

Abstract
The high-throughput nature of proteomics mass spectrometry is enabled by a productive combination of data acquisition protocols and the computational tools used to interpret the resulting spectra. One of the key components in mainstream protocols is the generation of tandem mass (MS/MS) spectra by peptide fragmentation using collision induced dissociation, the approach currently used in the large majority of proteomics experiments to routinely identify hundreds to thousands of proteins from single mass spectrometry runs. Complementary to these, alternative peptide fragmentation methods such as electron capture/transfer dissociation and higher-energy collision dissociation have consistently achieved significant improvements in the identification of certain classes of peptides, proteins, and post-translational modifications. Recognizing these advantages, mass spectrometry instruments now conveniently support fine-tuned methods that automatically alternate between peptide fragmentation modes for either different types of peptides or for acquisition of multiple MS/MS spectra from each peptide. But although these developments have the potential to substantially improve peptide identification, their routine application requires corresponding adjustments to the software tools and procedures used for automated downstream processing. This review discusses the computational implications of alternative and alternate modes of MS/MS peptide fragmentation and addresses some practical aspects of using such protocols for identification of peptides and post-translational modifications.
Technological and computational developments continue to expand the fundamental role of tandem mass spectrometry1 (MS2) in high throughput proteomics (1). Current protocols regularly identify thousands of proteins and post-translational modifications (PTMs (2)) per experiment and can deliver very high levels of reproducibility (3) (e.g. using targeted approaches). In the large majority of cases, these advances have been enabled by a combination of standard protocols based on trypsin digestion followed by protein identification from collision-induced dissociation (CID) MS2 spectra using database search tools (4). The efficiency and reliability of trypsin digestion remains one of the major reasons for the success of high-throughput proteomics. Protein digestion results in multiple peptides per protein and, in the limit, only one peptide needs to be significantly identified to be able to identify the corresponding protein (5, 6). Trypsin further contributes to spectrum identifiability in CID MS2 by cleaving C-term of K/R (Lysine/Arginine) and thus yielding rich and characteristic MS2 peptide fragmentation patterns (7, 8) for a substantial subset of all peptides. Incorporating knowledge of these peptide fragmentation patterns into algorithms for MS2 identification has led to a variety of database search software tools (9, 10) for peptide identification by scoring matches between acquired MS2 spectra and predicted spectra for peptide sequences extracted from a protein sequence database.
Despite the success of the popular trypsin/CID tandem mass spectrometry protocols, it is well known that this approach generally biases experiments toward the identification of certain types of peptides, such as doubly and triply charged peptide precursors from medium-length peptides (11–15). Historically this has been advantageous because CID generates identifiable spectra from these types of peptides (low charge, high m/z) with the greatest efficiency. But a disadvantage of relying upon a single specific protease like trypsin is that many digested peptides are too short and can thus lead to incomplete coverage of the proteome. In the case of trypsin, the enzyme cleaves K/R-rich regions into peptides that are too short for reliable identification (about half of tryptic yeast peptides are ∼6 residues long (15)). Alternative protease(s) can be used to obtain a wider distribution of peptides and increase coverage. For example, some have demonstrated the value of using of nonspecific proteases (16–19), but these can also decrease experimental reproducibility. Another limiting factor is that nonspecific proteases can greatly increase the computational search space when matching MS2 spectra to all possible peptides in the database. This not only increases the time required to search the database, but it can also decrease the sensitivity of spectrum identification at a given false discovery rate (FDR (6)). Others have had success using multiple specific proteases (e.g. LysC, AspN, GluC, and ArgC) in a more targeted approach (15, 20, 21). Compared with the use of a single specific protease, using multiple specific proteases allows for a greater percentage of the proteome (94% for yeast) to be covered by at least one peptide suitable for mass spectrometry sequencing technology (15). If a separate MS2 run is executed for each enzymatic digestion, there is little increase in computational complexity because MS2 spectra from each run can be separately matched to a set of peptides cleaved by a single specific protease.
