

Abstract
Mammalian hearing depends on the enhanced mechanical properties of the basilar membrane within the cochlear duct. The enhancement arises through the action of outer hair cells that act like force generators within the organ of Corti. Simple considerations show that underlying mechanism of somatic motility depends on local area changes within the lateral membrane of the cell. The molecular basis for this phenomenon is a dense array of particles that are inserted into the basolateral membrane and that are capable of sensing membrane potential field. We show here that outer hair cells selectively take up fructose, at rates high enough to suggest that a sugar transporter may be part of the motor complex. The relation of these findings to a recent candidate for the molecular motor is also discussed.
The inner ear of mammals has evolved to analyze sounds over a wide range of frequencies. In humans this range covers about 8 octaves, but it can be both more restricted (hearing in mice covers only 3.5 octaves) or, as in some cetaceans, extend over 10 octaves and use infrasound. Auditory specialists such as the echolocating bats may employ frequencies well into the ultrasonic. When considering how a hearing organ of the size of a pea or even smaller can perform such engineering feats, cochlear construction provides the clues. The cochlea in all these diverse mammalian species shows a remarkable conservation of design. A common feature of all mammalian cochleae is that, within the duct, the basilar membrane supports a propagated traveling wave. In mammals, the basilar membrane performs as a mechanical selector of sound frequencies and maps component frequencies in a complex sound onto a position in the duct. A second feature found throughout mammalian cochleae is the presence of two morphologically distinct sets of hair cells, inner (IHCs) and outer hair cells (OHCs). The cells lie within the organ of Corti, which runs along the full length of the basilar membrane and is shown in cross section in Fig. 1a. Hence the linkage between basilar membrane motion and the deflection of the sensory hair bundles of hair cells recodes sound frequency and intensity into a firing pattern of the afferents of the auditory nerve. The basilar membrane thus acts as a preconditioner of the sound signal. Other vertebrate genera use modified designs of hearing organ (using, for example, local filtering based on electrical resonance of their membranes or mechanical resonance of their hair cell stereocilia). The mammalian cochlear design ensures that even high frequencies can be detected by using material and mechanical properties of the macroscopic structures of the cochlea (1).
Figure 1.

OHCs in the cochlea. (a) Schematic cross section of the organ of Corti, showing site of inner hair cells (IHC) and OHCs. The primary stimulus is the shear delivered to the OHC stereocilia by the tectorial membrane (TM). OHC length changes (and therefore forces) are produced as arrowed. BM, basilar membrane. (b) OHC length change through electromotility, where membrane potential (Vo − Vm) alters cell surface area. The tight molecular packing in the lateral membrane allows the protein area change to have macroscopic effects. (c) OHC cell length change through cell volume change, where osmotic pressure (ΠO − ΠI) difference inside and outside requires water to follow solute entry.
A wide range of measurements have now shown that the basilar membrane has a mechanical pattern of vibration that is under physiological control. Optimally, threshold sound detection elicits a peak deflection of about 0.3 nm. This exceeds what a membrane of the same mechanical construction can achieve and an energy source is necessarily involved. Indeed, it was suspected more than 50 years ago that there had to be some “active” process in the cochlea to account for the known psychophysics of the auditory periphery (2). The process by which enhanced basilar membrane mechanics is generated has been termed “cochlear amplification.” How such amplification comes about has been the focus of much effort over the past two decades. Sensory hair cells are clearly implicated. There remains little doubt that inner hair cells are the primary sensory cells of the mammalian cochlea: they signal via a glutamatergic synapse to the auditory nerve. However, there are now many lines of evidence that OHCs of the cochlea are responsible for the enhancement of the basilar membrane motion, an idea that was proposed on the basis of ultrastructural evidence alone before direct physiological data became available (3). In addition, although OHCs also have an afferent innervation, the main neural pathway associated with them is a cholinergic efferent fiber system terminating on the cells. The organization is reminiscent of the innervation of an effector system. Most significantly, however, OHCs are positioned and coupled to the motion of the basilar membrane in a manner that does allow forces to be fed back into the dynamics of the cochlea.
