

Abstract
Swimming Escherichia coli cells are propelled by the rotary motion of their flagellar filaments. In the normal swimming pattern, filaments positioned randomly over the cell form a bundle at the posterior pole. It has long been assumed that the hook functions as a universal joint, transmitting rotation on the motor axis through up to ∼90° to the filament in the bundle. Structural models of the hook have revealed how its flexibility is expected to arise from dynamic changes in the distance between monomers in the helical lattice. In particular, each of the 11 protofilaments that comprise the hook is predicted to cycle between short and long forms, corresponding to the inside and outside of the curved hook, once each revolution of the motor when the hook is acting as a universal joint. To test this, we genetically modified the hook so that it could be stiffened by binding streptavidin to biotinylated monomers, impeding their motion relative to each other. We found that impeding the action of the universal joint resulted in atypical swimming behavior as a consequence of disrupted bundle formation, in agreement with the universal joint model.
INTRODUCTION
An Escherichia coli cell swimming is arguably one of the simplest examples of behavior in a living organism. As a consequence, much is understood about the system: the process of flagellum assembly (8), the motor's molecular architecture (25) and physiology (3, 32), and the physics of swimming (7, 29). The flagellum is a macromolecular complex made up of ∼30 different proteins (3), which are assembled in a particular order (8). It can be divided into three parts: the motor, the hook, and the filament. The motor is embedded in the cell envelope, spanning both the inner and outer membranes. In E. coli, motor rotation is powered by the diffusion of protons from the periplasm, down their electrochemical potential gradient, and into the cytoplasm (32). The motor is coupled via the hook to the filament. The hook consists of a flexible curved helical structure 55 nm long (19), made up of ∼120 copies of the FlgE protein which form 11 helical protofilaments (12, 13, 33). The filament also consists of a single protein (FliC) made into 11 protofilaments, but it is 10 to 15 μm long and rigid for its function as a propeller (3). An E. coli cell typically possesses ∼4 to 10 flagella which coalesce to form a bundle at the posterior pole when swimming. When one or more motors reverse direction in response to environmental cues, they break from the bundle, causing the cell to reorientate (10).
The hook is thought to act as a universal joint, translating rotation from the axis of rotation at the motor, through 90°, to the filament participating in the bundle. This idea was first postulated by Berg and Anderson in 1973 (4). Since then, models describing the universal joint have been developed based on ever-advancing structural information (17, 30, 31). FlgE is composed of four domains (D0, Dc, D1, and D2) positioned radially from the inside to the outside of the hook (17). The D0 domains form the inner core and are tightly packed together, providing the mechanical stability of the hook while allowing extension and compression of their helical array. The Dc and D2 domains form a mesh-like structure which may provide twisting rigidity, while the D1 domain is considered to be packed loose in all directions, allowing the hook to adopt highly curved conformations. Protofilaments on the inside of the curve have shorter repeat distances than those on the outside. As the hook rotates, the inside of the bend is successively occupied by different protofilaments, resulting in a kind of rolling rotation (30). This detailed structural information is supported by molecular dynamic simulations, lending further support to the universal joint hypothesis. However, direct investigation of the dynamics of the hook has been very limited (5, 6, 18, 28). In this study, we genetically modified the hook to contain a solvent-exposed AviTag sequence (GLNDIFEAQKIEWHE) (2), a 15-mer amino acid sequence that can be biotinylated by the E. coli enzyme biotin ligase, BirA (21). This enabled us to (i) reduce the torsional compliance of the hook and (ii) abolish the universal joint action, so that swimming cells failed to form bundles, simply by exposing the biotinylated hooks to streptavidin.
MATERIALS AND METHODS
Strain construction.
Five strains were made which carried a modified version of flgE on the genome. These strains produced FlgE proteins which had an AviTag sequence (GLNDIFEAQKIEWHE) in between the following amino acid positions: site A, T166 and V167; site B, A232 and N233; site C, I221 and A222; site D, K202 and T203; and site E, T227 and L228. DNA encoding the AviTag sequence plus some flgE flanking sequence (309 bp in total) was synthesized by Entelechon GmbH. This DNA was ligated into an AleI/ClaI-cut vector containing the flgE gene. These vectors were then used as a template to clone the modified full-length flgE gene into the suicide vector pDS132 (27). These were sequenced before replacing wild-type (WT) flgE in the E. coli RP437 chromosome by allelic exchange (23). Mutants were screened by PCR and had their DNA sequenced and strain codes applied (see Table S1 in the supplemental material).
Two deletion constructs were also required to produce a nonflagellate strain (ΔfliC) and a polyhook strain (ΔfliK). The constructs were made by amplifying ∼500 bases up- and downstream of the gene to be deleted. They were then incorporated into the chromosome by allelic exchange so that the fliC and fliK genes now encoded small polypeptides (4 and 5 amino acids long, respectively). This strategy ensured inactivation of the target gene without adversely affecting other genes in the operon. For the ΔfliC ΔfliK double mutant, ΔfliK was introduced into a background already containing ΔfliC.
