h (Fig. 12A), suggesting that additional adhesins are exposed in the absence of capsule, giving the bacteria an advantage to form an increased initial biomass in the absence of an immune response. This resulted in the formation of biofilms that formed more rapidly than those of the encapsulated parent strain and showed similar levels of matrix material at 48 h with no increased sensitivity to antibiotic treatment (Fig. 12D). This indicates that the biofilm matrix does not involve the pneumococcal capsule and confirms the results above that the extracellular matrix was formed from cells dying and lysing during the biofilm production. Cell death in the biofilms was directly confirmed using LIVE/DEAD staining of 48-h biofilms with both D39 and D39-Cps, which showed bacterial death randomly distributed throughout the biofilms (not shown). Cell death was also observed in biofilms with the LytA-negative strain that lacked matrix formation based on the inability of the dead cells to lyse. In fact, we observed consistently that biofilms formed from encapsulated strains on epithelial cells showed decreased expression of capsule (Fig. 13C) compared to broth-grown bacteria (Fig. 13A). To further confirm our visual observations, we measured cps2 expression using qRT-PCR under planktonic and biofilm growth conditions in 3 independent biofilms after 48 h of growth over a prefixed epithelial substratum. Expression of cps2 was downregulated in all biofilms relative to that under planktonic growth conditions, with an average of 32.7-fold relative reduction (P < 0.005) compared to that of planktonic cultures. These results agree with previous studies showing that cpsA is downregulated up to 10-fold during biofilm growth over an abiotic substratum, compared with planktonic cultures (34), and are also confirmed by the studies by Hammerschmidt et al. showing a decreased capsule formation when bacteria bind to epithelial cells (35).
Fig 13.

Decreased capsule expression during biofilm formation on epithelial cells. Scanning electron micrographs of D39 encapsulated bacteria grown in broth (A), the D39 unencapsulated mutant AM1000 grown in broth (B), and a section of a D39 biofilm after 48 h of growth (C) are shown. Bacteria growing in biofilm decrease their capsule expression.
These results suggest that epithelial cell substrata provide a more physiological environment for biofilm formation than abiotic surfaces and can better predict a role of colonization-associated factors during infection in vivo than biofilms formed on glass.
DISCUSSION
The results of the present study demonstrate that pneumococci form biofilms during carriage in a host and that this colonization pattern is associated with an increased resistance to antibiotic treatment. These are the first experimental data to show pneumococcal biofilm formation during nasopharyngeal carriage, and they support the fact that it is more difficult to eradicate pneumococcal colonization with antibiotics in patients than to eradicate infection (18, 19, 21–23, 26, 72). The studies also show the importance of continuous bacterial interactions with epithelial cells both to obtain biofilms with a structure similar to that seen during in vivo colonization and to produce pneumococcal biofilms whose characteristics directly correlate with in vivo colonization.
During nasopharyngeal colonization, the bacteria produced well-structured biofilms with a “honeycomb”-like initial interaction pattern that developed into tower structures with water channels, numerous fibrous connections between the bacteria, and embedment into an extracellular matrix. Interestingly, biofilm formation increased the more posterior in the nasopharyngeal tissue we observed, which would suggest either that the nasopharyngeal mucosa has different properties in different locations or, more likely, that the higher temperature in the more posterior regions better supports growth of biofilms.