Most computational models of peripheral hearing indicate cochlear amplification comes about by opposing dissipative forces within the cochlea. Such forces arise from the fluid content of the cochlear duct and the viscoelastic properties of the tissues of the basilar membrane. This situation places a number of constraints on how any mechanism dependent on the OHCs can operate. One proposal for the origin of OHC forces, for which there is evidence in nonmammalian species, is that the apical stereocilia act both as the sensors of the motion of the basilar membrane and as a motor source to amplify the motion (4). The uniformity of cochlear structure along the length of the duct implies that OHC parameters have to be tuned to match the mechanical impedance at each place (5).
The second proposal for how OHCs contribute to cochlear function invokes a novel cellular motor. This is the mechanism of “somatic electromotility.” The key observation is that cell hyperpolarization lengthens isolated cells, whereas depolarization leads to their shortening (6). This implies that OHCs are capable of generating forces. The mechanism is fast (7–9), and cells can be electrically responsive up to frequencies above 40 kHz. The sequence is that a shear displacement of the OHC stereocilia generate a change in membrane potential that in turn generates forces directed along the cell long axis. The process thus decouples the motor force from the shear forces (Fig. 1a). Computational models indicate that the range of frequencies presented as the input signal to any group of OHCs may be preselected by a broadly tuned mechanical resonance of the overlying tectorial membrane (10, 11). For this hypothesis there is now better experimental evidence (12). Thus during every cycle when the OHC is stimulated by displacement of its stereocilia, the OHC motor completes the force feedback loop to enhance the mechanical stimulus.
The mechanism of somatic electromotility copes readily with the phasing requirement on the force operating in the cochlea. Because deflection of the stereocilia causes current to enter the cell, 90° in advance of the voltage change, the motor, being voltage sensitive, responds with the right phase to oppose viscous forces. However, at high frequencies, most of the current passes through the membrane capacitance, and it is a contentious topic as to whether the transmembrane potential across the OHC membrane is sufficient to drive the motor. Although the membrane time constant effectively filters changes in OHC membrane potential, it has been proposed that the extracellular current that flows around an OHC from neighboring cells alters the effective field across the OHC basolateral membrane (8) and thereby provides sufficient driving potential for the motor (13). This mechanism requires further modeling once the electrical parameters of the cochlea become clear.
The underlying biophysics of electomotility is constrained by the requirement that OHC mechanisms generate forces at high frequencies. This allows OHCs to be involved in the acoustic processing of sound at frequencies in the kilohertz range. The simplest of the cellular models to describe electromotility is one in which the cell membrane area changes but the volume of the cell remains constant (14). As a result, a reduction of the unit area of lateral membrane will tend to make the cell shorter. In principle this is a dynamically favored mechanism for rapid length change, as no water needs to move (cyclically) back and forth across the membrane. Although an increase of volume at constant cell surface area will also change the length of a cylindrical cell, the movement of water would be anticipated to be a slower process (Fig. 1 b and c).
The lateral membrane of OHCs contains a high density of particles that can be seen either after deep-etch and low-angle rotary shadowing (15) or by freeze fracture of the membrane P-face (16). The observed particles are about 8 nm in diameter and close packed at 5,000 μm−2, making up a significant fraction of the membrane. The particles are candidates for the OHC motor. The electrophysiological signature of the motor is a gating charge that can be detected both in whole cell recording (17) and by wide-band patch-clamp recording (18). The gating charge can most simply be interpreted as a dipole reorientation of some component of the motor protein within the electrical field. The gating charge is about 10 times larger than the gating charge measured in the excitation contraction coupling of muscle. As in muscle, the dipole of the gating charge gives rise to a voltage-dependent capacitance of the cell membrane (19). In OHCs the apparent maximal membrane capacitance is found near −30 mV but is tension sensitive. The presence of this motor can almost double the geometric capacitance of the cell. By matching the electrophysiological measurements with the electron microscopy, each particle, tentatively identified with the motor, appears to be associated with the movement of about 1 electronic charge across the membrane field.