General procedures.
Frozen stocks of strains were made as follows. Cells were grown to stationary phase (Luria broth [LB] medium, with shaking at 37°C overnight) and diluted in glycerol (at a 20% final concentration) before being stored in aliquots at −80°C. Strains were then grown for 5.5 h at 30°C with shaking in tryptone broth to an optical density at 600 nm (OD600) of 0.6. Typically, 1 ml of cell culture was then spun down, although this volume was adjusted to keep the number of cells constant (an OD600 range of 0.5 to 0.7 was acceptable). Centrifugations were all carried out at 3,400 × g for 2 min. Experiments were all carried out in motility buffer (MB; 10 mM potassium phosphate, 0.1 mM EDTA [pH 7]), MB+ (10 mM potassium phosphate, 67 mM NaCl, 0.1 mM EDTA, 0.5% glucose, 0.002% Tween 20 [pH 7]), or MB-NaCl (10 mM potassium phosphate, 67 mM NaCl, 0.1 mM EDTA [pH 7.5]). Media were supplemented with an antibiotic (ampicillin at 100 μg/ml) and isopropyl β-d-1-thiogalactopyranoside (IPTG) when required.
Reagent stocks and storage.
Biotin (Sigma) was made up to 100 mM in dimethyl sulfoxide (DMSO), frozen in aliqouts at −20°C, and then diluted to 1 mM before use. ATP (Sigma) was made fresh at the start of each experiment at 100 mM in Milli-Q water. Purified BirA was stored in 20-μl aliquots at a 50 μM concentration. Streptavidin (Sigma) was made up to 200 μM in phosphate-buffered saline (PBS) and stored in aliquots at −20°C. l-Serine (Sigma) was made up to 1 M in MB immediately before use. Streptavidin-Alexa Fluor 532 (Invitrogen) was made up to 200 μM in PBS and stored in aliquots at −20°C. Cy3 monofunctional succinimidyl ester (Amersham/GE Healthcare; 5-mg pack) was made up to 10 mg/ml in aqueous DMSO and stored in 10-μl aliquots at 4°C.
Biotinylation.
We had two options for biotinylation: (i) endogenous biotinylation, achieved by expression of BirA from pET21a-BirA (see Table S3 in the supplemental material), which biotinylates the FlgE AviTag protein before being exported by the cell and incorporated into the hook, or (ii) exogenous biotinylation, achieved by exposing cells to purified BirA in vitro (see Howarth and Ting for a review of methods [21]). Exogenously biotinylated cells gave better labeling of hooks (as assessed using streptavidin-Alexa Fluor 532); thus, exogenous biotinylation was used throughout, except for the preparation of polyhooks prior to electron microscopy (EM) (see Fig. S2 in the supplemental material), in which cells were grown overnight in the presence of ampicillin, 180 μM IPTG, and 10 μM biotin.
BirA had to be purified before cells could be exogenously biotinylated. We used the procedure outlined in box 1 of reference 21, although we used BL21(DE3) RIPL cells (Stratagene) instead of BL21(DE3) pLysS E. coli cells.
Cells were then exogenously biotinylated as follows. One milliliter of culture grown to an OD600 of 0.6 was spun down, washed twice with 1 ml of MB-MgCl2 (MB supplemented with 5 mM MgCl2), and resuspended in 100 μl of MB-MgCl2. To this we added (i) 1 μl of biotin (10 μM final), (ii) 1 μl of ATP (1 mM final), and (iii) 4 μl of BirA (2 μM final) and then incubated the culture at room temperature on a shaker for 20 min. Cells at this stage, referred to herein as biotinylated cells, were then used for a number of different experiments.
Motility assays. (i) Swim plates.
Agar (0.25%) in LB was inoculated with 5 μl of stationary-phase culture and incubated for 8 h at 30°C.
(ii) Free swimming.
Biotinylated cells (strains RP437, MTB7, and MTB9; see Table S1 in the supplemental material) were washed twice with 1 ml of MB+ (see “General procedures” above) and resuspended in a final volume of 500 μl. These cells were then incubated with streptavidin (100 μM final), or the same volume of 0.5× PBS, and incubated for 20 min at room temperature. l-Serine (10 mM final) was added to the mix to suppress chemotactic tumbling immediately before the cells were injected into a tunnel slide. Swimming cells were observed using a Nikon Optiphot-150 phase-contrast microscope at ×20 magnification and images recorded using a DALSA Genie HM640 black and white camera at 50 Hz. Swimming direction and the longitudinal axis of the cell body were determined over time using automated tracking software (34).
Fluorescent labeling of hooks, imaging, and analysis.