To test the ability of antibiotics to eradicate colonization, we chose to use both the bactericidal antibiotic gentamicin and the more clinically relevant bacteriostatic penicillin G. Although the therapeutic concentrations of antibiotics in the tissues of treated mice could not be easily measured, the results definitively show that doses significantly higher than those that eradicate planktonic organisms in vitro were needed to eradicate colonization in the mice. For penicillin G, no significant reduction of nasopharyngeal tissue-associated bacteria was detected even at a concentration of 5,000 μg/ml. The data also showed that bacteria loosely associated with the tissues, and present in nasal washes, were more sensitive to antibiotic treatment than those more closely associated with the tissue, which were more likely to be integrated in biofilm structures. We do not believe that this difference in sensitivity is a matter of the antibiotic treatment not reaching the bacteria in the tissues, as we never observed bacteria invading the tissue. Rather, all bacteria and biofilm structures detected were observed on top of the ciliated epithelium, where they would be easily accessible to the antibiotic that was administered intranasally. The resistance to antimicrobial treatment further supports the notion that pneumococci make biofilms during colonization of the nasopharyngeal mucosa and suggests that biofilm formation during colonization may be one mechanism whereby the bacteria protect themselves against host defense mechanisms present on mucosal surfaces and an explanation for the clinically increased resistance to antibiotic treatment of pneumococcal carriage. Thus, prevention efforts focused on eradicating colonization as a means to reduce pneumococcal disease need to consider the decreased antimicrobial susceptibility associated with biofilm development of this organism.
Even though it is well known that pneumococci naturally colonize and survive in only the human host, few studies have been conducted to understand the roles of different mucosal structures in biofilm formation of pneumococci, and no studies have investigated the role of the continuous presence of epithelium during biofilm formation by pneumococci. There is a vast literature of pneumococcal biofilm studies on abiotic surfaces that show the importance of virulence factors, autoinducers, extracellular DNA, and extracellular matrix production in pneumococcal biofilm formation in vitro (15, 50, 56, 59, 67–69). The in vivo relevance of these studies is, however, not clear, as most of these studies were conducted over too short a time span to actually produce biofilms, defined as aggregated bacterial communities producing extracellular matrix. Thus, how these results relate to the respective strain's ability to form biofilms both in vitro and in vivo is unknown. For example, our study confirmed earlier results (49) showing that PspA-negative bacteria display decreased accretion on plastic surfaces at 6 h, but we show here that at 48 h this strain did not show any difference in biofilm formation compared with its parent strain, and no difference in early colonization has been seen (4), which correlates well with PspA's major role in septic infection.
Studies conducted for longer periods of time have also failed to show any association between the ability of clinical isolates to produce biofilms on abiotic surfaces in vitro and their virulence or isolation site in vivo (43, 65). This was also confirmed in our studies. Related to this is the controversy in the literature about the infectivity of pneumococci grown as biofilms on abiotic surfaces, with biofilm-grown bacteria shown to be both more likely (68) and less likely (59) to cause invasive disease in murine models.
The poor correlation between biofilm formation on abiotic surfaces and colonization efficiency, combined with our knowledge that pneumococci during their natural life cycle never form biofilms on abiotic surfaces and that biofilms formed on abiotic surfaces failed to show the structural and antibiotic resistance phenotypes that we observed during colonization, stimulated our interest in investigating the role of respiratory epithelial cells during pneumococcal biofilm formation in vitro. So far, two studies have included epithelial cells. Parker et al. showed that bacteria that adhere to epithelial cells and are detached by trypsin form more developed biofilms on abiotic surfaces than the original culture (56), and Sanchez et al. showed that after forming biofilms on abiotic surfaces, bacteria will bind better to epithelial cells (59). However, neither of these studies investigated how the presence of respiratory epithelial cells affects the formation of pneumococcal biofilms in vitro and how this correlates with pneumococcal colonization ability.
Here, we used an in vitro model to simulate the biology of the upper respiratory tract, and we were able to mimic the phenotypes associated with pneumococcal biofilm formation in vivo, including morphology and antibiotic resistance, in a way abiotic surfaces could not. This indicated an important role of mucosal cells in the biofilm formation process by potentially inducing optimal interbacterial signaling and expression of colonization-associated factors needed for biofilm formation through adherence to in vivo ligands. Furthermore, our results demonstrate that the development of biofilm-specific properties, including antibiotic resistance, during biofilm formation on epithelial cells correlates directly with the ability of both clinical strains and isogenic mutants lacking factors involved in colonization to colonize the murine nasopharynx. In support of this, we showed that clinical isolates from children (EF3030 and BG8826), which effectively colonize the murine nasopharynx (13, 42), formed biofilms of higher biomass and more developed architecture, with increased biofilm-specific antibacterial resistance on epithelial cells, than strains that colonize less effectively (D39, WU2, and SP670) and are more prone to disseminate to the bloodstream and cause septic infection (7, 32, 44, 48, 54). This difference in biofilm formation was not observed using abiotic surfaces. The correlation seen here between biofilm formation on epithelial cells and the colonization efficiency and virulence of clinical isolates is in contrast to the observed inability to correlate biofilm formation in vitro on abiotic surfaces with virulence in vivo (43, 65) and suggests that epithelial cell interaction during biofilm formation in vitro is essential and mimics the in vivo situation to a degree that can better predict virulence phenotypes in vivo.