It seems unlikely that the motor molecule in the OHC membrane is a modified (but necessarily nonconducting) ion channel. The voltage sensor in an ion channel, such as the S4 region, does not move within the membrane with sufficient speed. An alternative proposal is that the motor is a charged ion transporter. Cation transporters are known to be associated with rapid charge movements as part of the transport cycle and, although less well characterized, many anion transporters share this property.
A third class of candidate proteins includes those transporters that are electrically neutral. Such transporters may not reveal themselves electrophysiologically, as no net charge is carried during their transport cycle, but there is no a priori reason why such proteins cannot have a gating charge movement. This class includes those proteins implicated in water or in neutral solute transport. The evidence below suggests that the OHC membrane contains a transporter with similarities to the sugar transporter, GLUT5, whose normal substrate is fructose. Sugar molecules cannot cross lipid membrane by simple diffusion. GLUT5 is a member of a family of six facilitative hexose transporters (GLUT1–7) found in mammalian tissue. Each differs in its substrate specificity and tissue distribution. Sugars entering the cell can provide the energy required by OHCs for metabolic activities, but the only such transporter so far identified by immunohistochemistry is GLUT5 (20). Although originally identified in gerbils, the same epitope is also present in guinea-pig OHCs (21).
By identifying a fructose transporting mechanism in OHCs that is distributed along the length of the basolateral membrane (21), we have been led to explore the possibility that GLUT5 or a closely related isoform is also part of the motor complex. The experiments below examine quantitatively the uptake rates of different sugars into OHCs and the effect of known blockers for sugar transport and motility.
Methods
Adult guinea pigs (200–400 g) were killed by rapid cervical dislocation, and both bullae were removed. The organ of Corti was dissected in standard saline containing (in mM): NaCl, 142; KCl, 4; CaCl2, 1; MgCl2, 1.5; Hepes, 10; pH 7.35; osmolarity adjusted to 325 mosmol⋅kg−1 with approximately 30 mM sucrose. The tissue was then bathed in 0.25 mg⋅ml−1 trypsin (Sigma) for 10 min before gentle mechanical dissociation. The cells were transferred to a 1-ml chamber continuously perfused at a rate of 100 μl⋅min−1. Cells were used within 3 h of the dissection. Experiments were performed at room temperature (20–25°C).
Sugar transport was estimated as described previously (21) by measuring OHC length. Custom software was used to estimate the cell length changes produced by water entry. Sugars were added around the cell through a puffer pipette at different concentrations (3, 10, 20, and 30 mM) in standard saline solution, adjusted to 325 mosmol⋅kg−1 in all cases with sucrose. To compensate, 10 mM NaCl was removed from both bathing and perfusing solution in experiments where 50 mM glucose or fructose was tested. The superfusion rate from the applicator pipette was adjusted to 8–12 cm of H2O so that movement of the cell due to pressure was minimized. Cells were observed by a 1.3 numerical aperture 40× objective and recorded at 1 Hz by a high-resolution video camera using Axon Imaging Workbench software (Axon Instruments; Foster City, CA). The same software was used for image analysis and was enhanced by pixel estimation using matlab 5.3 (Mathworks; Natick, MA). In digitized images the pixel diameter was 114 nm. By analysis of the centroid of the 1-μm2 regions, we estimate that the errors in cell length and width measurements did not exceed 15%. Data are shown as mean ± SD and, where appropriate, were fitted to theoretical models by using a Marquadt–Levenberg algorithm.
Results
With the bath solution containing sucrose as the balancing osmolyte, solutions were applied containing isotonic glucose or fructose. With a 30-s exposure, both 30 mM fructose and 30 mM glucose produced a reversible shortening of the cell. Typical strains measured in apical cells 40–65 μm long were close to 5%, at 5.4% ± 0.7% (n = 6) in fructose and 4.9% ± 0.7% (n = 4) in glucose.
