

Abstract
Expression of conjugative transfer and virulence functions of the Enterococcus faecalis antibiotic resistance plasmid pCF10 is regulated by the interaction of the pheromone receptor protein PrgX with two DNA binding operator sites (XBS1 and XBS2) upstream from the transcription start site of the prgQ operon (encoding the pCF10 transfer machinery) and by posttranscriptional mechanisms. Occupancy of both binding sites by PrgX dimers results in repression of the prgQ promoter. Structural and genetic studies suggest that the peptide pheromone cCF10 functions by binding to PrgX and altering its oligomerization state, resulting in reduced occupancy of XBSs and increased prgQ transcription. The DNA binding activity of PrgX has additional indirect regulatory effects on prgQ transcript levels related to the position of the convergently transcribed prgX operon. This has complicated interpretation of previous analyses of the control of prgQ expression by PrgX. We report here the results of in vivo and in vitro experiments examining the direct effects of PrgX on transcription from the prgQ promoter, as well as quantitative correlation between the concentrations of XBSs, PrgX protein, and prgQ promoter activity in vivo. The results of electrophoretic mobility shift assays and quantitative analysis of prgQ transcription in vitro and in vivo support the predicted roles of the PrgX DNA binding sites in prgQ transcription regulation. The results also suggest the existence of other factors that impede PrgX repression or enhance its antagonism by cCF10 in vivo.
INTRODUCTION
Expression of genes required for conjugative transfer of Enterococcus faecalis plasmid pCF10 is controlled by cell-cell signaling. The plasmid encodes a sensing system to increase expression of the prgQ operon, which encodes the transfer machinery, in response to the peptide mating pheromone cCF10 (LVTLVFV) (16). cCF10 is excreted into the growth medium by plasmid-free E. faecalis cells that can serve as conjugative recipients for pCF10 transfer. Since cCF10 production is chromosomally encoded, pCF10 carries genes whose products block self-induction of donor cells by endogenous pheromone. These include prgY, whose protein product reduces pheromone production by the host cell (7), and prgQ, which encodes a 22-amino-acid polypeptide that is processed to an exported 7-amino-acid inhibitor peptide, iCF10 (AITLIFI) (25). The iCF10 peptide can be imported into donor cells, where it competitively inhibits binding of cCF10 and serves to neutralize any residual endogenous pheromone that escapes PrgY inhibition. For further review of pheromone peptide signaling in E. faecalis, see references 16 and 13.
Recent studies have implicated the prgQ promoter as the regulatory target for pheromone signaling in the pCF10 system, as illustrated in Fig. 1A. In the present model, transcription from the prgQ promoter PQ is repressed by PrgX. PrgX dimers bind specifically to two “operator” DNA binding sites (XBS1 and XBS2) in the intergenic region between prgX and prgQ (3). The lower-affinity XBS2 site overlaps PQ, and PrgX occupancy of XBS2 is proposed to inhibit transcription by steric hindrance of RNA polymerase (RNAP) binding to PQ. While XBS2 can be bound by a PrgX dimer in the absence of XBS1, our data (3) suggest that the affinity of the PrgX-XBS2 binary interaction is so low that it would not allow PrgX to compete effectively with RNA polymerase for binding to this region of pCF10 and repress prgQ transcription initiation. Genetic and structural data (19, 20, 27) suggest that protein-protein interactions between pairs of PrgX dimers bound to the XBSs and the resulting DNA loop formation may favor a repressing structure (state iv) shown in Fig. 1A. Both iCF10 and cCF10 bind to the same region of PrgX, and binding of either peptide has no direct effects on the structure of the DNA binding domain. Instead, the peptides have different effects on the structure of the C terminus of PrgX that cause opposing changes on the PrgX oligomerization state. It has been suggested that the repressing tetramer structure is enhanced by iCF10 binding, while cCF10 binding is predicted to favor conversion of tetramers to dimers and ultimately reduce PQ repression (20, 27).
Fig 1.

Regulatory circuits controlling the initiation of the E. faecalis pheromone response in the pCF10 system. (A) Potential states of occupancy of the PrgX DNA binding sites (XBSs) in the prgQ promoter region. (i) No PrgX present and maximum expression of prgQ. (ii) For a DNA target containing both binding sites, at low PrgX concentrations, dimers bind first to the higher-affinity site (>100× higher than the affinity of PrgX for XBS2) XBS1, but occupancy of this site has no direct effect on PQ. (iiia) Occupancy of both XBSs is predicted to reduce prgQ transcription by steric hindrance of RNA polymerase binding, but affinity for XBS2 is <1/100th that of the affinity for XBS1 (3). (iiib) Since PrgX tetramers have been observed in crystals (20, 27), it is theoretically possible to have XBS1 bound by a tetramer. (iv) Protein-protein interactions between pairs of PrgX dimers bound to the two XBSs results in a DNA loop that increases the stability of the repressing complex. Data presented in this paper and elsewhere (3) suggest that the affinity of the PrgX dimer for XBS2 is sufficiently low in the absence of DNA looping, that conversion between states ii and iv occurs rapidly, and states iiia and iiib, or a structure where only XBS2 is bound, probably do not exist at sufficient levels to be biologically important. Pheromone (cCF10) and inhibitor (iCF10) peptides do not directly alter the DNA binding regions of PrgX; instead they appear to affect the PrgX oligomerization state, with iCF10 predicted to drive the system toward state iv, with cCF10 having the opposite effect (20, 27). (B) Linear templates for in vitro transcription. The positions of the ends of the templates relative to the “+1” nucleotide of prgQ mRNA are indicated along with the locations of the XBSs and PQ regions. The long template (LT) contains both XBSs, whereas the short template (ST) contains only XBS2. The runoff transcript in both cases is 114 nt. (C) Sequences of the XBS regions of pCF10. Mutations used in this study are shown in boldface, and the sequence coordinates relative to the experimentally determined “+1” nucleotide of the prgQ transcript are indicated. WT, wild type.