Similarly, we found that deletion of PspA, which protects against complement attack in the bloodstream but shows no role in early colonization (4), had no effect on biofilm formation over 48 h, whereas PspC-, autolysin-, and pneumolysin-negative strains, which colonize poorly in vivo (4, 8, 53, 71), formed less-structured biofilms on epithelial cells than the wild-type strain. By quantifying biomass and biofilm-specific resistance to antibiotics after biofilm formation over a prefixed epithelial substratum, we could accurately predict virulence phenotypes using our in vitro model, whereas no difference was observed when biofilms were formed on abiotic surfaces.
The only discrepancy between colonization and biofilm formation in our studies was seen with the acapsular strain, which does not colonize effectively in vivo but formed advanced biofilms on both abiotic and epithelial surfaces in vitro. This is most probably associated with its increased ability to adhere to surfaces and cells in the absence of capsule, which was shown in this study and has been shown by others (49, 50). Even though it can be seen as an in vitro artifact, the role of capsule during colonization and biofilm formation has been addressed previously. First, it implies that the capsular material is not involved in forming the extracellular matrix surrounding the bacteria in the biofilm, as similar levels of matrix are produced by acapsular bacteria (Fig. 12). Rather, our study shows that the extracellular material requires lysis and death of bacteria in the biofilm, as autolysin-negative bacteria were completely devoid of extracellular material and made biofilms with poor organization, low biomass, and high antibiotic sensitivity (Fig. 11B). Second, there is evidence that capsule is downregulated when in intimate contact with epithelial cells (35), that the transparent phenotype that is associated with improved colonization of the nasopharynx has a reduced expression of capsular polysaccharide (74), and that biofilms produced on abiotic surfaces downregulate their capsular material (59), something we observed as well in our biofilms on epithelial cells. This would suggest that a downregulation of capsule is potentially important for optimal colonization and biofilm formation in vivo but that enough capsule must be present initially to circumvent host innate immunity.
In conclusion, this study shows for the first time that pneumococci form biofilms during carriage in mice and that carriage is associated with a significantly increased resistance to antibiotic treatment. The study also shows that only in the continuous presence of epithelial cells will pneumococci form biofilms with a structure and antibiotic resistance pattern similar to those of biofilms in vivo. Furthermore, the ability of both clinical bacterial strains and strains with mutations in potential virulence factors to form biofilms on epithelial cells directly correlates with their ability to colonize the murine nasopharynx in vivo, which is not achieved using abiotic surfaces. Thus, by modeling in vitro the essential epithelial-bacterial cell interactions that occur during biofilm formation in the respiratory tract, future studies may provide insights into the mechanisms that pneumococci use to adhere and colonize, compete with other microbial species, and survive as commensals in their native environment.
ACKNOWLEDGMENTS
This work was supported by funding from the Bill and Melinda Gates Foundation (grant 53085); the JR Oishei Foundation, Buffalo, NY; and The American Lung Association (grant RG-123721-N).
We thank Peter Bush for help and training with the scanning electron microscope and Robert Tyx for help with the colonization studies.