Isotonic replacement of sucrose with various concentrations of glucose or fructose was performed to assess substrate specificity. Fig. 2 shows that both sugars were transported into the cell in a graded and saturable manner characteristic of an uptake carrier. The affinity for fructose was higher than that for glucose, even though the maximal uptake rates were comparable; however, a significant difference between glucose- and fructose-induced strains was seen when these sugars were applied at 3 mM, 10 mM, or 20 mM. This result implies that the transport rate for glucose across the OHC membrane is less than for fructose.
Figure 2.

Glucose and fructose uptake by guinea-pig OHCs. (a) Fructose-induced cell shortening. Top, control; middle, with fructose; bottom, washout. Cell length, 55 μm. (b) Strains at these different concentrations. Sugars were applied at 3 (n = 6), 10 (n = 7), 20 (n = 8), 30 (n = 6), and 50 mM (n = 7). (c) Initial transport rate estimated by a linear fit between t = 0 s and t = 6 s of glucose (●) or fructose (■) application. Data were fitted with Vmax = 0.8% s−1 and Km = 15.8 ± 1.6 mM (fructose) and 34 ± 3.5 mM (glucose). (d) Effect of 30 mM deoxyglucose (▵) compared with 30 mM fructose (●) shortening. Stimulus timing is shown as a bar in b and d.
The data for the initial rate of cell shortening Vi were fitted by Michaelis–Menten kinetics for saturable uptake mechanism (Fig. 2c) with Vmax the maximum rate, [S] the sugar concentration, and Km the half-saturating concentration:
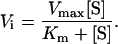 |
1 |
For fructose Km = 15.8 ± 1.6 mM and for glucose Km = 34.0 ± 3.5 mM (34 experiments, n = 23 cells). This is typical of values reported for GLUT5 in other cell systems—for example, in human enterocytes (22). These values for Km are related directly to sugar transport, since water equilibrated across the cell membrane at a faster rate than either of the sugars. The measured cell shortening rate obtained in hypotonic solution (switching to sucrose, 315 mosmol⋅kg−1 from 330 mosmol⋅kg−1) induced a faster change in length (0.71 ± 0.13 s−1) compared with 30 mM fructose [0.52 ± 0.18 s−1 (n = 3)].
Cytochalasin B is known to act as a reversible and noncompetitive inhibitor of glucose transport by GLUT1, -2, and -3. Cytochalasin B added (at 1 mg/ml) to the bath 10 min before experiments produced no significant difference on either the uptake rates or the steady-state strains in four cells. Additionally, OHCs did not respond to isotonic replacement of external sucrose with the sugar 2-deoxy-d-glucose in 30 mM solution (Fig. 2d). This sugar is also not transported by GLUT5 isoforms (23).
Acetylsalicylic acid blocks electromotility and OHC motor charge movement (24). If the sugar uptake carrier shares properties of the OHC motor protein, we would predict that the transport itself might be affected by salicylic acid. We tested this hypothesis by replacing glucose with fructose isotonically in the presence of 10 mM salicylate (Fig. 3). Cells were pretreated with salicylate for 1 min before fructose application. As salicylate entered the cell (as salicylic acid) the cell increased in volume and shortened, consistent with salicylate loading of the cell. After a rapid phase of shortening, the cell volume stabilized. No further change in cell length was observed when fructose was applied. Application of fructose before and after washout showed that fructose-induced strain was larger and faster than strain induced by salicylate alone.
Figure 3.

Salicylate blocks fructose uptake. Pretreatment of an OHC with 10 mM sodium salicylate induced noticeable cell swelling (○, black) with a stabilization after 80 s. Application of a fructose–salicylate solution on such a pretreated cell failed to induce any response. After washout, the same cell treated with fructose demonstrated a fast and large change in length (□, red). A cell length of 62 μm at the beginning of the experiment is taken as the reference for both recordings.