In the absence of pheromone, PrgX repression of prgQ operon transcription is incomplete, allowing for a basal level of expression of an ∼380-nucleotide (nt) RNA (Qs) whose 5′ segment encodes iCF10; induced cells contain increased levels of Qs, and longer transcripts that encode the conjugative transfer machinery (5, 9). The mRNA from which PrgX is translated is transcribed from a promoter (PX) located about 250 bp downstream of the prgQ transcription start site, but on the opposite strand: the first ∼100 nt of this countertranscript are processed into a stable small RNA that affects termination of nascent prgQ transcripts (4, 18, 28). Our cumulative results suggest that three important regulatory circuits act in this system: (i) repression of the prgQ promoter PQ by PrgX, (ii) RNA-RNA interactions between nascent and mature transcripts from the two opposing promoters, and (iii) interference resulting from collisions between opposing transcription complexes from the two promoters. Recently published mathematical modeling and quantitative analyses of the effects of each of these regulatory mechanisms on prgQ and prgX transcript levels suggest that they are all required for the system to function as a robust biological switch (8). Validation of this model requires accurate determination of the relative importance of each of these mechanisms in control of the system, necessitating development of experimental approaches to study each mechanism in the absence of the others. This has been particularly challenging in the case of control of prgQ promoter activity by PrgX, the mechanism that is directly impacted by the signaling peptides.
Problems with PrgX stability and with the solubility of both PrgX and the peptides in aqueous solutions, as well as nonspecific binding of all of these molecules to inert surfaces such as affinity matrices and dialysis membranes, have interfered with attempts to use biochemical approaches to confirm the features of the model for PrgX control of PQ. Furthermore, in vivo analyses of prgQ expression and its control by PrgX have been complicated by the fact that PrgX has a positive autoregulatory effect on expression of its own transcript (1, 19) and by the fact that prgQ expression is also controlled posttranscriptionally (18, 28). The purpose of the studies reported here was to determine quantitatively how PrgX occupancy of XBS1 and XBS2 affects prgQ transcription directly in the absence of other regulatory circuits. Since PrgX repression of PQ is the only regulatory mechanism directly controlled by pheromone binding, addressing this question is critical. We used electrophoretic mobility shift assays (EMSAs) to examine the interactions of PrgX with DNA probes containing various combinations of wild-type and mutated XBSs and compared these results with analyses of prgQ transcription from the same templates using runoff in vitro transcription (IVT) assays. We also examined direct effects of PrgX on prgQ transcription in vivo using Northern blot analysis of mRNA from E. faecalis strains carrying plasmids containing the DNA templates used for in vitro studies. For the in vivo studies, PrgX was provided in trans from a constitutively expressed prgX allele integrated into the chromosome to eliminate complicating effects of PrgX autoregulation.
MATERIALS AND METHODS
Strains, plasmids, and growth media.
The bacterial strains used in this study are listed in Table 1. E. faecalis strains OG1Sp (21) and 100-5 (14) were used for Northern analyses and for determination of intracellular PrgX concentrations and plasmid copy number. Strains were grown at 37°C in M9-YE (12) or on Todd-Hewitt agar plates supplemented with antibiotics at the following concentrations: chloramphenicol, 20 μg/ml; tetracycline, 10 μg/ml; spectinomycin,1,000 μg/ml; and fusidic acid, 25 μg/ml. Escherichia coli strain DH5α (Gibco, BRL) was used for plasmid propagation in Luria broth (LB) supplemented with antibiotics at the following concentrations: chloramphenicol, 20 μg/ml; erythromycin, 10 μg/ml. X-Gal (5-bromo-4 chloro-3indolyl-β-d-galactopyranoside) was used at a concentration of 40 μg/ml (Gold Biotechnology). Synthetic cCF10 was purchased from New England Peptides. The plasmids used in this study are listed in Table 1.
Table 1.
Bacterial strains and plasmids used in this studya
| Strain or plasmid | Relevant feature(s) | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | Cloning host | Lab stock |
| E. faecalis | ||
| OG1Sp | Spr | 21 |
| 100-5 | Spr; carries chromosomal copy of prgX | 14 |
| DM105 | Fusr; rpoC::His allele | This study |
| B. subtilis | rpoC::His | 26 |
| Plasmids | ||
| pGEM-T Easy | Crbr; TA cloning vector | Promega |
| pCF10 | Tcr; cCF10 conjugative plasmid | 12 |
| pCI3340 | Camr; vector used for all plasmids below | 17 |
| pBK1 | Camr | |
| pBK2 | Camr | |
| pDM1 | pCI3340 with prgX terminator | This study |
| pDM1.1 | pDM1 carrying LT −163 to +114 | This study |
| pDM1.2 | pDM1 carrying LT with B1m1 mutation | This study |
| pDM1.3 | pDM1 carrying LT with B1m2 mutation | This study |
| pDM1.4 | pDM1 carrying LT with B2m1 mutation | This study |
| pDM1.5 | pDM1 carrying LT with B2m2 mutation | This study |
| pDM1.6 | pDM1 carrying LT with B1m1 and B2m1 mutations | This study |
| pDM1.7 | pDM1 carrying ST −68 to +114 | This study |
| pDM1.8 | pDM1 carrying ST with B2m1 mutation | This study |
The pDM1.1 to -1.8 plasmids contain cloned pCF10-derived DNA fragments that were also used as templates for in vitro transcription. The sequences of the PrgX binding sites, as well as those of the B1 and B2 mutations analyzed in these studies, are shown in Fig. 1C.
Generation of templates for in vitro and in vivo transcription experiments. (i) Linear templates.
Linear long templates (LTs) (including pCF10 nt 8014 to 8290, i.e., −163 to +114 relative to the prgQ transcription start site) and short template (ST) (pCF10 nt from 8109 to 8290; −66 to +114 relative to the prgQ transcription initiation site) were generated by PCR using plasmid templates containing the prgX/prgQ region of pCF10 and primers complementary to the ends of the pCF10 sequences to be amplified. Binding site mutations (Fig. 1) were generated using overlap extension PCR. All templates generated were sequenced to confirm that no additional mutations were introduced during PCR amplification.
(ii) Plasmid templates.
The prgX terminator (pCF10 nt 6998 to 7049; accession no. AY855841) was PCR amplified and was introduced into pGEM-T Easy after the addition of 3′-dATP using Taq polymerase (NEB) to yield pGEMTerm8. This construct was digested with PstI and XhoI, and the 100-bp prgX terminator fragment was cloned into the same sites of pCI3340 (17), generating pCI3340Term3. In order to remove an additional EcoRI site that was introduced upstream of the terminator fragment, pCI3340Term3 was digested with NotI and SpeI and the ends were filled in with DNA polymerase I, treated with Klenow fragment (NEB), and self-ligated, to yield pDM1. DNA segments corresponding to the linear templates described above were then inserted adjacent to the terminator segment after each of the templates were cloned into pGEM-T Easy, digested with EcoRI, gel purified, and cloned into the same site in pDM1 to generate pDM1.1 to pDM1.8, corresponding to binding site mutations in (Fig. 1). The resulting plasmids were sequenced to confirm that they contained the desired inserts for analysis of prgQ transcription.