Footnotes
Published ahead of print 29 May 2012
REFERENCES
- 1. Abdi-Ali A, Mohammadi-Mehr M, Agha Alaei Y. 2006. Bactericidal activity of various antibiotics against biofilm-producing Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 27:196–200 [DOI] [PubMed] [Google Scholar]
- 2. Andersson B, et al. 1983. Identification of an active disaccharide unit of a glycoconjugate receptor for pneumococci attaching to human pharyngeal epithelial cells. J. Exp. Med. 158:559–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Avery OT, MacLeod CM, McCarty M. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Induction of transformation by a dexoxyribonuceic acid fraction isolated from pneumococcus type III. J. Exp. Med. 79:137–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Balachandran P, Brooks-Walter A, Virolainen-Julkunen A, Hollingshead SK, Briles DE. 2002. Role of pneumococcal surface protein C in nasopharyngeal carriage and pneumonia and its ability to elicit protection against carriage of Streptococcus pneumoniae. Infect. Immun. 70:2526–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bartoszewicz M, Rygiel A, Krzeminski M, Przondo-Mordarska A. 2007. Penetration of a selected antibiotic and antiseptic into a biofilm formed on orthopedic steel implants. Ortop. Traumatol. Rehabil. 9:310–318 [PubMed] [Google Scholar]
- 6. Bast DJ, et al. 2004. Novel murine model of pneumococcal pneumonia: use of temperature as a measure of disease severity to compare the efficacies of moxifloxacin and levofloxacin. Antimicrob. Agents Chemother. 48:3343–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benton KA, Everson MP, Briles DE. 1995. A pneumolysin-negative mutant of Streptococcus pneumoniae causes chronic bacteremia rather than acute sepsis in mice. Infect. Immun. 63:448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berry AM, Lock RA, Hansman D, Paton JC. 1989. Contribution of autolysin to virulence of Streptococcus pneumoniae. Infect. Immun. 57:2324–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berry AM, Yother J, Briles DE, Hansman D, Paton JC. 1989. Reduced virulence of a defined pneumolysin-negative mutant of Streptococcus pneumoniae. Infect. Immun. 57:2037–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bogaert D, De Groot R, Hermans PW. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect. Dis. 4:144–154 [DOI] [PubMed] [Google Scholar]
- 11. Bortoni ME, Terra VS, Hinds J, Andrew PW, Yesilkaya H. 2009. The pneumococcal response to oxidative stress includes a role for Rgg. Microbiology 155:4123–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bowers EF, Jeffries LR. 1955. Optochin in the identification of str. pneumoniae. J. Clin. Pathol. 8:58–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Briles DE, et al. 2003. Immunizations with pneumococcal surface protein A and pneumolysin are protective against pneumonia in a murine model of pulmonary infection with Streptococcus pneumoniae. J. Infect. Dis. 188:339–348 [DOI] [PubMed] [Google Scholar]
- 14. Briles DE, et al. 1981. Antiphosphocholine antibodies found in normal mouse serum are protective against intravenous infection with type 3 Streptococcus pneumoniae. J. Exp. Med. 153:694–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Camilli R, Pantosti A, Baldassarri L. 2011. Contribution of serotype and genetic background to biofilm formation by Streptococcus pneumoniae. Eur. J. Clin. Microbiol. Infect. Dis. 30:97–102 [DOI] [PubMed] [Google Scholar]
- 16. Carmen JC, et al. 2004. Ultrasonic-enhanced gentamicin transport through colony biofilms of Pseudomonas aeruginosa and Escherichia coli. J. Infect. Chemother. 10:193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chole RA, Faddis BT. 2003. Anatomical evidence of microbial biofilms in tonsillar tissues: a possible mechanism to explain chronicity. Arch. Otolaryngol. Head Neck Surg. 129:634–636 [DOI] [PubMed] [Google Scholar]