A disadvantage of using multiple proteases is that they often yield peptides that are longer and contain one or more internal basic residues, which are poorly fragmented by CID (22). But alternative fragmentation strategies such as higher-energy collision dissociation (HCD (23), also formerly known as higher-energy C-trap dissociation) and electron transfer dissociation (ETD (24)) are known to improve identification of long, highly charged peptides, peptides containing basic residues, and peptides containing many or highly labile PTMs (25–28). A popular strategy has been to use Lys-C in combination with ETD because compared with trypsin, Lys-C generates a larger portion of peptides amenable to efficient ETD fragmentation (15). The particular combination of ETD with Lys-N digestion yields peptide coverage complementary to trypsin and very simple fragmentation patterns that can aid in manual de novo peptide sequencing (29). Although the reduction of C-terminal ions in Lys-N ETD peptides reduces the difficulty of manual sequencing, it can also hinder automated approaches that use symmetry between N- and C-terminal ions. The complementarity of CID, HCD, and ETD dissociation strategies has been assessed in a variety of contexts (30–33) and the recognition that each improves identifications of different types of peptides underlies the decision tree (15, 33–35) approach for real-time selection of fragmentation mode(s) based on each precursor's m/z and charge. Alternatively, CID/ETD (36) or CID/ETD/HCD (31) alternating MS2 acquisition for every precursor have also been shown to substantially improve peptide identification and enable otherwise difficult analysis of PTMs (37). Underlying the utility of all tandem mass spectrometry approaches to high-throughput proteomics is the need to calculate false discovery rates, usually estimated using the Target/Decoy approach (TDA (38, 39)). This approach is a key control of statistical significance in high-throughput proteomics but, as discussed below, requires careful adjustments before it can be meaningfully applied to the analysis of MS2 data acquired with alternate peptide fragmentation modes.
Peptide Fragmentation Modes
Although there are several comparisons of peptide fragmentation modes in terms of the resulting numbers of spectrum and peptide identifications, only some (14, 36, 40–42) attempt to characterize the observed differences in terms of their underlying MS2 fragmentation statistics. These matter because the ability of a database search tool to identify MS2 spectra is proportional to how well it models MS2 fragmentation statistics and how it uses them to score Peptide Spectrum Matches (PSMs). The basic types of information captured in these peptide fragmentation models are illustrated here using spectra from a recent comparison (33) of how CID, HCD, and ETD MS2 acquisition modes affect Mascot (10) identifications. In brief, a HeLa tryptic digest was analyzed in three separate Thermo LTQ Orbitrap runs, one for each of CID, HCD, and ETD MS2 acquisition; survey scans were acquired in the Orbitrap with resolution 30,000 and MS2 spectra were acquired in the Orbitrap at resolution 7500. Peptide identifications were obtained here using MS-GFDB as previously described (36) and resulted in 17,378 CID PSMs (out of 33,586 spectra), 21,246 HCD PSMs (out of 37,810 spectra), and 12,834 ETD PSMs (out of 25,734 spectra). As shown in Table I, all fragmentation modes achieved comparable spectrum identification rates of ∼50% but also exhibit the expected correlations with precursor charge where ETD tends to perform better than CID or HCD on higher charge states. These show that ETD tends to achieve the highest identification rates for precursors of charge 3 or higher but yet also yields the lowest number of total identifications because of its slower scan rate (only ∼two-thirds as many MS2 spectra as in HCD acquisition). Longer acquisition time remains a disadvantage of ETD, however recent work suggests that optimizing ETD acquisition parameters can significantly reduce the scan rate without loss in coverage (43). The complementarity of the fragmentation modes is further illustrated in Fig. 1 and supported by the observation that most peptides are not identified by at least one acquisition mode (see supplementary Table S2). This is partly because of the different scan rates and the stochastic nature of data-dependent MS2 precursor selection but, as clearly illustrated for precursor charge 2 (for CID/HCD) and precursor charge 5 (for ETD), this also shows that different peptide fragmentation modes work best for different types of peptides. Of peptides that can be identified by all three acquisition modes, the breaks (observed cleavages along the peptide backbone, supported by either N- or C-terminal fragments) captured by ETD tend to complement those captured by CID and HCD, especially for precursors of charge 3 or higher. This can be seen in statistics from Fig. 2, which show how the union of observed peptide breaks increases by 24–72% from CID/HCD to CID/HCD/ETD for precursors of charge 3 or higher. Supplementary Table S1 details how many breaks are unique to every possible combination of fragmentation modes: ETD alone accounts for 19% of all possible peptide breaks in CID/HCD/ETD triplets, the intersection of breaks seen in CID and HCD accounts for 17%, and the intersection of breaks seen in CID, HCD, and ETD accounts for 30%.