Construction of an E. faecalis strain carrying an rpoC allele with a 10-His tag.
We amplified ∼1-kb segments of genomic DNA extending in both directions from the 3′ end of the rpoC gene (EF3237) using partially overlapping outward-reading primers containing restriction enzyme cleavage sites at their ends. These primers were engineered to add an in-frame 10-His tag to the 3′ end of rpoC and also contained BsaI sites. Following amplification, we digested the products of the initial PCRs with BsaI and ligated them into pGEM-T Easy and then recloned the insert from pGEM-T Easy into pCJK47 digested with XbaI and XmaI. This plasmid was then used to exchange the tagged allele for the wild-type allele in strain OG1F, which was made by selecting for spontaneous mutants on Todd-Hewitt broth (THB) plates containing 50 μg/ml fusidic acid. The resulting strain was designated DM105. The correct DNA sequences of the cloned DNA in the integration vector as well as the sequence of the EF3237 region of OG1F was confirmed experimentally. His-tagged purification of E. faecalis RNA polymerase purification was similar to that of B. subtilis RNA polymerase (RNAP) (26). Overnight cultures of DM105 were diluted 1:100 in 750 ml M9-yeast extract Casamino Acids (M9-YE CAA) and grown to an optical density at 600 nm (OD600) of ∼1.4. Cell pellets were resuspended in 10 ml lysozyme buffer (10 mM Tris [pH 8.0], 50 mM NaCl, 25% sucrose, 30 mg/ml lysozyme [Sigma], 250 U mutanolysin [Sigma]) for 20 min at 37°C. Cells were pelleted at 4°C and resuspended in 15 ml ice-cold yeast protein extraction reagent (Y-PER; Thermo Scientific) containing sonication buffer (SB) (50 mM Tris [pH 7.8], 300 mM NaCl, 5 mM MgCl2, 20% glycerol). We then added 1.0 ml Complete Mini, EDTA-free protease inhibitor tablet (Roche) dissolved in SB and sonicated. Sonicated cells were immediately pelleted at 13,000 rpm for 20 min at 4°C. We transferred the cleared lystate at 4°C to 1.5 ml of Ni-nitrilotriacetic acid (NTA) His bind resin (EMD) equilibrated with 3 volumes of SB. The column was washed with SB (8 volumes) and SB containing 60 mM imidazole (8 volumes). RNAP was eluted with 3.0 ml of 400 mM imidazole in SB (0.250 ml × 12). We collected two 0.5-ml fractions (fractions 1 and 2), four 0.250-ml fractions (fractions 3 to 6), and two 0.5-ml fractions (fractions 7 and 8). The column was capped and incubated 5 min before collection of fractions 2 and 3 and 2 min before collection of fraction 4. We added an equal volume of 60% glycerol to fractions 1, 7, and 8 and incubated the mixture at −20°C. Fractions 2 through 6 were dialyzed using Slide-A-Lyzer minidialysis devices, with a 3,5000-molecular-weight cutoff (MWCO) (Thermo Scientific) in 300 ml 2× storage buffer (20 mM Tris [pH 8.0], 20 mM MgCl2, 2 mM EDTA, 200 mM KCl, 20% glycerol) for 1 h (3 times) at 4°C. We stored dialyzed RNAP in an equal volume of 60% glycerol at −20°C.
EMSAs.
DNA templates containing both binding sites or mutations to one or both binding sites and the PCR products from the reactions described above were purified, and the 3′ end was labeled with digoxigenin (DIG [DIG-11-ddUTP]) according to the DIG gel shift kit instructions (Roche). EMSAs were performed using the DIG gel shift kit according to the manufacturer's instructions. Labeled probe (2.5 nM final concentration) was mixed with purified histidine-tagged PrgX (His-PrgX) at final concentrations of 0, 10, and 125 nM protein, and the reaction mixture was incubated at room temperature for 15 min. DNA/protein complexes were separated using 5% polyacrylamide gels containing 1× Tris-borate-EDTA (TBE; pH 7.9) at room temperature for 2 h at 140 V and transferred to nylon membranes (Roche) using the Genie electrophoretic blotter (Idea Scientific). DIG-labeled DNA was detected according to the immunological detection protocol provided by the manufacturer, using anti-DIG Fab fragments and CDP-STAR chemiluminescent substrate.
Qualitative and quantitative Western blot analysis.
Purified His-PrgX and cell lysates were subjected to denaturing electrophoresis on 12% polyacrylamide gels (SDS-PAGE) and transferred onto 0.2 μM nitrocellulose membrane (S&S) using Towbin buffer at 90 V for 70 min at 4°C. PrgX was detected by using the His-PrgX polyclonal antibody (1) at a dilution of 1:1,500 and horseradish peroxidase (HRP)-goat anti-rabbit IgG (Zymed) at 1:5,000. Detection was performed using SuperSignal West Pico chemiluminescent substrate according to the manufacturer's (Thermo Scientific) instructions. PrgX concentrations from the chemiluminescent autoradiographs were determined for the cell lysates from the smooth cubic spline standard curves (r ≥ 0.97), generated by the Spot Densitometry software on the AlphaImager 2200 (Alpha Innotech, Inc.). The intensities of immunoreactive PrgX bands from lysates were compared to those from standardization lanes containing 30, 20, 10, and 5 ng of purified His-PrgX. For each strain, cell lysates were isolated at least three times and the concentration was determined 4 times or more.
Plasmid copy number determination.
We used quantitative PCR (qPCR) to quantify plasmid copy number by comparing the amount of amplified product generated from genomic DNA from strain 100-5 (with a prgX gene in the chromosome) to that from isogenic strains carrying various plasmids containing the same prgX gene as described by Cook et al. (11). Cells were grown overnight (16 h) in M9-YE at 37°C with antibiotics. The next day, they were diluted 1:10 in fresh M9-YE with antibiotics and grown for 2.5 h at 37°C. For real-time qPCR, DNA was isolated from 100-5, OG1Sp(pCF10), OG1Sp(pBK2), and OG1Sp using the DNeasy blood and tissue kit according to the Gram-positive isolation procedure (Qiagen). DNA was digested completely with EcoRI (New England BioLabs) and precipitated prior to quantification of the concentration using the Quant-iT PicoGreen double-stranded DNA (dsDNA) kit (Invitrogen). Fluorescence of triplicate DNA samples was read on a Modulus Microplate reader (Turner Biosystems) using optical kit blue (excitation, 490 nm; emission, 510 to 570 nm).
Analysis of transcription in vivo by RT-PCR.