Biotinylated cells (strains RP437, MTB7, MTB8, MTB9, MTB10, MTB11; see Table S1 in the supplemental material) were washed twice with 1 ml of MB and resuspended in 100 μl of streptavidin-Alexa Fluor 532 (1 μM for epifluorescence experiments and variable concentrations for total internal reflection fluorescence [TIRF] experiments). Cells were incubated for 20 min at room temperature, washed five times with 1 ml of MB, and resuspended in 10 μl MB. One microliter of cells was placed on an agarose pad (1% agarose in Milli-Q water, wt/vol) and then imaged via epifluorescence or TIRF microscopy. Epifluorescence microscopy was carried out on a Nikon Eclipse TE2000-E microscope with illumination by a 100-W halogen lamp with yellow fluorescent protein (YFP) filters. TIRF microscopy was carried out on a custom-built inverted microscope with a 532-nm excitation wavelength and imaged with a 128- by 128-pixel, cooled, back-thinned electron-multiplying charge-coupled-device camera (iXon DV860-BI; Andor Technology) as described previously (11, 22). Images were sampled for ∼10 s, resulting in complete photobleaching of streptavidin-Alexa Fluor 532 molecules bound to hooks within the TIRF field, at 20-ms exposures and laser power at 720 μW. New custom-written software was used to determine the stoichiometry per spot by (i) locating spots using a Gaussian fit with fixed width (132 nm) to a composite image comprising the average of all images in the video sequence, (ii) calculating the intensity of the spot in each image by fitting the amplitude of a Gaussian fit with fixed width and position, (iii) calculating the initial intensity of the spot minus background, and (iv) identifying the unit step size by finding the first significant peak in the Fourier transform of the pairwise difference distribution function (PDDF, as in reference 22) of the filtered intensity trace (step-preserving L1PWC filter [24], with gamma set to 500). Note that the dye consists of 3 mol of Alexa Fluor 532 per mole of streptavidin (Invitrogen), which was accounted for in our calculations, and that the Fourier transform of the PDDF was evaluated exactly, without using a binned histogram, as follows:
Stiffness characterization.
Biotinylated cells (strains MTB38, MTB33, MTB34; see Table S1 in the supplemental material) were washed twice with 1 ml of MB and resuspended in 300 μl of MB. Cells were incubated in a tunnel slide turned upside down for 10 min so the cells were tethered to the coverslip. They were washed twice with 50 μl of MB and placed, with the ends of the slide left open, on a Nikon Optiphot-150 phase-contrast microscope at ×20 magnification. A field of view (FOV) with a high number of rotating cells was chosen and a short video recorded (DALSA Genie HM640 camera, 100 Hz) so rotating cells could be distinguished from nonmotile cells for the analysis. Twenty microliters of glutaraldehyde (0.1% in MB) was flushed through the channel, causing the cells to lock up within a few seconds. The FOV was then recorded for 5 min. Then 10 μl of streptavidin (100 μM in PBS) was wicked into the chamber, the slide sealed, and the recording continued for another 20 min. The total time to add streptavidin and continue recording was ∼90 s.
Only cells that were originally rotating before exposure to glutaraldehyde were analyzed. Custom software was used to determine the angle of the longitudinal axis of the cell body and the variance (square of the standard deviation) in angle determined over 1-s windows. The variance in body angle (V) was then converted into tether stiffness (Kθ) using the equipartition theorem (KθV/2 = kT/2) (where k is the Boltzmann constant and T is temperature) (5).
Flagellum and cell body staining.
Cells were biotinylated as normal except that MB-NaCl (rather than MB) was supplemented with MgCl2. Cells and filaments were then stained using the Cy3 protocol as follows (10). Biotinylated cells (strains RP437, MTB7, and MTB9; see Table S2 in the supplemental material) were washed twice with 1 ml of MB-NaCl and resuspended in 100 μl of MB-NaCl. Five microliters of 1 M sodium hydrogen carbonate was added to reach pH ∼8 before adding 10 μl of Cy3 monofunctional succinimidyl ester dye (1-mg/ml final concentration). Cells were then incubated for 90 min in the dark before being washed five times with MB+ and then resuspended in 500 μl of MB+. l-Serine (10 mM final concentration) was added just before the cells were injected into a tunnel slide and imaged using an OMX fluorescence microscope (Applied Precision). Excitation was provided by a 532-nm laser (at 300 mW), and 3-ms exposures were taken at 26 Hz (35 ms between frames) on a Cascade II:512 camera (Photometrics).
RESULTS AND DISCUSSION
Design and functionality of engineered strains.