Table I. Identified Peptide-Spectrum Matches (PSMs) for alternative fragmentation modes.
Each column shows the number of identified PSMs and the corresponding fraction of identified spectra per precursor charge state. As expected, the identification rate for ETD spectra is higher for precursors with charge states 3 or higher, in contrast with CID/HCD, which tend to perform best for doubly charged precursors. Nevertheless, the currently slower ETD scan rate (an issue that may be resolved soon (43)) still leads to lower numbers of identified spectra with precursor charges 3 and 4 even though its identification rate is consistently higher than those of CID and HCD spectra; this tradeoff seems to disappear at precursor charge states 5 and higher, where ETD consistently identified more spectra and a higher percentage of precursors than either CID or ETD.
| CID | HCD | ETD | ||||
|---|---|---|---|---|---|---|
| All Charges | 17378 | 51.7% | 21246 | 56.2% | 12834 | 49.9% |
| Charge 2 | 10031 | 56.0% | 1880 | 59.4% | 6058 | 44.1% |
| Charge 3 | 5719 | 49.2% | 7485 | 56.7% | 5076 | 57.0% |
| Charge 4 | 1408 | 43.6% | 1644 | 44.9% | 1391 | 56.8% |
| Charge 5 | 206 | 29.3% | 213 | 26.6% | 262 | 47.5% |
| Charge 6 | 13 | 12.9% | 23 | 20.2% | 42 | 51.9% |
Fig. 1.
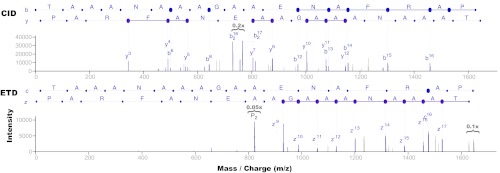
Complementary fragmentation in CID and ETD for peptide TAAANAAAGAAENAFRAP. CID (top) and ETD (bottom) spectra were separately identified against this C-terminal tryptic peptide at 1% FDR. Enough ions were separately detected in each spectrum to identify the peptide (65% of breaks in CID, 53% in ETD). But combining the two yields full coverage of all possible breaks, thus giving higher confidence to breaks observed in both spectra and possibly enabling full-length de novo sequencing. See Fig. 2 and supplementary Table S1 for evidence of CID/ETD complementarity over all identified spectra.
Fig. 2.
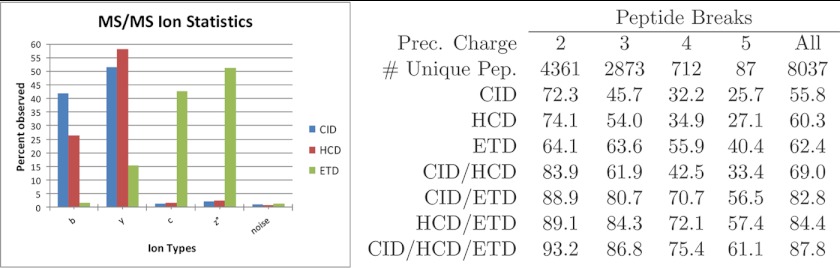
Ion statistics for alternative peptide fragmentation modes. (Left) Peptide MS2 ion statistics for alternative fragmentation modes - This shows the percentage of breaks observed by each ion type over all identified MS2 spectra with precursor charge 2 or 3 for each fragmentation method. z° corresponds to peaks at offset +H from z ions (94). Ions were counted if observed peak masses were within 20 ppm of expected ion masses. The “noise” ion corresponded to offset b+0.5, which was counted to show the level of noise in each type of MS2 spectra. (Right) Peptide break statistics for combinations of alternative fragmentation modes—peptide breaks were counted for all unique peptides identified by all three fragmentation modes. The six columns show the percentage of breaks detected by each fragmentation mode and combination of fragmentation modes per precursor charge state. In CID and HCD spectra, the presence of breaks was indicated by the presence of b or y ions. For ETD, c, z°, or z°+H ions indicated the presence of a break. Multiply charged ions (up to the spectrum's precursor charge) were also considered in each spectrum. Prior to this analysis, peak filtering was applied all CID, HCD, and ETD spectra such that each peak was retained only if its intensity was ranked fifth or higher over all neighboring peaks in a ±56 Da radius. If a peptide was identified by more than one CID, HCD, or ETD spectrum, a single representative spectrum was randomly chosen for each fragmentation mode.