We used previously published primers to quantify levels of prgX (11) and gyrB (6) mRNA. Real-time qPCR amplification and analysis were performed using the iQ5 cycler (Bio-Rad) with Optical System software version 2.0 (Bio-Rad) as described by Cook et al. (11). The real-time qPCRs were prepared using the Bio-Rad DNA Master SYBR green kit (Bio-Rad). Ten-fold serial dilutions of 100-5 were used to generate standard curves for prgX (pCF10 nt 7687 to 7874) and gyrB ranging from 3.3 × 10−7 to 3.3 × 10−3 copies/μl and the no-template control. Each reaction was run in triplicate, and the mixtures contained target samples OG1Sp(pBK2), OG1Sp(pCF10), and OG1Sp; serial dilutions ranged from 10 to 1.0, 0.1, and 0.01 ng. The range of slopes for the qPCR assays was from −3.7 to −3.9 (≥80% efficiency), and linearity (r2) values were all ≥0.99. A melting curve was run to confirm that a single PCR product was amplified and samples were also analyzed on 2% agarose gel to confirm size.
Analysis of transcription in vivo by Northern blotting.
Cells were grown overnight in M9-YE, diluted 1:10 in fresh medium, and allowed to grow for 2 to 2.5 h. RNAprotect bacterial reagent (Qiagen) was added to stabilize RNA before cell pellets were frozen at −80°. Prior to RNA isolation using the RNeasy RNA kit (Qiagen), cells were treated with 30 mg/ml lysozyme (Sigma) and 500 U/ml mutanolysin (Sigma) in 10 mM Tris HCl (pH 8.0)–1 mM EDTA (pH 8.0) for 15 min at 37°C. RNA samples were separated on 5% Criterion TBE-urea gels (Bio-Rad) and transferred to positively charged nylon membranes (Roche) using a Trans-Blot SD semidry electrophoretic transfer cell (Bio-Rad) at 395 mA 20 V for 15 min. The blots were probed with DIG-labeled anti-prgQ +1 to +110 probe, which was generated by in vitro transcription with SP6 RNA polymerase (Promega) according to the DIG RNA labeling kit (Roche). CDP-Star chemiluminescent substrate (Roche) was used for detection of RNA. Synthetic DIG-anti-5S oligonucleotide was purchased from Integrated DNA Technologies. AlphaImager 2200 software was also used for quantitative densitometric analysis of relative levels of in vivo prgQ transcripts in the Northern blots from the same templates in isogenic strains differing only in expression of PrgX; we used quantitative densitometry, and normalized loading by also quantifying relative levels of 5S RNA in different lanes. For each template, analysis of at least 3 independent cultures was carried out.
Runoff in vitro transcription assays.
To examine effects of PrgX binding on transcription from the prgQ promoter in vitro, we employed runoff transcription assays in 35-μl volumes with the linear, double-stranded DNA templates described above containing the prgQ promoter and various combinations of wild-type and mutated XBSs. For most assays, we used B. subtilis RNA polymerase purified by NTA-Ni2+ affinity chromatography (26) and reaction conditions essentially identical to those described previously (23, 29). The significant differences in the procedures used here included the substitution of the dinucleotide ApU (matching the first 2 bases of prgQ mRNA) as opposed to the ApC dinucleotide previously employed for transcription from the glyQS promoter. The storage buffer required to keep purified His-PrgX in concentrated solutions contains HEPES, LiCl, β-mercaptoethanol, EDTA, and Triton X-100 (3); to control for potential inhibitory effects of this buffer, each reaction mixture contained a 1/35 dilution of the PrgX storage buffer. For each reaction, all components of the IVT reaction mixture except the RNA polymerase were mixed at room temperature to allow for PrgX binding to template DNA. A 1-μl aliquot of RNA polymerase was then added to start the IVT reaction; each reaction was allowed to proceed for 15 min, before it was stopped by phenol-chloroform extraction, and the labeled RNA products were analyzed by denaturing polyacrylamide gel electrophoresis as previously described (23, 29). Nonlinear regression analysis was used to determine the concentration required for half-maximal termination (Graphpad Prism version 4.00 for Windows; Graphpad Software).
RESULTS
Effects of mutations in prgQ operator sequences on PrgX binding to IVT templates.
Our previous studies employed electrophoretic mobility shift assays (EMSAs) and DNase footprinting to identify the two XBSs and site-directed mutations to confirm the importance of specific nucleotides in the binding interactions (3). In order to determine the direct effects of PrgX occupancy of XBSs on prgQ transcription, we prepared linear, double-stranded DNA templates carrying either both XBSs (LT) (Fig. 1A) or only the secondary XBS (ST). Both templates contained the functional wild-type PQ and were predicted to generate identical 114-nt runoff mRNA products when transcribed (Fig. 1B). In addition to the templates carrying wild-type XBSs, we generated derivatives of LT and ST containing selected mutations in one or both XBSs (Fig. 1C).
We initially used EMSAs to ensure that XBS mutations had the expected effects on PrgX binding in vitro to the same DNA templates used for IVT studies. (The DNA fragments used in previously published studies [3] contained the same XBS regions but were different in size from the LT and ST). The PrgX-mediated shift in the electrophoretic mobility of the LT probe is shown in Fig. 2A. At PrgX concentrations in the range of 5 to 25 nM, a prominent shifted band (I) that is likely to represent the probe with one PrgX dimer bound to XBS1 (corresponding to state ii in Fig. 1A) was clearly evident. As the PrgX concentration was increased, a “supershifted” band (II) became more apparent. The supershifted band probably represents a probe molecule with two sets of PrgX dimers bound and in which the DNA has formed a loop via protein-protein interactions between the bound dimers (state iv in Fig. 1A). We believe that unlooped complexes in which both binding sites are occupied (state iiia in Fig. 1A) or in which a PrgX tetramer is bound to the primary binding site (state iiib in Fig. 1A), would be much less stable and would be unlikely to contribute significantly to the shifted probe bands seen in the assays; this is supported by the lack of larger shifted species with intensities more than 5% those of bands I and II in any reactions.
Fig 2.