There is no evidence that significant current can pass through the OHC lateral membrane (18, 25), most of the ion channels being localized at the base or apex of the cell. The lateral cell membrane, however, exhibits a voltage-dependent capacitance, as seen in Fig. 4. The bell-shaped curve is the nonlinear capacitance
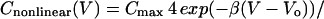 |
2 |
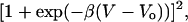 |
where the peak capacitance Cmax occurs at a membrane potential Vo and the parameter β is a measure of the voltage sensitivity of the charge movement. The voltage dependence of charge movement of the membrane depended on the species of sugar presented to the OHC (Fig. 4). The largest shift in Vo at the peak membrane capacitance occurred when fructose was present around the cell. Although hypoosmotic solutions also swell the cell, inducing membrane strain and a consequent positive shift in Vo of the voltage-dependent capacitance (26, 27), the effect of fructose was more pronounced than the osmotic stimulus that produced the same strain (Fig. 4b).
Figure 4.

OHC charge movement is modified by sugars. (a) Membrane capacitance determined by using a staircase ramp protocol. The maximum capacitance Cmax occurs at Vo. In this fit β = 0.036 mV−1. (b) Collected data of Vo shift vs. length change for several sugars (○). Cell capacitance was initially recorded in 330 mosmol⋅kg−1 glucose. Hypoosmotic solution (▵) also produced a Vo shift and length change. The effect of fructose shown in this plot was significantly different from that of other treatments.
Discussion
The results extend previous data suggesting the presence of a GLUT5-like transporter, selective for fructose in guinea-pig OHCs (21). Immunohistochemical evidence shows that a GLUT5 epitope is expressed in the OHC lateral membrane (28). The developmental pattern of expression runs parallel to the development of “motor” particles in the membrane (28). The data here show that OHCs have the capacity to take up fructose selectively over glucose. This uptake is not sensitive to cytochalasin B. Although maximal fructose- and glucose-induced strains are comparable, analysis of the transport kinetics shows that fructose is transported preferentially, with a half-saturation of about 16 mM. This falls within the range of values reported in other models: from 6 mM in human enterocytes (22) to 18 mM in rat enterocytes (29).
Little is known about sugar content in the perilymphatic space. Sugars are transferred into perilymph through a blood–tissue barrier by facilitative transporters within the cochlea. Fructose transport will be reduced, as blood itself does not contain more than 0.1 mM fructose, but perilymph contains about 2–4 mM glucose (30). Under these conditions, a molecule with the properties of GLUT5 cannot be using its normal substrate and it is tempting to suggest that GLUT5 is playing a different role in these cells. The most appealing hypothesis is that OHCs employ a protein in the lateral membrane in a role that is not functionally related to the protein's role in other tissues. Thus electromotility arises from (i) the high packing density of the protein and (ii) a generic property of the protein that allows it to generate areal forces in response to changes in transmembrane potential. As pointed out elsewhere (21), the rate of sugar uptake into OHCs is fast compared with other systems where it has been measured, usually with radioactive tracers. If sugar transport uptake rate is about 100 s−1 per carrier site, the transporter would have to be present in copy numbers exceeding 107 per cell to account for the rates of cell swelling. This number is comparable to the inferred number of motors in the OHC (17).
As a known blocker of the OHC electromotility and charge movement (24), salicylate also affects sugar transport in guinea-pig OHCs. It is not clear how this molecule interacts with membrane proteins, although curiously the shape of salicylic acid bears similarities to a hexose molecule. Once within the membrane, salicylate will affect intramembranous charged particles, as salicylate induces an increase in negative surface charge (31) and behaves as a lipid-soluble anion at neutral pH. Indeed, the swelling of the cell observed when salicylate was applied suggests that it readily penetrates the cells, but at a rate lower than that of sugar. It is probable that salicylate also affects the internal membranes of the OHC, as complete reversal of the shortening was difficult after salicylate exposure.