The connection between the identifications in Table I and the underlying MS2 peptide fragmentation statistics in Fig. 2 is easily established for MS-GFDB PSM scoring models. As previously described (44, 36), MS-GFDB uses the exact same computational model to learn MS2 fragmentation statistics for CID, HCD, and ETD and weigh different MS2 fragment ion types based on their observed propensity in spectra from each fragmentation mode. The predominant CID, HCD, and ETD ion types and their relative propensities are shown in Fig. 2. As expected, the most prominent differences in peptide fragmentation pertain to the contrast between the dominant b/y-ions in CID/HCD spectra and c/z-ions in ETD spectra. These ions are the most important in peptide identification because of their direct indication of peptide breaks; Fig. 2 compares the fractions of observed breaks on peptides identified on all three fragmentation modes. Not surprisingly, the per-precursor-charge fractions of observed breaks on CID, HCD, and ETD show a very high correlation with the relative identification rates in Table I as higher fractions of observed breaks almost always result in higher rates of identified spectra.
The complementarity of different peptide fragmentation modes in yielding identifications for different classes of peptides was the underlying principle behind the Decision Tree (DT) acquisition mode (34, 33) where MS2 acquisition modes are selected in real time based on precursor m/z and charge. Decision tree parameters are usually set to maximize the resulting number of peptide identifications and thus implicitly encode instrument-specific tradeoffs between scan rates and the expected rate of success in post-acquisition spectrum identification. Because the latter is highly dependent on the type of mass spectrometry experiment (e.g. shotgun proteomics (34) versus phosphoproteomics (30, 45)) and on the success rates of the software tools chosen for spectrum identification, it is important to note that optimal decision tree parameters may vary between instrument models and methods and depend on the choice of software tools used for peptide identification.
Peptide Identification
The complementarity of multiple peptide fragmentation modes can also be used to improve peptide identification by combining multiple spectra for the same peptides. One of the earliest such approaches was the utilization of MS/MS/MS (MS3) acquisition for de novo peptide sequencing (46), later automated with heuristic (47) and optimal (48) algorithms. Similar applications to de novo peptide sequencing were also introduced at about the same time (49) for paired CID and Electron Capture Dissociation (ECD) spectrum acquisition from the same precursors, also later automated for CID/ECD pairs (50) and CID/ETD pairs (51, 52). In all cases, automated de novo sequencing was improved by combining ions in the MS2/MS3 or in the paired CID/ExD spectra using either combined peak intensities (47, 49, 50, 52) or statistical scoring models (48, 51). Current de novo applications of CID/HCD/ETD and MS3 remain heavily dependent on manual interpretation with some assistance from automated methods (53).
Although database search tools developed for CID can be used for HCD and easily adapted to process ETD (by simply examining c/z° ion offsets instead of b/y), they would likely perform much worse than tools that are sensitive to unique features of HCD and ETD. Features of HCD that are not captured by CID scoring models include peaks in the low m/z range (including immonium ions), high fragment mass resolution (most CID spectra have low fragment mass resolution) and the presence of internal ions. Aside from c/z° ions, ETD spectra typically contain charge-reduced precursor peaks with high intensity, characteristic losses from charge-reduced precursors, and additional related ions at offsets ±H from c and z° ions. Thus, CID scoring models should be redesigned for HCD and ETD in order to be most effective. The difficulty of the adaptation depends on the algorithm being considered. For example, MS-GFDB (36) can be automatically retrained for new types of spectra with only 1000 PSMs from unique peptides (per precursor charge state). Most CID database search tools (4, 9, 54–59) have been extended to support identification of ETD MS2 spectra: OMSSA (60), ProteinProspector (61), MS-GFDB (36), PeaksDB (62), Trans-Proteomic Pipeline (63). However, database search using alternate fragmentation modes remains a less explored approach because of two key issues: (1) how to best combine multiple spectra from the same precursor and (2) how to calculate experiment-wide false discovery rates when CID, HCD, and ETD PSM scores may not be directly comparable.