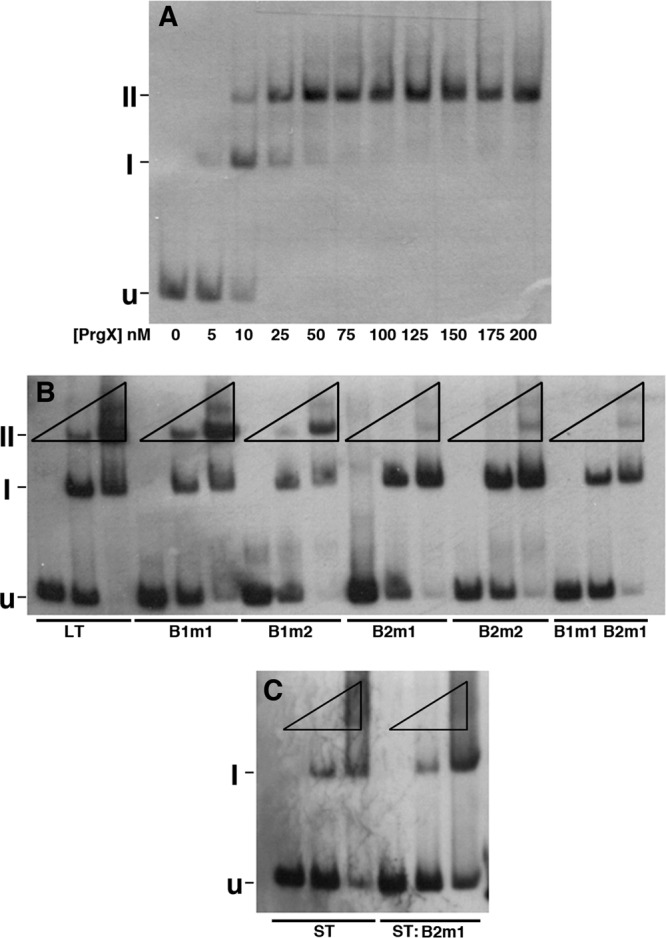
Binding of purified PrgX to pCF10 DNA templates as determined by electrophoretic mobility shift assay (EMSA). Binding reaction mixtures contained 2.5 nM digoxigenin-labeled DNA probes and various PrgX concentrations. In all three gels shown, “U” indicates the position of the unbound probe, “I” represents a shifted complex, and “II” represents a supershifted complex. Band I probably represents a PrgX dimer bound to XBS1, whereas band II represents a looped probe molecule with both XBSs occupied by a dimer. (A) Binding of PrgX to wild-type LT. Concentrations of PrgX (nM) are indicated below the respective lanes. (B) Binding of PrgX to LT derivatives with XBS mutations (indicated below the lanes). For each DNA, the mobility shifts were determined at PrgX concentrations of 0, 10, and 125 nM (indicated by the triangles above each set of 3 lanes run with the same DNA probe), since these produced, respectively, no shift, partial shift, and nearly complete shift of the LT probe. (C) Binding of PrgX to ST and ST:B2m1 probes (indicated below each lane). The same DNA and PrgX concentrations were used as those in panel B.
We also compared PrgX binding to the wild-type LT probe to that of probes with XBS mutations. The electrophoretic mobility of these probes was determined (i) in the absence of PrgX; (ii) at a concentration (10 nM), where about half of the wild-type LT probe XBS1 was shifted; and (iii) at 125 nM, where all of the LT probe was shifted. As seen in Fig. 2B, the B1m1 mutation had only a minor effect on PrgX binding to LT, with a slightly lower ratio of shifted/supershifted probe at both PrgX concentrations (lanes 4 to 6), while the B1m2 mutation (lanes 7 to 9) had a stronger impact on the occupancy of XBS1, as indicated by the relative ratios of I to unshifted and band I to II at 10 nM PrgX; however, at 125 nM PrgX, the majority of the probe was in the band II form, similar to the pattern observed with the wild-type LT probe. Neither mutation in XBS2 (lanes 10 to 15) affected the transition from unshifted to band I, but substantially inhibited the supershift, consistent with a minimal impact of the mutations on PrgX binding to XBS1, but a substantial reduction in the conformation where both sites are bound due to decreased affinity for XBS2. Mutation of both XBSs (Fig. 2B, lanes 16 to 18) resulted in decreases in the proportion of both shifted and supershifted probes, with the largest impact on the supershift, consistent with the results obtained with probes containing single XBS mutations. When the interaction of PrgX with an ST probe was examined (Fig. 2C), we found only a shifted species representing occupancy of XBS2 by a PrgX dimer, as expected, and this shift was reduced by the B2m1 mutation. (Note the differences in the relative amounts of band I between lanes 2 and 5 and the differences in the relative amounts of unshifted probe between lanes 3 and 6.) At 125 nM PrgX, we observed a reproducible smear of shifted material migrating more slowly than band I with both the ST and ST:B2m1 probes (Fig. 2C); this material might represent an unstable complex in which a PrgX tetramer was bound to XBS2 via one dimer pair.
Effects of PrgX on transcription of prgQ in vitro.
We next examined effects of PrgX on prgQ transcription in runoff IVT assays using purified B. subtilis RNA polymerase. Some of our IVT experiments included a reaction in which a linear double-stranded template of B. subtilis DNA was used that contained the well-characterized glyQS promoter (15) and produced a 119-nt runoff product; as expected, addition of PrgX to IVT reactions using this template did not inhibit transcription significantly (not shown). To ensure that inhibitory effects were due to PrgX itself rather than the storage buffer, we developed a protocol (Materials and Methods) that incorporates an equivalent dilution of PrgX buffer in all reactions, including the positive-control reaction mixture containing no PrgX. One microliter of the appropriate PrgX dilution was added to reaction tubes containing the DNA template, and a mixture containing the RNA polymerase and the other components of the reaction was then added to start transcription. Analysis of runoff transcripts produced in vitro from a typical experiment examining the effects of PrgX on transcription from the PQ promoter in the LT is shown in Fig. 3. In the absence of PrgX, an abundant runoff product of the expected size was produced, demonstrating the promoter activity of this template with B. subtiilis polymerase. PrgX reduced the amount of prgQ transcript in a dose-dependent fashion. Inhibition was observed at PrgX concentrations in the 10 nM range, and 50 nM PrgX resulted in 60 to 70% inhibition. Assuming that repression requires occupancy of both binding sites by PrgX dimers, the molar ratios of protein to binding sites were in the range of 1:1 at the low end of the inhibition curves and about 25:1 at the upper end. We carried out multiple replicate assays of the effects of PrgX on transcription of LT and ST and derivatives containing the same set of XBS mutations used in the EMSAs. The relative amounts of runoff products were quantified and averaged to generate inhibition curves.
Fig 3.

Effects of PrgX on prgQ transcription in vitro. Results of a typical IVT reaction with the LT template and various PrgX concentrations ranging from 0 to 1,374 nM. The picture on the left shows the gel, with PrgX concentrations indicated at the bottom of each lane and the 114-nt runoff transcript appearing as a distinct single band in the central region of the gel in each lane (arrowhead at left), while the plot on the right shows the inhibition curve generated from IMAGEQUANT analysis of the scanned gel. (The 1,374-nM PrgX concentration is not shown.)
