

Abstract
We propose two antigenic types of Helicobacter pylori lipopolysaccharides (LPS): highly antigenic epitope-carrying LPS (HA-LPS) and weakly antigenic epitope-carrying LPS (WA-LPS) based on human serum reactivity. Strains carrying WA-LPS are highly prevalent in isolates from gastric cancer patients. WA-LPS exhibits more potent biological activities compared to HA-LPS, namely, upregulation of Toll-like receptor 4 (TLR4) expression and induction of enhanced epithelial cell proliferation. The results of competitive binding assays using monosaccharides and methylglycosides, as well as binding assays using glycosidase-treated LPS, suggested that β-linked N-acetyl-d-glucosamine and β-linked d-galactose residues largely contributed to the highly antigenic epitope and the weakly antigenic epitope, respectively. WA-LPS exhibited greater binding activity to surfactant protein D (SP-D) in a Ca2+-dependent manner, and this interaction was inhibited by methyl-β-d-galactoside. The biological activities of WA-LPS were markedly enhanced by the addition of SP-D. Lines of evidence suggested that removal of β-N-acetyl-d-glucosamine residue, which comprises the highly antigenic epitope, results in exposure of the weakly antigenic epitope. The weakly antigenic epitope interacted preferentially with SP-D, and SP-D enhanced the biological activity of WA-LPS.
INTRODUCTION
Helicobacter pylori lipopolysaccharide (LPS) has markedly different characteristics compared to typical Gram-negative bacterial LPS, such as enterobacterial LPS. First, Helicobacter pylori LPS exhibits extremely low endotoxic activities, including lethality, pyrogenic activity, mitogenic activity, and induction of proinflammatory cytokine production (21, 26, 29). The low level of endotoxic activity is due to the variability in the chemical structures, i.e., the molecular species and the number of fatty acid residues of the lipid A portion (24). Some groups, including ours, have suggested that H. pylori LPS acts as a Toll-like receptor 2 (TLR2) agonist but not as a TLR4 agonist (16, 32, 41). In addition, there is a report that LPS derived from some strains can act as an antagonist of TLR4 (16). Conversely, other studies suggest that it acts as a TLR4 agonist (4, 8, 10, 27). Second, carbohydrate structures mimicking host Lewis antigens exist on the polysaccharide moiety (2, 19, 20). Lewis antigens are thought to be an escape mechanism from host immune defenses and can trigger an autoimmune response in the host (3, 7, 23). Third, we have identified two distinct antigenic epitopes on the polysaccharide moiety that are independent of Lewis antigen structures (35, 38, 39). We have termed these highly antigenic epitope and weakly antigenic epitope, respectively. Either highly antigenic epitope or weakly antigenic epitope appeared to be present on smooth-type LPS, namely, polysaccharide-carrying LPS. Strains that carry both epitopes or strains carrying neither epitope have yet to be identified among Japanese smooth-type isolates. Clinical isolates having weakly antigenic epitope-carrying LPS (WA-LPS) are the dominant strain isolated from gastric cancer patients. Isolates with the highly antigenic epitope-carrying LPS (HA-LPS) are frequently isolated from patients with chronic gastritis (35, 37, 38).
We previously showed that pretreatment with H. pylori LPS enhances Escherichia coli LPS-induced production of proinflammatory cytokines, such as interleukin-8 (IL-8), through induction of TLR4 expression in gastric epithelial cells (42). Furthermore, H. pylori LPS enhances the proliferation of gastric epithelial cells (4, 42). These events are believed to be mediated by activation of a transcription factor NF-Y via TLR2 and MEK1/2-ERK1/2 mitogen-activated protein (MAP) kinase signal transduction cascade (42). These activities of H. pylori LPS are significantly higher for WA-LPS than for HA-LPS. To date, the structures of these two epitopes have not been identified, even though the chemical structures of LPS derived from several strains have been determined (18). To clarify why the biological activities of HA-LPS and WA-LPS differ, it is essential to perform a characterization of the two dominant antigenic epitopes. In the present study, we tried to identify the carbohydrate residue(s) that contribute to the highly antigenic and the weakly antigenic epitopes using immunochemical approaches and examine the interaction of the H. pylori LPS with surfactant proteins, which are human lectins.
MATERIALS AND METHODS
H. pylori strains and human sera.
H. pylori clinical isolates were described previously (37). Sera derived from individuals infected with H. pylori were obtained as previously described (1, 37).
LPS.
LPS was prepared by hot phenol-water extraction and ultracentrifugation, as described previously (38). Highly purified LPS preparation used for biological assays was prepared previously (41). Briefly, the LPS preparation was treated with DNase I, RNase, and two lipoprotein lipases derived from bovine milk and Pseudomonas sp., followed by treatment with proteinase K. The resulting material was applied to an octyl-Sepharose column, and LPS was eluted with a linear gradient of 1-propanol. The antigenicity of the LPS (i.e., HA-LPS and WA-LPS) was determined by enzyme-linked immunosorbent assay (ELISA) using human sera, as described previously (37, 38). E. coli LPS derived from O111:B4 was purchased from Sigma-Aldrich (St. Louis, MO).
Recombinant surfactant protein A (SP-A) and SP-D.