Estimation of False Discovery Rates
As suggested by the MS2 fragmentation statistics in Fig. 2, database search tools trained to identify CID spectra will not perform well if given ETD or CID/ETD merged peak lists (45). A more careful combination of CID/ECD MS2 spectra for de novo sequencing was first described over a decade ago (49, 50) and later approaches (51, 36) have extended their statistical scoring models to incorporate CID- and ETD-specific ions into merged spectra that are then searched appropriately. In these cases PSM scores are directly comparable and standard FDR calculations (38) suffice to reveal substantial gains in peptide identification. An alternative approach to the creation of merged spectra is to separately search the multiple spectra from the same precursors and later merge the search results. This approach simplifies the reutilization of existing database search tools for additional alternate fragmentation modes but complicates the FDR calculations because, for example, a score threshold of 40 may yield a 1% FDR for CID matches but a 4% FDR for ETD matches. One possible way to address such discrepancies is to derive statistical e-value models for each type of search (64, 65) and use the resulting normalized values to combine search results, similarly to approaches devised to combine search results from multiple search tools (66). To avoid the need for score normalization, intersection-based approaches (40) address this issue by requiring matching identifications from both CID and ETD spectra to accept an identification for each precursor, but unfortunately this is known (36) to lower the number of resulting identifications by requiring significant matches from both CID and ETD spectra. Union-based approaches are often also mentioned where one imposes a 1% FDR on separate CID and ETD searches and reports the union of results; unfortunately this approach can result in a combined FDR higher than 1% because correct identifications will match the same peptide but incorrect identifications mostly accumulate. In practice, a simple strategy for 1% FDR estimation with k alternate acquisition modes would be to impose an FDR threshold of 1/k on each separate search, thus leading to merged results with accumulated false positives at ≤1% FDR. However, this is known to be a conservative strategy that is likely to be less sensitive than existing methods for combining search results from different search tools (59, 66–68), which can also be adapted for the identification of spectra from experiments using alternate fragmentation modes, including Decision Tree-based MS2 acquisition protocols (34). Another possible approach to this issue is to combine the search scores prior to FDR calculations. This approach was used in the first extension to multispectrum database search by deriving combined Mascot/probability scores for MS2/MS3 spectrum pairs (69). These approaches (70, 36) avoid the intersection/union difficulties by using combined scores to facilitate FDR calculations and allow identifications where one fragmentation mode results in a good spectrum even if the other mode results in a poor spectrum. It is expected that combining search engines for CID, HCD, and/or ETD also improves results, but it is important to combine these approaches with appropriate estimation of FDR (such as iProphet (68)).
Post-translational Modifications
Identification of post-translational modifications stands to benefit substantially from MS2 analysis with alternate fragmentation modes, especially those involving electron-based dissociation modes (71, 72). One of the earlier such approaches (73) is the still a popular phosphoproteomics protocol (74) in which MS3 acquisition is triggered by the dominant loss of phosphate from precursor ions observed in MS2 spectra. Alternating MS2 modes further improve identification of phosphorylated peptides (35, 45, 74) and, in addition, enable otherwise challenging experiments such as co-identification of glycans and peptides from glycosylated peptides using ETD (75) or alternating CID/ETD acquisition (37, 76). The complementarity of CID and ETD fragmentation modes is especially useful for PTMs such as glycosylation because CID leads to preferential fragmentation of the more labile glycosidic bonds and generally poor peptide fragmentation, thus facilitating glycan identification but complicating peptide identification. Conversely, ETD fragmentation of glycosylated peptides tends to result in series of c- and z-ions much like with unmodified peptides and thus facilitates peptide identification and localization of the site of glycosylation.