Normally the AviTag sequence is incorporated at the N or C terminus of the protein of interest, but the AviTag sequence had to be incorporated into the FlgE protein in a solvent-exposed site, which permitted normal assembly of the hook and retained the hook's functionality. The electron cryomicroscopy structure of the Salmonella enterica serovar Typhimurium hook (17), which is 88% homologous to the E. coli FlgE protein, shows that only a central segment from A146 to K284 (D2 domain, E. coli numbers) lies on the outside of the assembled hook structure. Since successful insertion of the AviTag in nonterminal regions has been achieved previously (1, 14, 26), we chose sites in positions that had limited similarity across different species (31). Based on this information, five insertion sites for the AviTag were chosen, three of which were in loops (Fig. 1a).
Fig 1.

Design and functionality of engineered strains. (a) Three-dimensional density maps of the hook, adapted from Structure (17) with permission from Elsevier. Shown is the structure of three FlgE units with the insertion sites for the AviTag marked in red and labeled A through E (Protein Data Bank [PDB] identifier 3A69). Scale bar, 50 Å. (b) Swim plate analysis of strains relative to the WT and a nonmotile control (ΔfliC). (c) Fluorescent images of streptavidin-Alexa Fluor 532 attached to exogenously biotinylated hooks; exposure, 300 ms; scale bar, 3 μm.
After making the strains, we tested whether the hooks (i) were functional and (ii) could be biotinylated and would bind streptavidin. Motility was assessed using the swim-plate assay; cells were inoculated onto semisolid agar and left to migrate. Figure 1b shows that strains with mutations in sites C and E were almost fully motile, the site A mutant was weakly motile (25% motility of the WT), and the remaining strains, with mutations in sites B and D, were nonmotile. Phenotypes were confirmed by observing free-swimming behavior under a light microscope.
The hook's ability to be biotinylated and bind streptavidin was assessed by exposing cells to streptavidin-Alexa Fluor 532 (Fig. 1c). Strains with mutations in sites A and C show distinct spots, in the range of 1 to 10 per cell, in agreement with the number of flagella per cell. Site B and E mutants show no fluorescence, indicating that the AviTag either could not be biotinylated or could be biotinylated but could not bind streptavidin. Finally, site D mutants produced fluorescence spots, but these were not localized to cells, suggesting that the hook was brittle in this mutant and was easily sheared off. We also used this assay to show that levels of exogenous biotinylation were higher than those of endogenous biotinylation (see Materials and Methods for details; see Fig. S1 in the supplemental material). Our aim was to create a hook that allows normal swimming, but with multiple streptavidin binding sites to allow investigation of the effects on swimming of disrupting normal hook dynamics. Based on these results, strains with site A and C mutations were selected for further characterization.
Hook stiffness.
As discussed in the introduction, the E. coli hook is a highly curved structure that undergoes continuous rolling rotation during which the protofilaments dynamically change their conformation as well as their repeat distance. An insertion of a 15-mer peptide might affect this natural flexibility and the torsional compliance of the hook. Binding of streptavidin molecules to the biotinylated hook could have an even greater impact on compliance through steric effects.
To test the effect of the presence of the AviTag and streptavidin binding on the hook's torsional compliance, the genes encoding the site A and site C FlgE proteins were put into a background that expressed FliCsticky (see Tables S1, S2, and S3 in the supplemental material for strain details). This enabled the biotinylated cells to be tethered to the coverslip by their flagella, which causes the cell body to rotate. Glutaraldehyde (0.1%) was used to lock the motors (5, 6), and the angle of the long axis of the cell body was measured over time. After 5 min, 100 μM streptavidin was added to the slide and the same single cells were recorded for a further 20 min. The variance in cell body angle (V) provides a good measure of the torsional compliance of the hook, where more compliant tethers lead to larger variances. Angle was converted to stiffness (Kθ) using the equipartition theorem (KθV/2 = kT/2) (5). Figure 2a shows that the presence of the AviTag in site A and C mutants does not affect the torsional compliance of the hook (1,300 pN·nm/rad2). However, upon addition of streptavidin, the site A and C mutant hooks become significantly stiffer than that of the WT (over a 2-fold increase to ∼3,000 pN·nm/rad2).
Fig 2.