The voltage dependence of nonlinear membrane capacitance is also dependent on the ionic strength of the external solution. In whole-cell recording conditions (32), reducing the external sodium concentration shifts the position of the peak Vo of the nonlinear capacitance reversibly by up to 30 mV in the negative direction, with a mean slope of 23 mV/decade. The effect appears to be specific for sodium, because simple replacement of sodium by N-methyl-d-glucamine showed no effect, and replacement of Cl by gluconate was without effects on the charge movement (J. E. Gale and J.F.A., data not shown). We therefore deduce that the voltage dependence of the nonlinear capacitance depends on external ionic strength. The most economical hypothesis to explain the data is that there is a negative surface charge on the OHC. To calculate the size of the charge requires application of the Grahame equation (33). We find that including the effect of external divalent ions as well as monovalent ions requires that the OHC has a surface charge of 1 e− per 16 nm2, the negative charge accounting for the leftward shift as ionic strength is reduced.
To relate the functional studies to the structure of protein candidates is uncertain, not least because the structures of very few integral membranes are known. Like other hexose transporters, GLUT5 is a protein containing 501 amino acids in the isoform found in mouse and is significantly hydrophobic. As an integral membrane protein it is predicted to contain 12 transmembrane α-helix regions. Analysis of the primary sequence suggests that there is a charge dipole associated with the second transmembrane helix from the N terminus of the protein. What is not known is whether such hydrophobic proteins also possess an intrinsic dipole moment. This dipole moment arises as a result of the charge associated with the peptide bonds of any α-helix (34). The dipole moment per helix is estimated to be equivalent to about 0.5 e− at either end and has been suggested to contribute to the packing and stability of some proteins in membranes (35). Both of these intrinsic dipoles of GLUT5 may form part of the voltage sensor structure. In a packed assembly of molecules the total dipole would be the result of vectorial addition of individual dipole moments from all of the transmembrane portions of the helix.
Uncancelled electric charge on the protein may be the origin of the surface charge estimate of 1 e− per 16 nm2. Since each α-helix is estimated to occupy an area of approximately 1.42 nm2, this surface charge would correspond to an assignment of 1 e− per molecule of GLUT5, with the same estimate for any comparably sized transporter. For the moment, such intrinsic dipole sensors have not been observed directly, but calculations suggest that a generic property of high-density protein arrays in membranes is that a cell can change surface area (36). The high insertion density of particles in the OHC membrane thus confers electromotility upon the cell. Fig. 5 shows how a hypothetical rearrangement a subset of helices, by allowing them to twist against each other in the membrane, might produce the fast area change required of the OHC motor.
Figure 5.

Hypothetical model for a motor structure in the OHC membrane. Only four of the transmembrane helices are shown. The net area change in the plane of the membrane, produced by helix tilt, can be sufficient to produce the 5% length change observed in an OHC when electrically stimulated.
By subtractive PCR hybridization from OHCs and functional expression in a kidney cell line, a molecule (named “prestin”) has also been identified as a candidate for the OHC motor (37). It has properties that mirror many of the anticipated properties of the hypothesized OHC motor. Isolated form a gerbil cochlear library, the protein contains 744 amino acids. This molecule, like GLUT5, does not have a characterized topology. The region identified with the charge sensor in prestin lies in a region of the molecule that, by hydrophobicity mapping alone, is in a relatively hydrophilic section of the molecule. Prestin shows homology to a family of proteins expressing a sulfate transporter motif, and thus might technically be described as an anion transporter. Physiological studies of related family members, for example the products of the pendrin gene, show that sulfate transport may be questionable (38) and that the family may be better described as chloride–iodide transporter (39). This molecule is a presumptive anion transporter. Nevertheless there is no evidence for sulfate transport in OHCs (J.F.A., unpublished data), and lowering extracellular chloride also has no effect on the motor (18). Neutral chloride transport can occur in OHCs, where it has been associated with bicarbonate transport (40), but at present it is unknown whether members of the “sulfate transporter” family can also use other substrates, such as a sugar.