Accurate localization of PTM sites is also an important area that stands to gain from alternate (30, 32, 74) and alternative (77) peptide fragmentation modes. As recently reviewed by Chalkley and Clauser (78), the problem of PTM site localization was first addressed with the AScore approach (79), which assigns a probabilistic score to a site assignment based on the number of observed ions distinguishing the top-scoring site assignment from the runner-up site assignment. More recent approaches have slightly adapted this concept to assign site assignment scores based on the difference of database search scores between the top and runner-up peptide-spectrum matches to each spectrum from a modified peptide. In all cases, the key factor determining the significance of site assignments is the presence or absence of MS2 ions in between the possible sites of post-translational modification. Because it is clear from Fig. 2 that alternate fragmentation modes tend to increase observation of peptide breaks, it is expected that the use of such modes will result in different numbers and quality of site assignments (30, 80), even though strategies for estimation of false-positive site assignments (False Localization Rates (78)) are still in their infancy. In addition to improving PTM site localization by increasing the numbers of observed b/y-ions, one especially interesting feature of HCD MS2 acquisition is its generation of x-ions that are very specific indicators of phosphorylation sites (81). These are hypothesized to derive from the phosphoric acid being a much better leaving group than water on serines and threonines and hence was observed to be a precise indicator of phosphorylation sites, though it was found in only 33% of all phoshphorylated peptides.
Conclusions and Outlook
The substantial advantages of peptide identification with alternate fragmentation modes have been clearly demonstrated in a variety of contexts (31, 36, 37, 49, 51, 82–84) but their widespread adoption remains limited by two significant hurdles: (1) the scan rate tradeoff between increasing the chances of identifying each peptide versus just acquiring spectra for more distinct peptides and (2) the evolving but limited support of peptide identification tools for taking advantage of alternate fragmentation modes.
The scan rate tradeoff is a challenge that will continue to be addressed with technological developments such as those that brought us the current generation of mass spectrometry instruments, some of which are already able to generate tens of thousands of MS2 spectra per hour (85). Nevertheless, alternate fragmentation has already been shown to be useful in key areas or potential therapeutic relevance (86–89) such as peptidomics (31) and post-translational modifications analysis such as glycosylation (37), phosphorylation identification, and site localization (30, 84) and histone modifications (83).
The challenge of limited software support is well on its way to resolving itself as more and more peptide identification tools (36, 41, 51, 61–63) add support for alternative MS2 fragmentation modes. Still, too few tools support integrated analysis of multiple fragmentation modes (36). Although the concept of combining fragmentation modes is over a decade old (49), most existing scoring functions for peptide identification process CID, HCD, and/or ETD spectra individually. But possibly the biggest hurdle in the development of new software tools is the recurring limited public availability of mass spectrometry data. For example, only five out of over 120 published papers on CID/ETD have deposited their raw data (34, 36, 45, 90, 91) on Tranche/ProteomeCommons (92) or PeptideAtlas (93). This is a limitation that disproportionably affects the most novel and least common types of mass spectrometry experiments. We hope that the growing trend of making raw data publicly available will catch up with the dominant public availability of software tools and continue the self-reinforcing cycle toward better and more robust peptide identification strategies.
Supplementary Material
Footnotes
* This work was partly supported by the National Institutes of Health grant 1-P41-RR024851 from the National Center for Research Resources.
 This article contains supplemental Tables S1 and S2.
This article contains supplemental Tables S1 and S2.
1 The abbreviations used are:
- MS2
- tandem MS
- PTM
- post-translational modification
- CID
- collision-induced dissociation
- FDR
- false discovery rate
- HCD
- higher-energy collision dissociation
- ETD
- electron transfer dissociation
- TDA
- target/decoy approach.