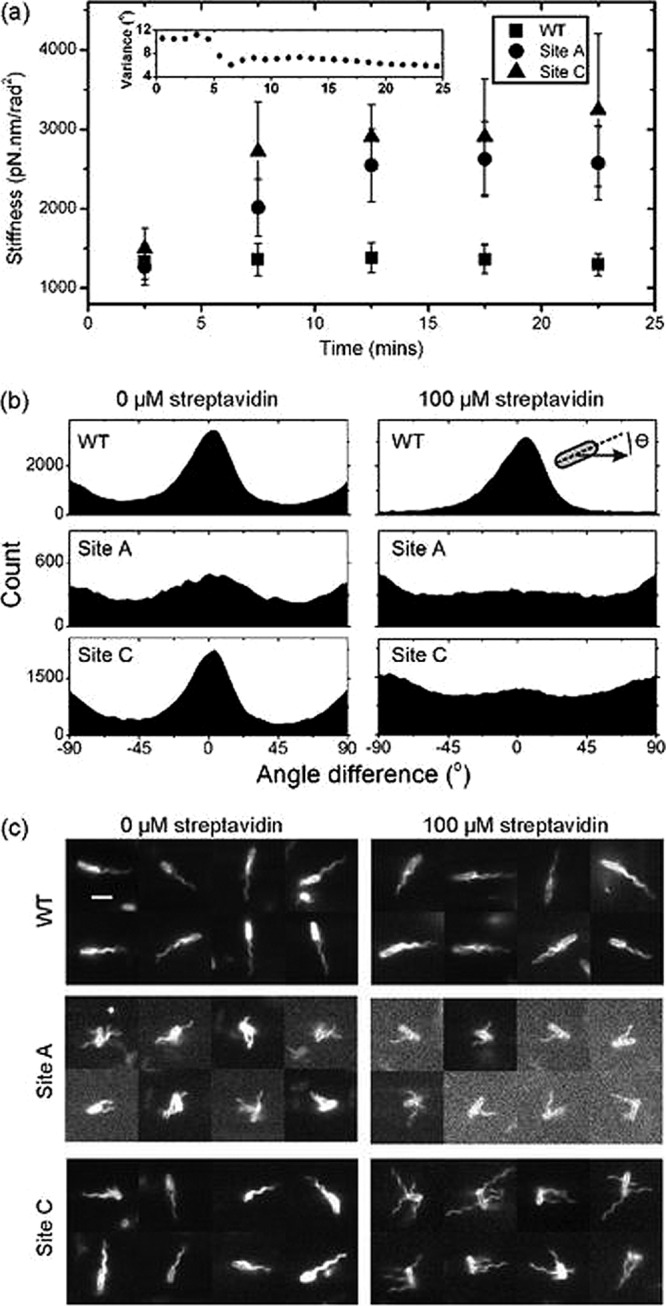
Biophysical characterization of site A and site C AviTag mutants in response to streptavidin. (a) Hook stiffness of single tethered cells over time calculated using the equipartition theorem (see the text for details). Streptavidin (100 μM) was added at t = 5 min; stiffnesses are means ± standard errors of 28, 31, and 27 hooks from WT, site A, and site C strains, respectively. The inset shows the variance in body angle of a single cell from the site A data set versus time. (b) Analysis of free-swimming cells (see Videos S1, S2, and S3 in the supplemental material); histograms of the difference in angle between the cells' swimming trajectory and the angle of the cell body (illustrated by the inset). A cell swimming along its long axis gives an angle of 0°. (c) Selected video frames (3-ms exposure) of swimming cells dyed with Cy3 monofunctional succinimidyl ester showing the position of filaments. Scale bar, 3 μm (see Videos S4, S5, and S6).
These experiments were designed to measure relative changes in hook stiffness. Nonetheless, we note that our WT E. coli measurements are over three times stiffer than a previous E. coli measurement of 400 pN·nm/rad2 (5). One possible explanation is that we tethered cells by hydrophobic sticky filaments, which is expected to give a tighter attachment to the surface than the antibody tethering used in the previous measurement. This might restrict extra degrees of freedom of the tether (18), other than twisting of the hook, that add to the measured compliance with antibody tethering. It might also force the part of the hook nearest to the filament to be parallel to the glass, preventing it from contributing to the measured torsional compliance. The only other published values for comparison come from Rhodobacter sphaeroides, a bacterium propelled by a single filament attached to a much stiffer, straighter hook (3,800 pN·nm/rad2) (28). At ∼3,000 pN·nm/rad2, the streptavidin-bound AviTag E. coli hooks are approaching this stiffness.
To investigate whether the stiffening of the hook by addition of streptavidin may lead to a straight hook phenotype, we made appropriate polyhook strains that that could be endogenously biotinylated (see Tables S1, S2, and S3 for strain details). Polyhooks are made when the fliK gene, which controls hook length, is disrupted, leading to the production of a hook that has the correct helical packing but is much longer than normal (6). Due to large culture sizes, we endogenously biotinylated the cells, purified the polyhooks using the shearing method, and imaged the polyhooks by electron microscopy (EM) (see SI Methods in the supplemental material) (35). Figure S2 shows the average contour lengths and curvatures of site A and site C polyhooks, with and without streptavidin. Site A polyhooks were shorter and straighter than those of the wild type, independent of streptavidin. This is consistent with reduced swimming of this strain (Fig. 1b), indicating disruption and possibly weakening of the hook. Site C polyhooks were shorter and more curved than those of the wild type, but only with streptavidin.
It was also possible that stiffening the hook may have adversely affected the motor's ability to efficiently transmit torque to the load, as predicted by theoretical work (16). To test this, we measured the speeds of 0.75-μm-diameter beads attached to the filament of biotinylated cells with and without streptavidin, but we found only small differences in speed between the samples, suggesting that torque transmission is unaffected (see Fig. S3 in the supplemental material).