The 1.13-kb cDNA for human SP-A1 and the 1.181-kb cDNA for human SP-D were inserted into the pEE14 plasmid vectors, and recombinant human SP-A and SP-D were expressed in CHO-K1 cells using the glutamine synthetase gene amplification system. Recombinant proteins were purified using a mannose-Sepharose 6B column, as described previously (14, 30). The endotoxin content in the SP-D preparation was <0.3 pg/μg of protein as determined by the Limulus amebocyte assay.
Binding of surfactant proteins and LPS.
LPS (5 μg/ml) was immobilized in the wells of a 96-well microplate. After blocking the wells with phosphate-buffered saline (PBS) containing 2% bovine serum albumin (BSA), SP-A or SP-D protein (10 μg/ml) dissolved in PBS containing 5 mM CaCl2 and 2% BSA was dispensed into the wells, followed by incubation at 37°C for 1 h. After washing the wells with PBS containing 5 mM CaCl2 and 0.05% Tween 20 [PBS(+)T], rabbit anti-SP-A or SP-D polyclonal antibodies (30) (1 μg/ml) were added, and then the plates were incubated at 37°C for 90 min. After washing the wells with PBS(+)T, horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulins antibodies (Biosource International, Camarillo, CA) were added as a secondary antibody, and the plates were incubated at 37°C for 90 min. After being washed with PBS(+)T, the plates were developed using tetramethylbenzidine (TMB) solution (KPL, Gaithersburg, MD). Reactions were terminated with 1 M phosphoric acid, and the absorbance at 450 nm (A450) was measured using a plate reader.
Competitive enzyme-linked immunosorbent assay (ELISA).
d-Glucose (d-Glc), d-galactose (d-Gal), d-mannose (d-Man), l-fucose (l-Fuc), d-fucose (d-Fuc), l-rhamnose (l-Rha), N-acetyl-d-glucosamine (d-GlcNAc), N-acetyl-d-galactosamine (d-GalNAc), N-acetyl-d-mannosamine (d-ManNAc), methyl-α-d-glucoside (d-Glc-α-OMe), methyl-β-d-glucoside (d-Glc-β-OMe), methyl-α-d-galactoside (d-Gal-α-OMe), methyl-β-d-galactoside (d-Gal-β-OMe), methyl-α-d-mannoside (d-Man-α-OMe), methyl-β-d-mannnoside (d-Man-β-OMe), methyl-α-l-fucoside (l-Fuc-α-OMe), methyl-β-l-fucoside (l-Fuc-β-OMe), methyl-α-N-acetyl-d-glucosaminide (d-GlcNAc-α-OMe), and methyl-β-N-acetyl-d-glucosaminide (d-GlcNAc-β-OMe) were purchased from Nacalai Tesque (Kyoto, Japan), Sigma-Aldrich, and Pfanstiel Laboratories (Waukegen, IL). Competitive ELISA using monosaccharides and methylglycosides was performed as described previously (40). Briefly, human serum, diluted at a concentration giving A450 of ∼1.0 in ELISA, was mixed with competitor and then incubated at 37°C for 1 h. The resulting mixture was applied to ELISA using LPS-coated microplates, as described previously (37, 40).
Glycosidase treatment of LPS.
β-N-Acetylglucosaminidase derived from Streptococcus pneumoniae (reaction conditions: 37°C in 50 mM sodium phosphate buffer [pH 5.0]) and β-galactosidase derived from Aspergillus oryzae (reaction conditions: 30°C in 50 mM sodium phosphate buffer [pH 4.5]) were purchased from Sigma-Aldrich. Other glycosidases were described previously (38). Glycosidase solution (1 mU/ml) was dispensed into LPS-coated wells of a microplate, followed by incubation for 12 h. The wells were washed with PBST, and then the reactivity of human sera was analyzed by ELISA, as described previously (37, 38).
Western blotting.
Rabbit anti-TLR2 and -TLR4 polyclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Preparation of cell lysates, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and Western blotting were carried out as previously described (31). The resulting protein bands were scanned on a flatbed scanner and quantified by using the ImageJ program (National Institutes of Health, Bethesda, MD).
Induction of IL-8 from gastric epithelial cells.
Gastric carcinoma cell lines (MKN28 and MKN45) were obtained from the Japan Collection of Research Biosources (Ibakaki, Japan). IL-8 production by gastric carcinoma cells was examined as described previously (41, 42). Typically, H. pylori LPS and SP-D were mixed and incubated in PBS containing 5 mM CaCl2 at 37°C for 30 min. The mixture or untreated H. pylori LPS was added to the cultures of gastric epithelial cells, and then the cells were incubated for 24 h. After washing the cells with PBS, the cells were cultured in the presence of E. coli LPS (100 ng/ml) for an additional 24 h. Concentrations of IL-8 in the culture supernatants were determined by ELISA (ELISA development kit; R&D Systems, Minneapolis, MN).
Cell proliferation assay.
H. pylori LPS or a mixture of H. pylori LPS and SP-D, prepared as described above, was added to gastric epithelial cells, and the cells were incubated for 18 h. Cell proliferation was determined by the uptake of 5-ethynil-2′-deoxyuridine (EdU) into DNA, using a Click-iT EdU microplate assay kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. EdU incorporated into DNA was coupled to Oregon Green-azide and then detected using an HRP-conjugated anti-Oregon Green antibody and Amplex UltraRed. Fluorescence (expressed as relative fluorescence units [RFU]) was detected at an excitation/emission wavelength of 490/585 nm and was taken as the cell proliferation rate.
RESULTS
Monosaccharides and methylglycosides compete for binding of human sera to H. pylori LPS.