- 18. Cohen R, et al. 1997. Change in nasopharyngeal carriage of Streptococcus pneumoniae resulting from antibiotic therapy for acute otitis media in children. Pediatr. Infect. Dis. J. 16:555–560 [DOI] [PubMed] [Google Scholar]
- 19. Cohen R, et al. 1999. One dose ceftriaxone vs. ten days of amoxicillin/clavulanate therapy for acute otitis media: clinical efficacy and change in nasopharyngeal flora. Pediatr. Infect. Dis. J. 18:403–409 [DOI] [PubMed] [Google Scholar]
- 20. Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322 [DOI] [PubMed] [Google Scholar]
- 21. Dabernat H, et al. 1998. Effects of cefixime or co-amoxiclav treatment on nasopharyngeal carriage of Streptococcus pneumoniae and Haemophilus influenzae in children with acute otitis media. J. Antimicrob. Chemother. 41:253–258 [DOI] [PubMed] [Google Scholar]
- 22. Dagan R, Klugman KP, Craig WA, Baquero F. 2001. Evidence to support the rationale that bacterial eradication in respiratory tract infection is an important aim of antimicrobial therapy. J. Antimicrob. Chemother. 47:129–140 [DOI] [PubMed] [Google Scholar]
- 23. Dagan R, et al. 1998. Dynamics of pneumococcal nasopharyngeal colonization during the first days of antibiotic treatment in pediatric patients. Pediatr. Infect. Dis. J. 17:880–885 [DOI] [PubMed] [Google Scholar]
- 24. Domenech M, Garcia E, Moscoso M. 2009. Versatility of the capsular genes during biofilm formation by Streptococcus pneumoniae. Environ. Microbiol. 11:2542–2555 [DOI] [PubMed] [Google Scholar]
- 25. Donlan RM, Costerton JW. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15:167–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garcia-Rodriguez JA, Fresnadillo Martinez MJ. 2002. Dynamics of nasopharyngeal colonization by potential respiratory pathogens. J. Antimicrob. Chemother. 50(Suppl. S2):59–73 [DOI] [PubMed] [Google Scholar]
- 27. Gray BM, et al. 1981. Epidemiologic studies of Streptococcus pneumoniae in infants: antibody response to nasopharyngeal carriage of types 3, 19, and 23. J. Infect. Dis. 144:312–318 [DOI] [PubMed] [Google Scholar]
- 28. Griffith F. 1928. The significance of pneumococcal types. J. Hyg. (Lond.) 27:113–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hakansson A, et al. 1996. Aspects on the interaction of Streptococcus pneumoniae and Haemophilus influenzae with human respiratory tract mucosa. Am. J. Respir. Crit. Care Med. 154:S187–S191 [DOI] [PubMed] [Google Scholar]
- 30. Hakansson A, Zhivotovsky B, Orrenius S, Sabharwal H, Svanborg C. 1995. Apoptosis induced by a human milk protein. Proc. Natl. Acad. Sci. U. S. A. 92:8064–8068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hakenbeck R, et al. 1991. Antigenic variation of penicillin-binding proteins from penicillin-resistant clinical strains of Streptococcus pneumoniae. J. Infect. Dis. 164:313–319 [DOI] [PubMed] [Google Scholar]
- 32. Hakenbeck R, et al. 1999. Penicillin-binding proteins in beta-lactam-resistant streptococcus pneumoniae. Microb. Drug Resist. 5:91–99 [DOI] [PubMed] [Google Scholar]
- 33. Hall-Stoodley L, et al. 2006. Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. JAMA 296:202–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hall-Stoodley L, et al. 2008. Characterization of biofilm matrix, degradation by DNase treatment and evidence of capsule downregulation in Streptococcus pneumoniae clinical isolates. BMC Microbiol. 8:173 doi:10.1186/1471-2180-8-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hammerschmidt S, et al. 2005. Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect. Immun. 73:4653–4667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoa M, Syamal M, Sachdeva L, Berk R, Coticchia J. 2009. Demonstration of nasopharyngeal and middle ear mucosal biofilms in an animal model of acute otitis media. Ann. Otol. Rhinol. Laryngol. 118:292–298 [DOI] [PubMed] [Google Scholar]