Free swimming and bundle formation.
Next we observed the behavior of free-swimming biotinylated cells before and after exposure to streptavidin (see Videos S1, S2, and S3 in the supplemental material). Site A mutant strains could barely swim with or without streptavidin, indicating that the presence of the AviTag alone was sufficient to disrupt the action of the universal joint. Site C mutant cells, however, were indistinguishable from the WT without streptavidin, but in the presence of streptavidin cells, they typically swam end over end rather than pole forward as normal (see Videos S1, S2, and S3). To quantify this observation, we used automated tracking software (34) to obtain histograms of the angle between swimming direction and the long axis of the cell in 20-s-long videos of swimming cells (Fig. 2b). Wild-type cells normally swim along their long axis, giving a peak at 0° in Fig. 2b. This peak is absent for site A cells and site C cells with streptavidin, indicating disruption of the normal swimming pattern. Interestingly, the automated tracking software revealed that despite swimming in an unstreamlined fashion without a bundle, site C cells with streptavidin swam just as fast as WT cells (see Fig. S6 in the supplemental material). This is consistent with a previous finding that one filament propels an E. coli cell as fast as many (10). The small 90° peaks present in the panels representing WT and site C mutants without streptavidin may be an artifact of the tracking software. We also explored more subtle effects on behavior by titrating streptavidin into the sample (see Fig. S4 in the supplemental material). Traptavidin, a streptavidin variant with a 10-fold-slower biotin dissociation (9), was also included for interest. We found that normal swimming was disrupted at concentrations greater than or equal to 5 μM streptavidin.
Next, we investigated why the site C mutant could not swim normally following exposure to streptavidin. We fluorescently labeled the flagella and cell body using Cy3 monofunctional succinimidyl ester, which binds to any free amino group (e.g., on lysine residues) (10), and recorded videos with 3-ms exposure times, enabling us to see individual rotating filaments (see Videos S4, S5, and S6 in the supplementary material). Figure 2c shows that site C mutant filaments no longer form bundles when exposed to streptavidin; instead, they tend to stick straight out from the cells. Meanwhile, site A mutant cells cannot form bundles even in the absence of streptavidin.
Mechanism by which streptavidin affects the site C mutant hook.
The simplest explanation for increasing hook stiffness and abolition of universal joint action in the presence of streptavidin is that streptavidin binds to the D2 domains at sufficiently high densities that steric interference between bound streptavidin molecules inhibits the interdomain rearrangements that normally occur in WT hooks. Alternatively, a single streptavidin molecule (containing four biotin binding sites) might be able to bind to two adjacent monomers simultaneously. The minimum distance between two monomers on the same protofilament, on the inside of the curve, is predicted to be 35 Å (30), while the maximum distance between two sites on a streptavidin molecule is 34 Å (15). Thus, it is just possible that streptavidin might cross-link such monomers and hinder the protofilament length changes that are necessary for the universal joint action. To test this possibility, we conducted a free-swimming experiment with monovalent streptavidin (a streptavidin tetramer with only one functional biotin binding subunit) (20). We found that monovalent streptavidin gives the same phenotype as the normal streptavidin with four binding sites (see Fig. S5 in the supplemental material), showing that binding between monomers is not the dominant mechanism which abolishes universal joint action.
Finally, we estimated the number of streptavidin molecules bound to the hook. To do this, we prepared the cells using the streptavidin-Alexa Fluor 532 procedure as described for Fig. 1c, but this time we imaged cells using a custom-made total internal reflection fluorescence (TIRF) microscope fitted with a high-sensitivity camera. Fluorescent spots were photobleached to obtain stepwise photobleaching fluorescence intensity traces. For each spot, the unitary step size corresponding to the bleaching of a single fluorophore was identified, and the number of dye molecules per hook was estimated by dividing the initial intensity (minus the background) by the unitary step size (11, 22). This number was then divided by 3, the average number of dye molecules per streptavidin molecule (manufacturer's data sheet). Our best estimate of the streptavidin stoichiometry (Fig. 3a, inset) is ∼36 streptavidins per hook.
Fig 3.