To characterize the highly antigenic and weakly antigenic epitopes in detail, we performed competition binding assays using ELISA to assess the binding of human serum antibodies to LPS in the presence of monosaccharides and methylglycosides (Fig. 1). In the case of HA-LPS (derived from strain GU2), sera derived from five individuals were used, including sera containing antibodies against only the highly antigenic epitope (HA-LPS-specific sera) and both epitopes (sera react to both HA-LPS and WA-LPS). Similar results were obtained for all five sera, so mean values and standard deviations calculated are shown in Fig. 1-A. Among the monosaccharides, the reactivity of human sera to HA-LPS was strongly suppressed by d-GlcNAc and to a lesser extent by d-GalNAc. Among the methylglycosides, d-GlcNAc-OMe, particularly the β-linked one, was the most potent, inhibiting binding to HA-LPS at concentrations as low as 1/10 that of other competitors, such as d-GlcNAc. In the case of WA-LPS (derived from strain CA2), sera derived another five individuals were used, including sera containing antibodies against only the weakly antigenic epitope (WA-LPS-specific) and both epitopes, were examined for competitive ELISA using WA-LPS as an antigen. These sera gave similar results. Binding of sera to WA-LPS was significantly inhibited by d-Gal, d-GalNAc, and d-Gal-β-OMe to similar extents (Fig. 1B). For the weakly antigenic epitope, the galacto-configuration and the β-linked sugar moiety seemed to be immunodominant.
Fig 1.

Competitive ELISA using monosaccharides and methylglycosides to assess the binding of human sera to H. pylori LPS. Competitors were used at a concentration of 100 mM, unless otherwise specified described in the graph. (A) Binding of human sera to HA-LPS (derived from strain GU2). Values represent means of five individual sera containing antibodies against only the highly antigenic epitope and against both epitopes. (B) Binding of human sera to WA-LPS (derived from strain CA2). Values were means of five individual sera containing antibodies against only the weakly antigenic epitope and against both epitopes. The data are expressed relative to values without competitor (which is set at 100%), and they represent means ± the standard deviation from five individual sera. **, P < 0.01; *, P < 0.05 (compared to the binding in the absence of competitors).
Effect of glycosidase treatment of H. pylori LPS on the reactivity of human sera.
Among several β-N-acetyl-glucosaminidases examined, only S. pneumoniae-derived β-N-acetyl-glucosaminidase altered the reactivity of human sera to H. pylori LPS (Fig. 2A). Treatment of HA-LPS with this enzyme reduced the binding of highly antigenic epitope-specific sera (Fig. 2Aa). On the other hand, the binding of weakly antigenic epitope-specific sera to HA-LPS significantly increased by the treatment (Fig. 2Ab). β-N-Acetylglucosaminidases other than the S. pneumoniae-derived one examined had no effect on the reactivity of human sera to LPS. These results suggested that S. pneumoniae-derived β-N-acetyl-glucosaminidase uniquely recognizes carbohydrate structures containing β-linked d-GlcNAc residue, which comprise the highly antigenic epitope. However, the detailed substrate specificities of these β-N-acetylglucosaminidases have not been clarified. Thus, the highly antigenic epitope contains β-d-GlcNAc residue as an immunodominant sugar residue.
Fig 2.

Effect of glycosidase treatment of H. pylori LPS on binding of human sera. (A) Treatment of HA-LPS (derived from strain GU2) with β-N-acetylglucosaminidase derived from S. pneumoniae. (a) Reactivity of human serum containing antibodies against the highly antigenic epitope only. (b) Reactivity of human serum containing antibodies against the weakly antigenic epitope only. (B) Treatment of WA-LPS (derived from strain CA2) with β-galactosidase derived from A. oryzae. (a) Reactivity of human serum containing antibodies against the highly antigenic epitope only. (b) Reactivity of human serum containing antibodies against the weakly antigenic epitope only. Values represent means ± the standard deviation of triplicate experiments. **, P < 0.01.
Treatment of WA-LPS with β-galactosidase derived from A. oryzae significantly decreased the binding of the weakly antigenic epitope-specific sera (Fig. 2Bb). Treatment with this enzyme did not alter the binding of the highly antigenic epitope-specific sera to the WA-LPS (Fig. 2Ba). These results suggested that β-linked d-Gal residue was involved in the weakly antigenic epitope and that this epitope is exposed by the removal of the β-d-GlcNAc residue, which comprises the highly antigenic epitope.
Interaction of H. pylori LPS with SP-D.
We anticipated that the binding specificities of different types of LPS, namely, HA-LPS, WA-LPS and rough-type LPS, to host lectin(s) were different. So we examined interaction of the LPS with SP-A and SP-D. Bindings to SP-A were not significantly different among the three types of LPS (Fig. 3A). Notably, the interaction of SP-D with WA-LPS was significantly stronger that those with HA-LPS and rough-type LPS (Fig. 3B), and the binding was significantly inhibited by the addition of 10 mM d-Gal-β-OMe (Fig. 4), and the Ca2+ chelator ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) (Fig. 5). Binding of SP-D to HA-LPS and rough LPS was not altered by the addition of EGTA. The treatment with β-galactosidase derived from A. oryzae significantly reduced the binding of WA-LPS to SP-D (Fig. 6). These results suggested that WA-LPS specifically interacts with SP-D via the weakly antigenic epitope, which includes β-linked d-Gal residue, in a Ca2+-dependent manner.