When the various templates containing XBS mutations or deletions were compared to the LT, we found that all of them showed decreased susceptibility to PrgX inhibition (Fig. 4). Although the IVT assays are not completely comparable to the EMSAs, the effects of the mutations on the ability of PrgX to repress prgQ transcription in vitro were consistent with the effects of the same mutations on DNA binding (Fig. 2). When the relative amounts of PrgX inhibition of transcription in concentrations between 25 and 50 nM (50% inhibition of transcription from LT was observed consistently in this concentration range) were compared for different templates, all of the XBS mutations reduced PrgX inhibition by at least 60% (Fig. 4, left panel). PrgX inhibition of transcription from ST containing the wild-type XBS2 was comparable to that of the most severe single XBS mutations in LT (compare left and right panels for Fig. 4). The B2m1 mutation in the context of ST essentially abolished PrgX repression (Fig. 4, right panel), whereas this mutation reduced repression of transcription from LT by 60 to 70% (Fig. 4, left panel). When the LT contained one mutation at each XBS, repression was also abolished (not shown; see next section). Taken together, these results suggest that cooperative interactions between the DNA/protein complexes at the two binding sites partially overcame the deleterious effects of an XBS2 mutation when the template also contained wild-type XBS1.
Fig 4.

Effects of XBS mutations on PrgX repression. The inhibition curves for various templates containing the indicated XBS mutations relative to that observed for the wild-type LT as determined by IMAGEQUANT analysis are shown in the left panel. The right panel shows the relative inhibition obtained with LT, ST, and the B2m1 mutation in the ST context. These curves were generated by nonlinear regression analysis from multiple replicate reactions and the bars represent the standard deviations.
We attempted several experiments to examine the effects of the cCF10 pheromone and iCF10 inhibitor peptides on PrgX repression in vitro with the expectation that iCF10 would enhance repression and cCF10 would block repression. We did not obtain meaningful results due to nonspecific inhibition of RNA polymerase activity from residual organic solvents used to dissolve these extremely hydrophobic peptides, and we were unable to use dialysis or other methods to completely remove the organic solvents while keeping the peptides in solution (data not shown).
PrgX repression of prgQ transcription in vivo.
In order to examine direct effects of PrgX binding to the XBSs on prgQ transcription in vivo, we constructed plasmids carrying DNA templates containing the same pCF10 sequences used for the in vitro studies described above and used quantitative Northern blot analysis to measure prgQ transcription in E. faecalis cells. Because runoff transcript analysis is not feasible in vivo, we isolated a strong transcription terminator sequence derived from the 3′ end of the prgX gene, attached it to the templates used for the in vitro studies, and cloned the constructs into a plasmid vector that is stably maintained in E. faecalis (Materials and Methods). Isogenic strains carrying all of the plasmids used in these studies (pDM1.1 to pDM1.8) (Table 1) produced PQ-derived mRNAs of identical size in vivo; based on comparisons to RNA size standards, the transcripts produced from the plasmid templates were of a size (∼197 nt) expected from the addition of the prgX terminator to the prgQ DNA templates used in the for IVT reactions. To determine the effects of PrgX on transcription of these templates in vivo, we compared prgQ RNA levels produced from each plasmid in strain OG1S to those obtained in the isogenic strain 100-5, which expresses PrgX constitutively from the chromosome (8). In order to minimize effects of random variation of replicate cultures, RNA isolation, loading of gel lanes, and efficiency of transfer of RNAs from gels to hybridization membranes, we isolated RNA from multiple cultures of each strain and ran multiple gels with each RNA sample, varying the lanes in which samples were loaded. In all cases where PQ was active, we detected a single hybridizing band of ∼200 nt, suggesting that in vivo termination was efficient and there were not high levels of processed prgQ transcripts; our blots were also probed for 5S rRNA to normalize for loading differences. The upper part of Fig. 5 shows a typical Northern blot analysis of prgQ expression from templates containing the XBS mutations in LT that were used for IVT, and the cumulative results for in vivo effects of PrgX on transcription of all the templates are shown in the lower portion of the same figure. In the presence of PrgX, transcription from LT was reduced by over 90%, an effect comparable to the strongest repression observed in vitro at very high PrgX concentrations. Interestingly, mutations of XBS1 in LT had less dramatic effects on PrgX repression in vivo than in vitro, but the XBS2 mutations had similar effects in both assays.
Fig 5.

Effects of XBS mutations on PrgX repression of prgQ transcription in vivo. (Top) Appearance of a typical Northern blot used for this analysis. Each blot was probed for 5S RNA and the predicted prgQ transcript. No bands other than those shown in the figure were ever detected, so the region of the gel between these two bands was deleted in the photograph. “200nt” designates the relative migration of a 200-nt digoxigenin-labeled RNA marker. The templates are listed across the top of the gel. Expression from each template was assayed in isogenic E. faecalis cells either expressing PrgX (+; strain 100-5) or not (−; strain OG1Sp). (Bottom) Quantitative analysis of effects of XBS mutations on PrgX repression, as determined by densitometric scanning of the prgQ transcripts in each strain. Transcripts from each template were isolated from at least two independent cultures and were run in multiple gels with 2 to 6 replicates analyzed to generate the data shown.
In order to relate the prgQ expression levels determined in vivo to those observed in IVT experiments, we also determined intracellular levels of PrgX (using quantitative Western blot analysis) and plasmid copy numbers (using qPCR analysis) in the strains described above as well as in strains carrying pCF10. For plasmid copy number determination, we used prgX-specific primers for PCR and normalized the products obtained from strains carrying prgX-containing plasmids to those obtained from strain 100-5, in which the prgX gene exists in single copy in the chromosome. This eliminated variations due to loss of plasmid DNA by random nicking during isolation, as well as potential differences in efficiencies of PCR amplification that might occur when using different plasmid-encoded and chromosomal genes as starting templates for qPCR. The results of these studies are summarized in Table 2. This analysis showed that the PrgX concentrations in the strains used for the in vivo studies (recall that these strains contained an integrated prgX allele whose expression was driven by a constitutive promoter) were very similar to those found in cells containing pCF10, while the template concentrations were somewhat lower than those used in the IVT reactions. The in vivo PrgX concentrations were much higher than the concentrations that produced complete inhibition of transcription in vitro, making the in vitro assays more sensitive indicators of the effects of XBS mutations on PrgX repression. However, when we ranked the relative effects of various XBS mutations on PrgX DNA binding in vitro and prgQ transcription repression in vitro and in vivo, all three assays gave similar results.