Quantification of the number of streptavidin molecules on the hook. (a) Stoichiometry of streptavidin-Alexa Fluor 532-labeled hooks via single molecule stepwise photobleaching. The top panel shows TIRF (false-color) images taken every 100 ms; the first image has the corresponding bright-field image superimposed. Scale bar, 1 μm. At the bottom is a histogram of data collected at saturating levels of streptavidin-Alexa Fluor 532 (≥10 μM; number of cells = 50 and number of spots = 114). Inset, stoichiometry at different concentrations of streptavidin-Alexa Fluor 532 fit with a Michaelis-Menten curve (Km = 2 μM; Vmax = 36). Measurements were made at concentrations of 0.1 μM (28, 50), 1 μM (19, 45), 5 μM (5, 5), 10 μM (15, 36), 50 μM (4, 17), 75 μM (13, 26), and 100 μM (18, 35); numbers in parentheses are numbers of cells and spots. (b) Schematic of streptavidin bound to the hook, based on the model of Samatey and coworkers (adapted by permission from Macmillan Publishers Ltd.: Nature [30]) (PDB, 1WLG). The α-carbon atoms of the curved hook are displayed as a solvent-accessible surface, with the D1 domains shown in white and the D2 domains shown in blue. Streptavidin molecules (PDB, 1SWE [15]) are shown as gray surfaces. The top panel shows how streptavidin binding along the 11-start helix may lead to steric clashes (red). The bottom panel shows an alternative packing strategy that avoids steric clashes, where the streptavidins are positioned in alternating arrangements along the 11-start helices. Images were generated using PyMOL.
Figure 3b shows a schematic of the hook and associated streptavidin molecules. Each streptavidin was positioned by aligning the protein such that the opening of one of the biotin binding cavities faced directly toward the insertion site at the surface of the D2 domain, with the carbon atom of the carboxylic acid of the biotin at a distance approximately 16 Å away from the insertion site in each case. In the top panel, the red surfaces show all atoms from one streptavidin that are within 5 Å of any atom of its neighbor, i.e., indicating a clash. The compression of the interdomain distances on the inner side of the curved hook results in considerable overlap of adjacent streptavidins and leads to severe steric clashes when attempting to pack streptavidins in this fashion. Even the outer curve cannot be fully occupied by streptavidins without an increase in curvature. The observed increased curvature of site C polyhooks with streptavidin (see Fig. S2) is consistent with streptavidin binding preferentially to the outer curve, which would not only stiffen the hooks but also give them a “permanent” preferred direction of curvature. When streptavidins are docked in an arrangement where overlaps are not tolerated (bottom panel), we predict an absolute maximum occupancy of 55 streptavidins for the complete hook. This is in broad agreement with ∼36 streptavidins per hook obtained by single-molecule microscopy. Bringing the two numbers together, we estimate that approximately a third of the 120 AviTag sites present per hook will be occupied, at saturating levels of streptavidin.
Of the five AviTag FlgE mutants made, two were subjected to biophysical tests. The incorporation of the AviTag at one site impeded the action of the universal joint, without affecting its torsional compliance, resulting in very poor motility. The other strain was unaffected by the presence of the AviTag. In this strain we were able to increase the torsional stiffness of the hook and inhibit the action of the universal joint by adding streptavidin, which disrupted bundle formation and the normal swimming pattern. Cells swam at similar speeds with and without bundles, indicating that the function of the bundle is not to increase swimming speed. Rather, we speculate that bundles may be necessary to allow cell reorientation when any of the motors in the cell switch direction (10). In addition, there may be some selective advantage associated with the normal pattern in which cells swim along their long axis. Thus, we provide experimental confirmation of the hypothesis made over 35 years ago (4), that the universal joint model accurately describes the action of the hook.
Supplementary Material
ACKNOWLEDGMENTS
We thank the following people: Keichi Namba for use of his PyMOL model of the curved hook and advice regarding AviTag site selection, Christian Bell for help with initial AviTag site selection, Ian Dobbie for setting up the OMX microscope, and Mark Howarth and Claire Chivers for advice and material regarding BirA and streptavidin protocols.
This work was supported by the Biotechnology and Biological Sciences Research Council, grant code BB/F021224/1.