Fig 3.

Binding of H. pylori LPS to SP-A and SP-D. H. pylori LPS (50 μl of a 5-μg/ml solution) derived from 16 clinical isolates were immobilized on a microplate. (A) Binding of SP-A to LPS. SP-A (10 μg/ml) was dispensed into the H. pylori LPS-coated wells, followed by rabbit anti-SP-A antibodies. (B) Binding of SP-D to LPS. SP-D (10 μg/ml) was dispensed into the H. pylori LPS-coated wells, followed by rabbit anti-SP-A antibodies. The binding of anti-SP antibodies was detected using HRP-conjugated anti-rabbit immunoglobulin antibodies, followed by incubation with TMB substrate solution. Reactions were terminated by the addition of 1 M phosphoric acid, and A450 was measured. Values represent the mean values of triplicate experiments. Hatched line indicates the value given without surfactant proteins. **, P < 0.01.
Fig 4.

Effect of methylglycosides on the binding of SP-D to H. pylori WA-LPS. Methylglycosides (10 mM) were dispensed into microplate wells coated with WA-LPS derived from strain CA2 (50 μl of a 5-μg/ml solution), followed by incubated at 37°C for 30 min. SP-D (10 μg/ml) was added, followed by incubation for 37°C for 1 h. Binding SP-D was detected with subsequence incubation with rabbit anti-SP-D antibodies (1 μg/ml), HRP-conjugated goat anti-rabbit immunoglobulin antibody, and TMB substrate solution. Reactions were terminated by the addition of 1 M phosphoric acid, and the A450 was measured. Values represent means ± the standard deviation of triplicate experiments. *, P < 0.05; **, P < 0.01 (compared to the value in the absence of competitors).
Fig 5.

Effect of EGTA on the binding of SP-D to H. pylori LPS. SP-D (10 μg/ml) was dispensed into H. pylori LPS (50 μl of a 5-μg/ml solution)-coated microplate wells in the presence of 5 mM Ca2+ or 5 mM EGTA. Bound SP-D was detected by serial incubation with rabbit anti-SP-D antibodies, HRP-conjugated goat anti-rabbit immunoglobulin antibody, and TMB substrate solution. Reactions were terminated by the addition of 1 M phosphoric acid, and the A450 was measured. Values are means of triplicate experiments. *, P < 0.01; ns, not significant.
Fig 6.
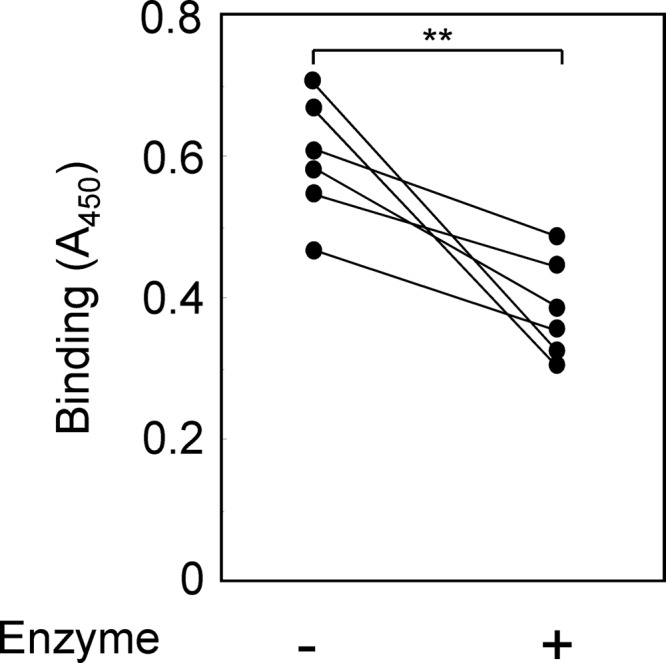
Effect of β-galactosidase derived from A. oryzae on the binding of SP-D to H. pylori WA-LPS. WA-LPS (50 μl of a 5-μg/ml solution) derived from six clinical isolates were immobilized on a microplate. The immobilized LPS was treated with A. oryzae β-galactosidase (1 mU/ml) at 30°C for 12 h. SP-D (10 μg/ml) was added, followed by incubation for 37°C for 1 h. Binding SP-D was detected with subsequence incubation with rabbit anti-SP-D antibodies (1 μg/ml), HRP-conjugated goat anti-rabbit immunoglobulin antibody, and TMB substrate solution. Reactions were terminated by the addition of 1 M phosphoric acid, and the A450 was measured. Values represent mean values of triplicate experiments. **, P < 0.01 between samples with or without enzyme treatment.
Effect of SP-D on H. pylori WA-LPS-induced TLR4 expression and enhancement of E. coli LPS-induced IL-8 production in gastric epithelial cell lines.
Previously, we reported that pretreatment of H. pylori LPS enhanced E. coli-LPS-induced IL-8 production in gastric epithelial cells, which express TLR4 at a very low level (41, 42). The enhancing activity is due to the upregulation of TLR4, which is an E. coli LPS receptor. The upregulation of TLR4 and enhanced IL-8 production are induced more strongly by WA-LPS than by HA-LPS. We examined the effect of SP-D on these biological activities of H. pylori LPS using gastric epithelial cell lines MKN28 and MKN45, because WA-LPS specifically interacted with SP-D. SP-D was not detected by ELISA (detection limit of 1.56 ng/ml) in culture supernatants and cell lysates derived from 106 cells, but it was detected by reverse transcription-PCR in both cell lines (data not shown). Thus, endogenous SP-D was not a significant variable in the experiments using MKN28 and MKN45 cells.