Table 2.
PrgX concentrations and plasmid copy numbers in strains used for in vivo prgQ expression studies and in OG1Sp(pCF10)
| Straina | Plasmid | Plasmid copy no./chromosome (approx no. of molecules/cell)b | PrgX |
|
|---|---|---|---|---|
| No. of molecules/cell | Concn (μM) | |||
| OG1Sp | None | 0 | 0 | 0 |
| 100-5 | None | 0 | 715 | 3.8 |
| OG1Sp | pCF10 | 2.2 (4-5) | 696 | 3.7 |
| OG1Sp | pCI3340c | 11.2 (4.6) | 0 | 0 |
All strains shown here are derived from OG1Sp.
Assuming that the average volume of the cells used in these experiments is 0.31 μM3 and that the average cell contains 2 chromosomes. All copy number determinations were carried out with at least 3 independent cultures, and at least 3 replicate qPCRs carried out on each DNA sample; the standard deviations were all <15% for pCI330 and <20% for pCF10.
Plasmid pBK2 and all plasmids used for in vivo analysis of PrgX repression (Fig. 4) used pCI3340 as the cloning vector; we detected no effects of cloned pCF10/prgQ DNA on copy number.
Since the experimentally determined levels of PrgX in pCF10-containing cells were very high (>3 μM), and certain templates (e.g., ST) seemed less susceptible to inhibition in vivo than in vitro, we considered the possibility that B. subtilis RNA polymerase might be inherently more susceptible to PrgX inhibition than the native E. faecalis polymerase. Therefore, we generated an E. faecalis strain containing an rpoC allele encoding a 10-His tag, purified active RNA polymerase from this strain as described in Materials and Methods, and used it in IVT assays. In these experiments (not shown), PrgX concentrations in the range of 62.5 to 250 nM inhibited transcription in vitro from the LT by 70 to 90%, which was nearly identical to the results obtained with B. subtilis polymerase, and additional experiments examining PrgX effects on transcription by the E. faecalis RNA polymerase from templates containing XBS mutations also gave similar results to those obtained with B. subtilis polymerase. Therefore, it is unlikely that the differences in PrgX repression in vitro versus in vivo are due to differences in the RNA polymerases.
DISCUSSION
The available genetic and biochemical data (1–3, 19, 20, 27) strongly implicate PrgX-mediated repression of transcription initiation from the PQ promoter as the primary determinant for control of pheromone-inducible conjugative transfer of pCF10; it is also likely that other pheromone-inducible enterococcal conjugation systems are controlled in a similar fashion (10). In spite of this evidence, the multiple regulatory mechanisms that impact expression of the prgQ operon (13) and limitations in enterococcal biochemical and genetic tools have precluded thorough examination of the direct effects of PrgX on the prgQ promoter. In the present study, we used both in vitro and in vivo systems to determine the effects of PrgX binding to specific DNA target sites on transcription from PQ. These data demonstrated direct PrgX-mediated repression of prgQ transcription and implicated the two XBS sequences identified previously as the functional targets for the PrgX binding activity required for repression. Since in vitro transcription systems employing E. faecalis RNA polymerase have not been previously reported, we initiated IVT studies using B. subtilis RNA polymerase; PQ proved to be a good substrate for transcription initiation with this enzyme, and the PrgX-mediated repression of this promoter observed in vitro was very similar to those with B. subtilis and E. faecalis RNA polymerases. These data support previous suggestions (3, 27) that PrgX repression involves steric hindrance of RNA polymerase binding via PrgX occupancy of XBS2. The results reported here also suggest potential cooperativity between PrgX molecules bound to the two XBS operator sites, in agreement with a DNA-looping model for PQ repression by PrgX based on previous genetic, molecular, and structural data (27). Structural studies suggested that Apo-PrgX has repressor activity, in agreement with the in vitro results presented here. (Previous structural studies also indicated that PrgX purified from E. coli contains no bound peptides.) We predict that iCF10 would enhance this repressor activity, while cCF10 would inhibit it. Unfortunately, the IVT system used here did not allow us to directly assess the functional effects of the cCF10 pheromone and iCF10 inhibitor peptides on PrgX function since the addition of either peptide abolished the activity of the RNA polymerase nonspecifically. It is possible that a different method of preparing PrgX/peptide complexes could circumvent this problem, and experiments to test this possibility are under way. Another interesting question that emerges from these results relates to the high level of sensitivity of E. faecalis donor cells carrying pCF10 to induction by exogenously supplied cCF10. Initial studies of the sensitivity of these cells to synthetic cCF10 suggested that a detectable response could be observed at concentrations corresponding to <5 molecules of cCF10/responder cell (24), and this was confirmed in subsequent studies (22). Quantitative analyses of the PrgX concentrations and PrgX repression reported here indicate a remarkably high intracellular concentration of PrgX and high molar ratios of PrgX to XBSs in cells carrying the plasmids used for our in vivo studies. Thus, in donor cells exposed to low pheromone concentrations, there should still be significant pools of apo-PrgX available for prgQ repression, even if all of the exogenous pheromone were efficiently internalized and bound to PrgX. We propose that this suggests the existence of unknown features of the system that could serve to amplify the effects of pheromone on induction. These could include subcellular localization of repressed PrgX/pCF10 complexes to sites where pheromone is imported, targeted degradation of PrgX molecules following pheromone binding, or additional host or plasmid factors associated with repression of PrgX/pCF10 complexes that enhance the disruption of these complexes by cCF10.
ACKNOWLEDGMENTS
This research was supported by PHS grants to G.M.D. (GM49530) and T.M.H. (GM47823) from the NIH.