Footnotes
Published ahead of print 20 April 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Bates IR, et al. 2006. Membrane lateral diffusion and capture of CFTR within transient confinement zones. Biophys. J. 91:1046–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beckett D, Kovaleva E, Schatz PJ. 1999. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 8:921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berg HC. 2003. The rotary motor of bacterial flagella. Annu. Rev. Biochem. 72:19–54 [DOI] [PubMed] [Google Scholar]
- 4. Berg HC, Anderson RA. 1973. Bacteria swim by rotating their flagellar filaments. Nature 245:380–382 [DOI] [PubMed] [Google Scholar]
- 5. Block SM, Blair DF, Berg HC. 1989. Compliance of bacterial flagella measured with optical tweezers. Nature 338:514–518 [DOI] [PubMed] [Google Scholar]
- 6. Block SM, Blair DF, Berg HC. 1991. Compliance of bacterial polyhooks measured with optical tweezers. Cytometry 12:492–496 [DOI] [PubMed] [Google Scholar]
- 7. Chattopadhyay S, Moldovan R, Yeung C, Wu X. 2006. Swimming efficiency of bacterium Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 103:13712–13717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chevance FFV, Hughes KT. 2008. Coordinating assembly of a bacterial macromolecular machine. Nat. Rev. Microbiol. 6:455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chivers CE, et al. 2010. A streptavidin variant with slower biotin dissociation and increased mechanostability. Nat. Methods 7:391–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Darnton NC, Turner L, Rojevsky S, Berg HC. 2007. On torque and tumbling in swimming Escherichia coli. J. Bacteriol. 189:1756–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Delalez NJ, et al. 2010. Signal-dependent turnover of the bacterial flagellar switch protein FliM. Proc. Natl. Acad. Sci. U. S. A. 107:11347–11351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DePamphilis M, Adler J. 1971. Fine structure and isolation of the hook-basal body complex of flagella from Escherichia coli and Bacillus subtilis. J. Bacteriol. 105:384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DePamphilis M, Adler J. 1971. Purification of intact flagella from Escherichia coli and Bacillus subtilis. J. Bacteriol. 105:376–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Digby GJ, Sethi PR, Lambert NA. 2008. Differential dissociation of G protein heterotrimers. J. Physiol. 586:3325–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freitag S, Le Trong I, Klumb L, Stayton PS, Stenkamp RE. 1997. Structural studies of the streptavidin binding loop. Protein Sci. 6:1157–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fricks J, Wang H, Elston TC. 2006. A numerical algorithm for investigating the role of the motor-cargo linkage in molecular motor-driven transport. J. Theor. Biol. 239:33–48 [DOI] [PubMed] [Google Scholar]
- 17. Fujii T, Kato T, Namba K. 2009. Specific arrangement of a-helical coiled coils in the core domain of the bacterial flagellar hook for the universal joint function. Structure 17:1485–1493 [DOI] [PubMed] [Google Scholar]
- 18. Hashimoto M, Mashimo T, Hirano T, Yamaguchi S, Aizawa SI. 2008. Functional roles of the hook in a rotating tethered cell. J. Mol. Biol. 375:367–375 [DOI] [PubMed] [Google Scholar]
- 19. Hirano T, Yamaguchi S, Oosawa K, Aizawa S. 1994. Roles of FliK and FlhB in determination of flagellar hook length in Salmonella typhimurium. J. Bacteriol. 176:5439–5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Howarth M, et al. 2006. A monovalent streptavidin with a single femtomolar biotin binding site. Nat. Methods 3:267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Howarth M, Ting AY. 2008. Imaging proteins in live mammalian cells with biotin ligase and monovalent streptavidin. Nat. Protoc. 3:534–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leake MC, et al. 2006. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature 443:355–358 [DOI] [PubMed] [Google Scholar]
- 23. Link AJ, Phillips D, Church GM. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 179:6228–6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Little MA, et al. 2011. Steps and bumps: precision extraction of discrete states of molecular machines. Biophys. J. 101:477–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Minamino T, Imada K, Namba K. 2008. Molecular motors of the bacterial flagella. Curr. Opin. Struct. Biol. 18:693–701 [DOI] [PubMed] [Google Scholar]
- 26. Oddershede L, Dreyer JK, Grego S, Brown S, Berg-Sørensen K. 2002. The motion of a single molecule, the l-receptor, in the bacterial outer membrane. Biophys. J. 83:3152–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Philippe N, Alcaraz JP, Coursange E, Geiselmann J, Schneider D. 2004. Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51:246–255 [DOI] [PubMed] [Google Scholar]
- 28. Pilizota T, et al. 2009. A molecular brake, not a clutch, stops the Rhodobacter sphaeroides flagellar motor. Proc. Natl. Acad. Sci. U. S. A. 106:11582–11587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Purcell EM. 1977. Life at low Reynolds number. Am. J. Phys. 45:3–11 [Google Scholar]
- 30. Samatey FA, et al. 2004. Structure of the bacterial flagellar hook and implication for the molecular universal joint mechanism. Nature 431:1062–1068 [DOI] [PubMed] [Google Scholar]
- 31. Shaikh TR, et al. 2005. A partial atomic structure for the flagellar hook of Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 102:1023–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sowa Y, Berry RM. 2008. Bacterial flagellar motor. Q. Rev. Biophys. 41:103–132 [DOI] [PubMed] [Google Scholar]
- 33. Wagenknecht T, DeRosier DJ. 1982. Flagellar hook structures of Caulobacter and Salmonella and their relationship to filament structure. J. Mol. Biol. 162:69–87 [DOI] [PubMed] [Google Scholar]
- 34. Wood TM, Yates CA, Wilkinson DA, Rosser G. 2012. Visual tracking of bacteria using the Gaussian Mixture Probability Hypothesis Density Filter. IEEE Trans. Circuits Syst. Video Technol. 22:702–713 [Google Scholar]
- 35. Woods RD, et al. 2007. Bifunctional nanotube scaffolds for diverse ligands are purified simply from Escherichia coli strains coexpressing two functionalized flagellar genes. Nano Lett. 7:1809–1816 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.