The addition of SP-D enhanced TLR4 upregulation induced by WA-LPS, but not by HA-LPS, in MKN28 cells (Fig. 7) and MKN45 cells (data not shown) based on Western blotting. SP-D alone did not alter the expression levels of TLR4. On the other hand, expression levels of TLR2 were not significantly altered by SP-D, LPS and the mixture of LPS and SP-D. The level of induction of TLR4 will influence E. coli-LPS induced IL-8 production, since these gastric cell lines express TLR4 at very low levels under unstimulated conditions. SP-D significantly enhanced H. pylori WA-LPS-induced weak IL-8 production. Furthermore, SP-D markedly enhanced E. coli-LPS-induced IL-8 production in the presence of WA-LPS, but not HA-LPS production, in a dose-dependent manner (Fig. 8).
Fig 7.

Effect of SP-D on H. pylori-LPS-induced TLR4 expression determined by Western blotting. (A) MKN28 cells were treated with WA-LPS or HA-LPS (100 ng/ml each) in the presence or absence of SP-D (10 μg/ml). Cell lysates were applied to SDS-PAGE on a 7.5% polyacrylamide slab gel. After transfer to a polyvinylidene difluoride membrane, specific proteins were detected using antibodies against TLR4, TLR2, or actin. TLR2 and actin were assessed as controls. (B) The resulting bands were scanned and quantified. Values represent means ± the standard deviation of triplicate experiments. **, P < 0.01 (compared to untreated cells).
Fig 8.

Effect of SP-D on the enhancement of E. coli-LPS-induced IL-8 production by H. pylori LPS. H. pylori LPS (W, WA-LPS [derived from strain CA2]; or H, HA-LPS [derived from strain GU2]) (100 ng/ml) and SP-D (1 or 10 μg/ml) were mixed and incubated for 30 min. MKN28 cells were treated with H. pylori LPS (100 ng/ml), SP-D, or a mixture of H. pylori LPS and SP-D (1 or 10 μg/ml) for 24 h. After the cells were washed, the resulting cells were treated with 100 ng of E. coli LPS/ml for an additional 24 h. The IL-8 concentration in the culture supernatant was determined by ELISA. The data represent means ± the standard deviations of triplicate experiments. **, P < 0.01.
Effect of SP-D on H. pylori LPS-enhanced epithelial cell proliferation.
H. pylori LPS significantly upregulates the cell proliferation of MKN28 and MKN45 cells, and WA-LPS is more potent than HA-LPS in this respect (42). The addition of SP-D significantly enhanced WA-LPS-induced cell proliferation in MKN28 cells (Fig. 9) and MKN45 cells (data not shown). Treatment with HA-LPS plus SP-D or SP-D alone did not significantly alter the rate of cell proliferation.
Fig 9.

Effect of SP-D on the enhancement of MKN28 cell proliferation by H. pylori LPS. H. pylori LPS (W, WA-LPS [derived from strain CA2; or H, HA-LPS [derived from strain GU2]) (1 μg/ml) and SP-D (10 μg/ml) were mixed, followed by incubation for 30 min. MKN28 cells were treated with H. pylori LPS (1 μg/ml), SP-D (10 μg/ml), or a mixture of H. pylori LPS and SP-D for 18 h. Cell proliferation was determined by uptake of EdU into DNA. The data represent means ± the standard deviations of triplicate experiments. **, P < 0.01.
DISCUSSION
We propose that H. pylori smooth-type LPS can be classified into two antigenic types, HA-LPS and WA-LPS (35, 38). H. pylori clinical strains with smooth-type LPS isolated in Japan have one or the other epitope, but not both. These two epitopes are suggested to exist in the O-polysaccharide chain near the joint region of the core oligosaccharides (38). In the present study, we demonstrated that the highly antigenic epitope involved β-linked d-GlcNAc as an immunodominant carbohydrate residue. In contrast, the weakly antigenic epitope appeared to involve β-linked d-Gal as an immunodominant residue. Treatment with β-N-acetylglucosaminidase derived from S. pneumoniae resulted in removal of the β-d-GlcNAc residue, the immunodominant residue of the highly antigenic epitope, and exposure of the weakly antigenic epitope. Thus, the presence or absence of the β-d-GlcNAc residue determines the antigenicity of the two types of LPS.