Footnotes
Published ahead of print 27 April 2012
REFERENCES
- 1. Bae T, Clerc-Bardin S, Dunny GM. 2000. Analysis of expression of prgX, a key negative regulator of the transfer of the Enterococcus faecalis pheromone-inducible plasmid pCF10. J. Mol. Biol. 297:861–875 [DOI] [PubMed] [Google Scholar]
- 2. Bae T, Dunny GM. 2001. Dominant negative mutants of prgX: evidence for a role of PrgX dimerization in negative regulation of pheromone-inducible conjugation. Mol. Microbiol. 39:1307–1320 [DOI] [PubMed] [Google Scholar]
- 3. Bae T, Kozlowicz B, Dunny GM. 2002. Two targets in pCF10 DNA for PrgX binding: their role in production of Qa and prgX mRNA and in regulation of pheromone-inducible conjugation. J. Mol. Biol. 315:995–1007 [DOI] [PubMed] [Google Scholar]
- 4. Bae T, Kozlowicz BK, Dunny GM. 2004. Characterization of cis-acting prgQ mutants: evidence for two distinct repression mechanisms by Qa RNA and PrgX protein in pheromone-inducible enterococcal plasmid pCF10. Mol. Microbiol. 51:271–281 [DOI] [PubMed] [Google Scholar]
- 5. Bensing BA, Manias DA, Dunny GM. 1997. Pheromone cCF10 and plasmid pCF10-encoded regulatory molecules act post-transcriptionally to activate expression of downstream conjugation functions. Mol. Microbiol. 24:295–308 [DOI] [PubMed] [Google Scholar]
- 6. Bourgogne A, et al. 2007. EbpR is important for biofilm formation by activating expression of the endocarditis and biofilm-associated pilus operon (epbABC) of Enterococcus faecalis OG1RF. J. Bacteriol. 189:6490–6493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chandler JR, Flynn AR, Bryan EM, Dunny GM. 2005. Specific control of endogenous cCF10 pheromone by a conserved domain of the pCF10-encoded regulatory protein PrgY in Enterococcus faecalis. J. Bacteriol. 187:4830–4843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chatterjee A, et al. 2011. Convergent transcription confers a bistable switch in Enterococcus faecalis conjugation. Proc. Natl. Acad. Sci. U. S. A. 108:9721–9726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung JW, Dunny GM. 1995. Transcriptional analysis of a region of the Enterococcus faecalis plasmid pCF10 involved in positive regulation of conjugative transfer functions. J. Bacteriol. 177:2118–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clewell DB, Dunny GM. 2002. Conjugation and genetic exchange in enterococci, p 265–300 In Gilmore MS, et al. (ed), The enterococci: pathogenesis, molecular biology and antibiotic resistance. American Society for Microbiology Press, Washington, DC [Google Scholar]
- 11. Cook L, et al. 2011. Biofilm growth alters regulation of conjugation by a bacterial pheromone. Mol. Microbiol. 81:1499–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dunny G, Funk C, Adsit J. 1981. Direct stimulation of the transfer of antibiotic resistance by sex pheromones in Streptococcus faecalis. Plasmid 6:270–278 [DOI] [PubMed] [Google Scholar]
- 13. Dunny GM, Johnson CM. 2011. Regulatory circuits controlling enterococcal conjugation: lessons for functional genomics. Curr. Opin. Microbiol. 14:174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fixen KR, et al. 2007. Analysis of the amino acid sequence specificity determinants of the enterococcal cCF10 sex pheromone in interactions with the pheromone-sensing machinery. J. Bacteriol. 189:1399–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grundy FJ, Henkin TM. 2004. Kinetic analysis of tRNA-directed transcription antitermination of the Bacillus subtilis glyQS gene in vitro. J. Bacteriol. 186:5392–5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haemig HAH, Dunny GM. 2008. New Insights into pheromone control and response in Enterococcus faecalis pCF10, p 31–49 In Bassler BL, Winans SC. (ed), Chemical communication among bacteria, 2nd ed ASM Press, Washington, DC [Google Scholar]
- 17. Hayes F, Daly C, Fitzgerald GF. 1990. Identification of the minimal replicon of Lactococcus lactis subsp. lactis UC317 plasmid pCI305. Appl. Environ. Microbiol. 56:202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson CM, et al. 2010. Direct evidence for control of the pheromone-inducible prgQ operon of Enterococcus faecalis plasmid pCF10 by a countertranscript-driven attenuation mechanism. J. Bacteriol. 192:1634–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kozlowicz BK, Bae T, Dunny GM. 2004. Enterococcus faecalis pheromone-responsive protein PrgX: genetic separation of positive autoregulatory functions from those involved in negative regulation of conjugative plasmid transfer. Mol. Microbiol. 54:520–532 [DOI] [PubMed] [Google Scholar]
- 20. Kozlowicz BK, et al. 2006. Molecular basis for control of conjugation by bacterial pheromone and inhibitor peptides. Mol. Microbiol. 62:958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kristich CJ, Chandler JR, Dunny GM. 2007. Development of a host genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57:131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leonard BA, Podbielski A, Hedberg PJ, Dunny GM. 1996. Enterococcus faecalis pheromone binding protein, PrgZ, recruits a chromosomal oligopeptide permease system to import sex pheromone cCF10 for induction of conjugation. Proc. Natl. Acad. Sci. U. S. A. 93:260–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McDaniel BA, Grundy FJ, Kurlekar VP, Tomsic J, Henkin TM. 2006. Identification of a mutation in the Bacillus subtilis S-adenosylmethionine synthetase gene that results in derepression of S-box gene expression. J. Bacteriol. 188:3674–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mori M, et al. 1988. Structure of cCF10, a peptide sex pheromone which induces conjugative transfer of the Streptococcus faecalis tetracycline resistance plasmid, pCF10. J. Biol. Chem. 263:14574–14578 [PubMed] [Google Scholar]
- 25. Nakayama J, Ruhfel RE, Dunny GM, Isogai A, Suzuki A. 1994. The prgQ gene of the Enterococcus faecalis tetracycline resistance plasmid, pCF10, encodes a peptide inhibitor, iCF10. J. Bacteriol. 176:7405–7408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qi Y, Hulett FM. 1998. PhoP-P and RNA polymerase sigmaA holoenzyme are sufficient for transcription of Pho regulon promoters in Bacillus subtilis: PhoP-P activator sites within the coding region stimulate transcription in vitro. Mol. Microbiol. 28:1187–1197 [DOI] [PubMed] [Google Scholar]
- 27. Shi K, et al. 2005. Structure of peptide sex pheromone receptor PrgX and PrgX/pheromone complexes and regulation of conjugation in Enterococcus faecalis. Proc. Natl. Acad. Sci. U. S. A. 102:18596–18601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shokeen S, et al. 2010. Structural analysis of the anti-Q-Qs interaction: RNA-mediated regulation of E. faecalis plasmid pCF10 conjugation. Plasmid 64:26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tomsic J, McDaniel BA, Grundy FJ, Henkin TM. 2008. Natural variability in S-adenosylmethionine (SAM)-dependent riboswitches: S-box elements in Bacillus subtilis exhibit differential sensitivity to SAM In vivo and in vitro. J. Bacteriol. 190:823–833 [DOI] [PMC free article] [PubMed] [Google Scholar]