Both of the candidate OHC motor molecules are too small by themselves to be the 8-nm-diameter motor particle observed in the freeze-etch replicas of the lateral membrane. On the basis of the size of the particles, and allowing for tissue preparation artifacts, the observed particle type is thus likely to represent a cluster of membrane-inserted proteins with a combined molecular mass of 200–250 kDa. This would match either a tetramer of GLUT5 (56 kDa) or a trimer of prestin (81 kDa). Alternatively, both molecules may be required to form a stable multiunit heteromer. In support of the latter possibility, freeze-fracture replicas of the P-face of OHCs show structures that indicate a multimeric unit (16). Although the structural topology of the motor is unclear, how the OHC inserts such high densities of protein into the basolateral membrane remains another issue that deserves attention. The insertion, assembly, and regulation of this array of molecular motors make OHCs able to carry out their role as force generators at high frequencies within the cochlear partition. How to span the gap between protein sequence and a molecular description of function must be one of the postgenomic challenges for the future.
Acknowledgments
We thank Jonathan Gale for discussion. This work was supported by the Wellcome Trust and the Medical Research Council.
Abbreviation
- OHC
outer hair cell
Footnotes
This paper was presented at the National Academy of Sciences colloquium “Auditory Neuroscience: Development, Transduction, and Integration,” held May 19–21, 2000, at the Arnold and Mabel Beckman Center in Irvine, CA.
References
- 1.Manley G A. Proc Natl Acad Sci USA. 2000;97:11736–11743. doi: 10.1073/pnas.97.22.11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gold T. Proc R Soc London Ser B. 1948;135:492–498. [Google Scholar]
- 3.Flock A, Cheung H. J Cell Biol. 1977;75:339–343. doi: 10.1083/jcb.75.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hudspeth A J, Choe Y, Mehta A D, Martin P. Proc Natl Acad Sci USA. 2000;97:11765–11772. doi: 10.1073/pnas.97.22.11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zweig G. J Acoust Soc Am. 1991;89:1229–1254. doi: 10.1121/1.400653. [DOI] [PubMed] [Google Scholar]
- 6.Brownell W E, Bader C R, Bertrand D, De Ribaupierre Y. Science. 1985;227:194–196. doi: 10.1126/science.3966153. [DOI] [PubMed] [Google Scholar]
- 7.Ashmore J F. J Physiol. 1987;388:323–347. doi: 10.1113/jphysiol.1987.sp016617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dallos P, Evans B N. Science. 1995;267:2006–2009. doi: 10.1126/science.7701325. [DOI] [PubMed] [Google Scholar]
- 9.Frank G, Hemmert W, Gummer A W. Proc Natl Acad Sci USA. 1999;96:4420–4425. doi: 10.1073/pnas.96.8.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neely S T, Kim D O. J Acoust Soc Am. 1986;79:1472–1480. doi: 10.1121/1.393674. [DOI] [PubMed] [Google Scholar]
- 11.Mammano F, Nobili R. J Acoust Soc Am. 1993;93:3320–3332. doi: 10.1121/1.405716. [DOI] [PubMed] [Google Scholar]
- 12.Hemmert W, Zenner H P, Gummer A W. Biophys J. 2000;78:2285–2297. doi: 10.1016/S0006-3495(00)76775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dallos P, Evans B N. Science. 1995;267:2006–2009. doi: 10.1126/science.7701325. [DOI] [PubMed] [Google Scholar]
- 14.Holley M C, Ashmore J F. Proc R Soc London Ser B. 1988;232:413–429. doi: 10.1098/rspb.1988.0004. [DOI] [PubMed] [Google Scholar]