Previously, we showed that WA-LPS is more potent in terms of TLR4 upregulation and enhancement of cell proliferation than HA-LPS (42). We used 10 to 100 ng of LPS/ml in our in vitro study. The numbers of infected H. pylori in gastric tissues estimate 106 CFU/g of tissue of H. pylori-infected and urease-test positive patients (9). The dry weight of one bacterial cell is 3 × 10−13 g (in terms of E. coli) (33), and the LPS content of dry cells is ca. 10%, so the H. pylori LPS in gastric tissues is estimated to be several 10 ng/g of tissue. It suggests that the amounts of LPS used in our in vitro study are reasonable. We anticipated that these differences in the biological activities of the two types of LPS were associated with host factor(s). The results of present study indicated that WA-LPS interacts specifically with a host C-type lectin SP-D, but not SP-A. The data from competition binding assays with methylglycosides indicated that the interaction is mediated by a β-linked d-Gal residue, the most likely immunodominant carbohydrate residue of the weakly antigenic epitope of WA-LPS. However, a higher concentration of d-Gal-β-OMe was required for competition of the binding of human sera and SP-D to the WA-LPS. Carbohydrate structure proximity to the β-d-Gal residue of the WA-LPS might be also contributed to binding of human sera and SP-D. Inhibition of binding by EGTA, a Ca2+ chelator, suggested that WA-LPS interacted with the carbohydrate recognition domain (CRD) of SP-D. The interaction between H. pylori LPS and SP-D has been reported previously (12, 22). Khamri et al. reported a variant strain of H. pylori, which exhibited no detectable binding to SP-D, derived from strain J178. In the variant strain, fucosylation occurs on the GlcNAc residue of H type 1 structure in the O polysaccharide (12). Thus, the carbohydrate specificity of SP-D binding revealed in the current study differed from that reported by Khamri et al. for the strain J178.
Whereas surfactant proteins and their biological significance in pulmonary tissues have been studied extensively (15, 17), those in gastric mucosa had not been clear yet. Murray et al. reported that SP-D also expresses in gastric mucosa, particularly those infected with H. pylori at higher levels (22). In the present study, we detected SP-D mRNA but not SP-D protein by ELISA, probably because it is under the detection limit in gastric cell lines MKN28 and MKN45. However, we observed that SP-D expression was upregulated by treatment with H. pylori cells. The upregulation levels were similar by both cells carrying WA-LPS and HA-LPA (unpublished results). Thus, SP-D should be involved in an innate immunity against H. pylori in gastric mucosa. SP-D acts as an opsonin for H. pylori via bacterial cell agglutination (12), which facilitates phagocytosis by host phagocytes, and inhibits motility of H. pylori (12). Khamri et al. reported that SP-D knockout mice are more susceptible to low-dose challenge of Helicobacter felis, with a higher density of colonization by H. felis in the stomachs of knockout mice than control mice (13).
WA-LPS-mediated upregulation of TLR4 expression in gastric epithelial cells and enhancement of proliferation of gastric epithelial cells were further enhanced by SP-D. Two mechanisms of enhancement of WA-LPS activity by SP-D are possible. The first involves a cross-linking effect. SP-D molecule has a multivalent CRD (5, 15). Thus, the large multivalent complex formed by interaction of H. pylori WA-LPS with SP-D, and the resulting high avidity may amplify signal transduction from TLR2. The second is a modulatory effect on signal transduction. SP-D interacts with the extracellular domains of TLR4, TLR2, MD2, and the complex of TLR4-MD2 via its CRD (25, 28, 34). SP-D can inhibit LPS-induced signal transduction via TLRs independent of the presence of TLR ligands (34). In addition, via its CRD, SP-D binds directly to signal inhibitory regulatory protein α (SIRPα) in the absence of microbes, and blocks downstream signaling through Src family kinases and p38 MAP kinase via the activation of tyrosine phosphatase SHP-1 (6). H. pylori WA-LPS interacted with CRD of SP-D; thus, this interaction could suppress the inhibitory activities of SP-D by blocking the interaction of the SP-D CRD with host receptors.
We propose the following model based on the results of the present study. H. pylori carrying the HA-LPS is dominant during relatively early periods of infection, and high titers of antibodies to this epitope are elicited. This is supported by the fact that high titers of anti-highly antigenic epitope antibodies are found in almost all individuals infected with H. pylori regardless of antigenic type of infected H. pylori (1, 36). H. pylori carrying HA-LPS is considered to escape from innate immunity involving SP-D, because HA-LPS does not interact significantly with the SP-D via its CRD. During prolonged infection, H. pylori carrying HA-LPS may be cleared from the host by the specific antibodies. As a result, H. pylori that lacks the highly antigenic epitope, namely, orgainsms in which the weakly antigenic epitope is exposed, remains. This is supported by the observation that antibody titers against the weakly antigenic epitope are significantly higher in H. pylori-positive gastroduodenal patients compared to asymptomatic H. pylori-positive individuals (36), in contrast to antibody titers against anti-highly antigenic epitope. This switch from HA-LPS- to WA-LPS-carrying strains can be caused by (i) antigenic conversion of the same strain, (ii) selection from mixed infection of HA-LPA-carrying strains and WA-LPS-carrying strains, or (iii) infection with other strains which are WA-LPS-carrying. Although it is unclear which mechanism is at work, antigenic conversion, which can result from the loss or lack of only one β-linked d-GlcNAc residue, is more likely. This phenomenon in turn serves as an escape mechanism for H. pylori to evade the host antibody-mediated immune response.
In replacement of the evasion from antibody-mediated rejection, the exposed weakly antigenic epitope is recognized by an extracellular pattern recognition molecule, SP-D, representing a host innate immune defense. On the other hand, we demonstrate that WA-LPS, which is generated through the loss or lack of the highly antigenic epitope, has alternative biological activities. WA-LPS exhibited more potent upregulation of TLR4 expression and enhancement of epithelial cell proliferation (42). These activities might lead to inflammation and tumorigenesis. H. pylori WA-LPS interacts with SP-D, and the WA-LPS/SP-D complex enhances the biological activities of WA-LPS, namely, it enhanced inflammatory cytokine production and increases proliferation of gastric epithelial cells.