- 37. Hogberg L, et al. 2007. Age- and serogroup-related differences in observed durations of nasopharyngeal carriage of penicillin-resistant pneumococci. J. Clin. Microbiol. 45:948–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johansen HK, Jensen TG, Dessau RB, Lundgren B, Frimodt-Moller N. 2000. Antagonism between penicillin and erythromycin against Streptococcus pneumoniae in vitro and in vivo. J. Antimicrob. Chemother. 46:973–980 [DOI] [PubMed] [Google Scholar]
- 39. Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288–301 [DOI] [PubMed] [Google Scholar]
- 40. Karp PH, et al. 2002. An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods Mol. Biol. 188:115–137 [DOI] [PubMed] [Google Scholar]
- 41. Lewis K. 2008. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 322:107–131 [DOI] [PubMed] [Google Scholar]
- 42. Lipsitch M, et al. 2000. Competition among Streptococcus pneumoniae for intranasal colonization in a mouse model. Vaccine 18:2895–2901 [DOI] [PubMed] [Google Scholar]
- 43. Lizcano A, Chin T, Sauer K, Tuomanen EI, Orihuela CJ. 2010. Early biofilm formation on microtiter plates is not correlated with the invasive disease potential of Streptococcus pneumoniae. Microb. Pathog. 48:124–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Magee AD, Yother J. 2001. Requirement for capsule in colonization by Streptococcus pneumoniae. Infect. Immun. 69:3755–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marshall KJ, Musher DM, Watson D, Mason EOJ. 1993. Testing of Streptococcus pneumoniae for resistance to penicillin. J. Clin. Microbiol. 31:1246–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McDaniel LS, et al. 1987. Use of insertional inactivation to facilitate studies of biological properties of pneumococcal surface protein A (PspA). J. Exp. Med. 165:381–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mitchell TJ. 2003. The pathogenesis of streptococcal infections: from tooth decay to meningitis. Nat. Rev. Microbiol. 1:219–230 [DOI] [PubMed] [Google Scholar]
- 48. Mizrachi-Nebenzahl Y, et al. 2003. Differential activation of the immune system by virulent Streptococcus pneumoniae strains determines recovery or death of the host. Clin. Exp. Immunol. 134:23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moscoso M, Garcia E, Lopez R. 2006. Biofilm formation by Streptococcus pneumoniae: role of choline, extracellular DNA, and capsular polysaccharide in microbial accretion. J. Bacteriol. 188:7785–7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Munoz-Elias EJ, Marcano J, Camilli A. 2008. Isolation of Streptococcus pneumoniae biofilm mutants and their characterization during nasopharyngeal colonization. Infect. Immun. 76:5049–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Brien KL, et al. 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902 [DOI] [PubMed] [Google Scholar]
- 52. Oggioni MR, et al. 2006. Switch from planktonic to sessile life: a major event in pneumococcal pathogenesis. Mol. Microbiol. 61:1196–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ogunniyi AD, et al. 2007. Contributions of pneumolysin, pneumococcal surface protein A (PspA), and PspC to pathogenicity of Streptococcus pneumoniae D39 in a mouse model. Infect. Immun. 75:1843–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Orihuela CJ, et al. 2003. Organ-specific models of Streptococcus pneumoniae disease. Scand. J. Infect. Dis. 35:647–652 [DOI] [PubMed] [Google Scholar]
- 55. Palaniappan R, et al. 2005. Differential PsaA-, PspA-, PspC-, and PdB-specific immune responses in a mouse model of pneumococcal carriage. Infect. Immun. 73:1006–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Parker D, et al. 2009. The NanA neuraminidase of Streptococcus pneumoniae is involved in biofilm formation. Infect. Immun. 77:3722–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reid SD, et al. 2009. Streptococcus pneumoniae forms surface-attached communities in the middle ear of experimentally infected chinchillas. J. Infect. Dis. 199:786–794 [DOI] [PubMed] [Google Scholar]