- 15.Kalinec F, Kachar B. Biophys J. 1992;62:A166. [Google Scholar]
- 16.Forge A. Cell Tiss Res. 1991;265:473–483. doi: 10.1007/BF00340870. [DOI] [PubMed] [Google Scholar]
- 17.Ashmore J F. In: Sensory Transduction. Corey D P, Roper S D, editors. New York: Rockefeller Univ. Press; 1992. pp. 395–412. [Google Scholar]
- 18.Gale J E, Ashmore J F. Nature (London) 1997;389:63–66. doi: 10.1038/37968. [DOI] [PubMed] [Google Scholar]
- 19.Santos-Sacchi J. J Neurosci. 1991;11:3096–3110. doi: 10.1523/JNEUROSCI.11-10-03096.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakazawa K, Spicer S S, Schulte B A. Hear Res. 1995;82:93–99. doi: 10.1016/0378-5955(94)00162-j. [DOI] [PubMed] [Google Scholar]
- 21.Geleoc G S, Casalotti S O, Forge A, Ashmore J F. Nat Neurosci. 1999;2:713–719. doi: 10.1038/11174. [DOI] [PubMed] [Google Scholar]
- 22.Burant C F, Takeda J, Brot-Laroche E, Bell G I, Davidson N O. J Biol Chem. 1992;267:14523–14526. [PubMed] [Google Scholar]
- 23.Concha I I, Velasquez F V, Martinez J M, Angulo C, Droppelmann A, Reyes A M, Slebe J C, Vera J C, Golde D W. Blood. 1997;89:4190–4195. [PubMed] [Google Scholar]
- 24.Tunstall M J, Gale J E, Ashmore J F. J Physiol. 1995;485:739–752. doi: 10.1113/jphysiol.1995.sp020765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santos-Sacchi J. J Neurosci. 1992;12:1906–1916. doi: 10.1523/JNEUROSCI.12-05-01906.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwasa K H. Biophys J. 1993;64:102. doi: 10.1016/S0006-3495(93)81053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gale J E, Ashmore J F. J Physiol. 1993;467:274P. [Google Scholar]
- 28.Nakazawa K, Spicer S S, Schulte B A. Hear Res. 1995;82:93–99. doi: 10.1016/0378-5955(94)00162-j. [DOI] [PubMed] [Google Scholar]
- 29.Corpe C P, Bovelander F J, Hoekstra J H, Burant C F. Biochim Biophys Acta. 1998;1402:229–238. doi: 10.1016/s0167-4889(97)00155-9. [DOI] [PubMed] [Google Scholar]
- 30.Dancer A. Audiology. 1992;31:301–312. [PubMed] [Google Scholar]
- 31.Spedding M. Br J Pharmacol. 1984;83:211–220. doi: 10.1111/j.1476-5381.1984.tb10137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gale J E, Ashmore J F. Nature (London) 1997;389:63–66. doi: 10.1038/37968. [DOI] [PubMed] [Google Scholar]
- 33.Hille B, Woodhull A M, Shapiro B I. Philos Trans R Soc London B. 1975;270:301–318. doi: 10.1098/rstb.1975.0011. [DOI] [PubMed] [Google Scholar]
- 34.Hol W G. Prog Biophys Mol Biol. 1985;45:149–195. doi: 10.1016/0079-6107(85)90001-x. [DOI] [PubMed] [Google Scholar]
- 35.Ben-Tal N, Honig B. Biophys J. 1996;71:3046–3050. doi: 10.1016/S0006-3495(96)79498-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raphael R M, Popel A S, Brownell W E. Biophys J. 2000;78:2844–2862. doi: 10.1016/S0006-3495(00)76827-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng J, Shen W, He D Z, Long K B, Madison L D, Dallos P. Nature (London) 2000;405:149–155. doi: 10.1038/35012009. [DOI] [PubMed] [Google Scholar]
- 38.Bogazzi F, Bartalena L, Raggi F, Ultimieri F, Martino E. J Endocrinol Invest. 2000;23:170–172. doi: 10.1007/BF03343701. [DOI] [PubMed] [Google Scholar]
- 39.Scott D A, Karniski L P. Am J Physiol. 2000;278:C207–C211. doi: 10.1152/ajpcell.2000.278.1.C207. [DOI] [PubMed] [Google Scholar]
- 40.Ikeda K, Saito Y, Nishiyama A, Takasaka T. J Physiol. 1992;447:627–648. doi: 10.1113/jphysiol.1992.sp019021. [DOI] [PMC free article] [PubMed] [Google Scholar]