We observed that several Western strains with smooth-type LPS contain neither the highly antigenic nor the weakly antigenic epitope (1a). It is possible that these epitopes may be distributed among Japanese (or East Asian) strains, but not Western strains. Kawai et al. reported that East Asian strains appear to differ greatly from the European strains in comparison of complete genome sequences (11). By extension, the structure and antigenicity of LPS would also differed between East Asian strains (including Japanese strains) and Western strains. These researchers also reported that a fucosyltransferase gene (futA/futB), which is involved in biosynthesis of LPS, diverges considerably between East Asian and European strains (11). Now, we will focus on exploring the diversity among H. pylori strain in terms of biosynthesis gene(s) involved in formation of the highly antigenic epitope, namely, β-linked d-GlcNAc residue.
ACKNOWLEDGMENT
This study was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science.
Footnotes
Published ahead of print 21 May 2012
REFERENCES
- 1. Amano K, et al. 1998. Utilization of proteinase K-treated cells as lipopolysaccharide antigens for the serodiagnosis of Helicobacter pylori infections. Microbiol. Immunol. 42:509–514 [DOI] [PubMed] [Google Scholar]
- 1a. Amano K, Yokota S, Monteiro MA. 2012. Comparison of the serological reactivity of lipopolysaccharides from Japanese and Western strains of Helicobacter pylori to sera from H. pylori-positive humans. ISRN Microbiol. Article ID 162816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Appelmelk BJ, Monteiro MA, Martin SL, Moran AP, Vandenbroucke-Grauls CM. 2000. Why Helicobacter pylori has Lewis antigens. Trends Microbiol. 8:565–570 [DOI] [PubMed] [Google Scholar]
- 3. Appelmelk BJ, et al. 1996. Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect. Immun. 64:2031–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chochi K, et al. 2008. Helicobacter pylori augments growth of gastric cancers via the lipopolysaccharide-Toll-like receptor 4 pathway, whereas its lipopolysaccharide attenuates antitumor activities of human mononuclear cells. Clin. Cancer Res. 14:2909–2917 [DOI] [PubMed] [Google Scholar]
- 5. Crouch E, Persson A, Chang D, Heuser J. 1994. Molecular structure of pulmonary surfactant protein D (SP-D). J. Biol. Chem. 269:17311–17319 [PubMed] [Google Scholar]
- 6. Gardai SJ, et al. 2003. By binding SIRPα or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 115:13–23 [DOI] [PubMed] [Google Scholar]
- 7. Heneghan MA, McCarthy CF, Janulaityte D, Moran AP. 2001. Relationship of anti-Lewis x and anti-Lewis y antibodies in serum samples from gastric cancer and chronic gastritis patients to Helicobacter pylori-mediated autoimmunity. Infect. Immun. 69:4774–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ishihara S, et al. 2004. Essential role of MD-2 in TLR4-dependent signaling during Helicobacter pylori-associated gastritis. J. Immunol. 173:1406–1416 [DOI] [PubMed] [Google Scholar]
- 9. Kamiya S, et al. 1993. Evaluation of rapid urease test for detection of Helicobacter pylori in gastric biopsy specimens. Eur. J. Epidemiol. 9:450–452 [DOI] [PubMed] [Google Scholar]
- 10. Kawahara T, et al. 2001. Type I Helicobacter pylori lipopolysaccharide stimulates Toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect. Immun. 69:4382–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawai M, et al. 2011. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol. 11:104 doi:10.1186/1471-2180-11-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khamri W, et al. 2005. Variations in Helicobacter pylori lipopolysaccharide to evade the innate immune component surfactant protein D. Infect. Immun. 73:7677–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khamri W, et al. 2007. Helicobacter infection in the surfactant protein D-deficient mouse. Helicobacter 12:112–123 [DOI] [PubMed] [Google Scholar]
- 14. Kingstone RE, Kaurman RJ, Bebbington CR, Rolfe MR. 1992. Amplification using CHO cell expression vectors, p 16.14.11–16.14.13 In Ausubel F, et al. (ed), Current protocols in molecular biology. John Wiley & Sons, Inc, New York, NY: [DOI] [PubMed] [Google Scholar]
- 15. Kuroki Y, Takahashi M, Nishitani C. 2007. Pulmonary collectins in innate immunity of the lung. Cell. Microbiol. 9:1871–1879 [DOI] [PubMed] [Google Scholar]
- 16. Lepper PM, Triantafilou M, Schumann C, Schneider EM, Triantafilou K. 2005. Lipopolysaccharides from Helicobacter pylori can act as antagonists for Toll-like receptor 4. Cell. Microbiol. 7:519–528 [DOI] [PubMed] [Google Scholar]
- 17. McCormack FX, Whitsett JA. 2002. The pulmonary collectins, SP-A and SP-D, orchestrate innate immunity in the lung. J. Clin. Invest. 109:707–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Monteiro MA, et al. 2000. Expression of histo-blood group antigens by lipopolysaccharides of Helicobacter pylori strains from Asian hosts: the propensity to express type 1 blood-group antigens. Glycobiology 10:701–713 [DOI] [PubMed] [Google Scholar]
- 19. Moran AP. 2008. Relevance of fucosylation and Lewis antigen expression in the bacterial gastroduodenal pathogen Helicobacter pylori. Carbohydr. Res. 343:1952–1965 [DOI] [PubMed] [Google Scholar]