- 58. Roush SW, Murphy TV. 2007. Historical comparisons of morbidity and mortality for vaccine-preventable diseases in the United States. JAMA 298:2155–2163 [DOI] [PubMed] [Google Scholar]
- 59. Sanchez CJ, et al. 2011. Streptococcus pneumoniae in biofilms are unable to cause invasive disease due to altered virulence determinant production. PLoS One 6:e28738 doi:10.1371/journal.pone.0028738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sanchez CJ, et al. 2010. The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog. 6:e1001044 doi:10.1371/journal.ppat.1001044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sanderson AR, Leid JG, Hunsaker D. 2006. Bacterial biofilms on the sinus mucosa of human subjects with chronic rhinosinusitis. Laryngoscope 116:1121–1126 [DOI] [PubMed] [Google Scholar]
- 62. Shah P, Briles DE, King J, Hale Y, Swiatlo E. 2009. Mucosal immunization with polyamine transport protein D (PotD) protects mice against nasopharyngeal colonization with Streptococcus pneumoniae. Exp. Biol. Med. 234:403–409 [DOI] [PubMed] [Google Scholar]
- 63. Spangler SK, Jacobs MR, Appelbaum PC. 1997. Time-kill studies on susceptibility of nine penicillin-susceptible and -resistant pneumococci to cefditoren compared with nine other beta-lactams. J. Antimicrob. Chemother. 39:141–148 [DOI] [PubMed] [Google Scholar]
- 64. Stoodley P, Sauer K, Davies DG, Costerton JW. 2002. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 56:187–209 [DOI] [PubMed] [Google Scholar]
- 65. Tapiainen T, et al. 2010. Biofilm formation by Streptococcus pneumoniae isolates from paediatric patients. APMIS 118:255–260 [DOI] [PubMed] [Google Scholar]
- 66. Trappetti C, et al. 2011. The impact of the competence quorum sensing system on Streptococcus pneumoniae biofilms varies depending on the experimental model. BMC Microbiol. 11:75 doi:10.1186/1471-2180-11-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Trappetti C, et al. 2009. Sialic acid: a preventable signal for pneumococcal biofilm formation, colonization, and invasion of the host. J. Infect. Dis. 199:1497–1505 [DOI] [PubMed] [Google Scholar]
- 68. Trappetti C, Ogunniyi AD, Oggioni MR, Paton JC. 2011. Extracellular matrix formation enhances the ability of Streptococcus pneumoniae to cause invasive disease. PLoS One 6:e19844 doi:10.1371/journal.pone.0019844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Trappetti C, Potter AJ, Paton AW, Oggioni MR, Paton JC. 2011. LuxS mediates iron-dependent biofilm formation, competence, and fratricide in Streptococcus pneumoniae. Infect. Immun. 79:4550–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tyx RE, Roche-Hakansson H, Hakansson AP. 2011. Role of dihydrolipoamide dehydrogenase in regulation of raffinose transport in Streptococcus pneumoniae. J. Bacteriol. 193:3512–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van Rossum AM, Lysenko ES, Weiser JN. 2005. Host and bacterial factors contributing to the clearance of colonization by Streptococcus pneumoniae in a murine model. Infect. Immun. 73:7718–7726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Varon E, et al. 2000. Impact of antimicrobial therapy on nasopharyngeal carriage of Streptococcus pneumoniae, Haemophilus influenzae, and Branhamella catarrhalis in children with respiratory tract infections. Clin. Infect. Dis. 31:477–481 [DOI] [PubMed] [Google Scholar]
- 73. Waite RD, Struthers JK, Dowson CG. 2001. Spontaneous sequence duplication within an open reading frame of the pneumococcal type 3 capsule locus causes high-frequency phase variation. Mol. Microbiol. 42:1223–1232 [DOI] [PubMed] [Google Scholar]
- 74. Weiser JN. 2010. The pneumococcus: why a commensal misbehaves. J. Mol. Med. (Berl.) 88:97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wolcott RD, Ehrlich GD. 2008. Biofilms and chronic infections. JAMA 299:2682–2684 [DOI] [PubMed] [Google Scholar]