- 20. Moran AP, Prendergast MM. 2001. Molecular mimicry in Campylobacter jejuni and Helicobacter pylori lipopolysaccharides: contribution of gastrointestinal infections to autoimmunity. J. Autoimmun. 16:241–256 [DOI] [PubMed] [Google Scholar]
- 21. Muotiala A, Helander IM, Pyhala L, Kosunen TU, Moran AP. 1992. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect. Immun. 60:1714–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murray E, et al. 2002. Expression of surfactant protein D in the human gastric mucosa and during Helicobacter pylori infection. Infect. Immun. 70:1481–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Negrini R, et al. 1996. Antigenic mimicry between Helicobacter pylori and gastric mucosa in the pathogenesis of body atrophic gastritis. Gastroenterology 111:655–665 [DOI] [PubMed] [Google Scholar]
- 24. Netea MG, van Deuren M, Kullberg BJ, Cavaillon JM, Van der Meer JW. 2002. Does the shape of lipid A determine the interaction of LPS with Toll-like receptors? Trends Immunol. 23:135–139 [DOI] [PubMed] [Google Scholar]
- 25. Nie X, et al. 2008. Pulmonary surfactant protein D binds MD-2 through the carbohydrate recognition domain. Biochemistry 47:12878–12885 [DOI] [PubMed] [Google Scholar]
- 26. Nielsen H, Birkholz S, Andersen LP, Moran AP. 1994. Neutrophil activation by Helicobacter pylori lipopolysaccharides. J. Infect. Dis. 170:135–139 [DOI] [PubMed] [Google Scholar]
- 27. Ogawa T, et al. 2003. Endotoxic and immunobiological activities of a chemically synthesized lipid A of Helicobacter pylori strain 206-1. FEMS Immunol. Med. Microbiol. 36:1–7 [DOI] [PubMed] [Google Scholar]
- 28. Ohya M, et al. 2006. Human pulmonary surfactant protein D binds the extracellular domains of Toll-like receptors 2 and 4 through the carbohydrate recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry 45:8657–8664 [DOI] [PubMed] [Google Scholar]
- 29. Perez-Perez GI, Shepherd VL, Morrow JD, Blaser MJ. 1995. Activation of human THP-1 cells and rat bone marrow-derived macrophages by Helicobacter pylori lipopolysaccharide. Infect. Immun. 63:1183–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sawada K, et al. 2010. Pulmonary collectins protect macrophages against pore-forming activity of Legionella pneumophila and suppress its intracellular growth. J. Biol. Chem. 285:8434–8443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shimizu T, et al. 2004. Membrane-anchored CD14 is important for induction of interleukin-8 by lipopolysaccharide and peptidoglycan in uroepithelial cells. Clin. Diagn. Lab. Immunol. 11:969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith SM, et al. 2011. Tribbles 3: a novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J. Immunol. 186:2462–2471 [DOI] [PubMed] [Google Scholar]
- 33. Sundararaj S, et al. 2004. The CyberCell Database (CCDB): a comprehensive, self-updating, relational database to coordinate and facilitate in silico modeling of Escherichia coli. Nucleic Acids Res. 32:D293–D295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yamazoe M, et al. 2008. Pulmonary surfactant protein D inhibits lipopolysaccharide (LPS)-induced inflammatory cell responses by altering LPS binding to its receptors. J. Biol. Chem. 283:35878–35888 [DOI] [PubMed] [Google Scholar]
- 35. Yokota S, Amano K, Fujii N. 2011. Helicobacter pylori lipopolysaccharide as a possible pathogenic factor for gastric carcinogenesis, p 243–258 In Tonino P. (ed), Gastritis and gastric cancer: new insights in gastroprotection, diagnosis, and treatment. InTech, Rijeka, Croatia [Google Scholar]
- 36. Yokota S, Amano K, Fujii N, Yokochi T. 2000. Comparison of serum antibody titers to Helicobacter pylori lipopolysaccharides, CagA, VacA and partially purified cellular extracts in a Japanese population. FEMS Microbiol. Lett. 185:193–198 [DOI] [PubMed] [Google Scholar]
- 37. Yokota S, Amano K, Hayashi S, Fujii N. 1997. Low antigenicity of the polysaccharide region of Helicobacter pylori lipopolysaccharides derived from tumors of patients with gastric cancer. Infect. Immun. 65:3509–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yokota S, et al. 2000. Two distinct antigenic types of the polysaccharide chains of Helicobacter pylori lipopolysaccharides characterized by reactivity with sera from humans with natural infection. Infect. Immun. 68:151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yokota S, Amono K, Chiba S, Fujii N. 2003. Structures, biological activities and antigenic properties of Helicobacter pylori lipopolysaccharide, p 251–267 In Pandalai SG. (ed), Recent research developments in microbiology, vol 7, part II Research Signpost, Trivandrum, India [Google Scholar]
- 40. Yokota S, Noguchi H. 1994. Epitopes for human monoclonal antibodies and serotyping antisera against the O-specific polysaccharide of Pseudomonas aeruginosa O11. Carbohydr. Res. 261:57–66 [DOI] [PubMed] [Google Scholar]
- 41. Yokota S, et al. 2007. Highly purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol. Med. Microbiol. 51:140–148 [DOI] [PubMed] [Google Scholar]
- 42. Yokota S, Okabayashi T, Rehli M, Fujii N, Amano K. 2010. Helicobacter pylori lipopolysaccharides upregulate Toll-like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect. Immun. 78:468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]