REFERENCES
- 1. Nilsson T., Mann M., Aebersold R., Yates J. R., 3rd, Bairoch A., Bergeron J. J. (2010) Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods 7, 681–685 [DOI] [PubMed] [Google Scholar]
- 2. Larsen M. R., Trelle M. B., Thingholm T. E., Jensen O. N. (2006) Analysis of posttranslational modifications of proteins by tandem mass spectrometry. Bio. Technique, 40, 790–798 [DOI] [PubMed] [Google Scholar]
- 3. Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 4. Eng J. K., Searle B. C., Clauser K. R., Tabb D. L. (2011) A face in the crowd: recognizing peptides through database search. Mol. Cell. Proteomics 10, 10.1074/mcp.R111.009522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gupta N., Pevzner P. A. (2009) False discovery rates of protein identifications: a strike against the two-peptide rule. J. Proteome Res. 8, 4173–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nesvizhskii A. I. (2010) A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics 73, 2092–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paizs B., Suhai S. (2005) Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 24, 508–548 [DOI] [PubMed] [Google Scholar]
- 8. Gucinski A. C., Dodds E. D., Li W., Wysocki V. H. (2010) Understanding and exploiting peptide fragment ion intensities using experimental and informatic approaches. Methods Mol. Biol. 604, 73–94 [DOI] [PubMed] [Google Scholar]
- 9. Eng J. K., McCormack A. L., Yates J. R. (1994) An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 10. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 11. Tabb D. L., Huang Y., Wysocki V. H., Yates J. R., 3rd (2004) Influence of basic residue content on fragment ion peak intensities in low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 76, 1243–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Z. (2004) Prediction of low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 76, 3908–3922 [DOI] [PubMed] [Google Scholar]
- 13. Zhang Z. (2005) Prediction of low-energy collision-induced dissociation spectra of peptides with three or more charges. Anal. Chem. 77, 6364–6373 [DOI] [PubMed] [Google Scholar]
- 14. Zubarev R. A., Zubarev A. R., Savitski M. M. (2008) Electron capture/transfer versus collisionally activated/induced dissociations: solo or duet? J. Am. Soc. Mass Spectrom. 19, 753–761 [DOI] [PubMed] [Google Scholar]
- 15. Swaney D. L., Wenger C. D., Coon J. J. (2010) Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res. 9, 1323–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aebersold R., Leavitt J., Saavedra R. A., Hood L. E., Kent S. B. H. (1987) Internal amino-acid sequence-analysis of proteins separated by one-dimensional or two-dimensional gel-electrophoresis after insitu protease digestion on nitrocellulose. Proc. Natl. Acad. Sci., 84, 6970–6974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. MacCoss M. J., McDonald W. H., Saraf A., Sadygov R., Clark J. M., Tasto J. J., Gould K. L., Wolters D., Washburn M., Weiss A., Clark J. I., Yates J. R. (2002) Shotgun identification of protein modifications from protein complexes and lens tissue. Proc. Natl. Acad. Sci. U.S.A. 99, 7900–7905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schlosser A., Vanselow J. T., Kramer A. (2005) Mapping of phosphorylation sites by a multi-protease approach with specific phosphopeptide enrichment and nanoLC–MS/MS Analysis. Anal. Chem. 77, 5243–5250 [DOI] [PubMed] [Google Scholar]
- 19. Wang B., Malik R. E., Nigg A., Kor̈ner R. (2008) Evaluation of the low-specificity protease elastase for large-scale phosphoproteome analysis. Anal. Chem. 80, 9526–9533 [DOI] [PubMed] [Google Scholar]
- 20. Biringer R. G., Amato H., Harrington M. G., Fonteh A. N., Riggins J. N., Hühmer A. F. (2006) Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap LC-MS/MS. Brief Funct. Genomic Proteomic 5, 144–153 [DOI] [PubMed] [Google Scholar]
- 21. Choudhary G., Wu S. L., Shieh P., Hancock W. S. (2003) Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary LC with ion trap MS/MS. J. Proteome Res. 2, 59–67 [DOI] [PubMed] [Google Scholar]
- 22. Huang Y., Triscari J. M., Tseng G. C., Pasa-Tolic L., Lipton M. S., Smith R. D., Wysocki V. H. (2005) Statistical characterization of the charge state and residue dependence of low-energy cid peptide dissociation patterns. Anal. Chem. 77, 5800–5813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olsen J. V., Macek B., Lange O., Makarov A., Horning S., Mann M. (2007) Higher-energy c-trap dissociation for peptide modification analysis. Nat. Methods 4,709–712 [DOI] [PubMed] [Google Scholar]
- 24. Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coon J. J., Ueberheide B., Syka J. E., Dryhurst D. D., Ausio J., Shabanowitz J., Hunt D. F. (2005) Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 102, 9463–9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chalkley R. J., Thalhammer A., Schoepfer R., Burlingame A. L. (2009) Identification of protein o-glcnacylation sites using electron transfer dissociation mass spectrometry on native peptides. Proc. Natl. Acad. Sci. U.S.A. 106, 8894–8899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Young N. L., Dimaggio P. A., Garcia B. A. (2010) The significance, development and progress of high-throughput combinatorial histone code analysis. Cell. Mol. Life Sci. 67, 3983–4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim M. S., Pandey A. (2012) Electron transfer dissociation mass spectrometry in proteomics. Proteomics Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taouatas N., Heck A. J., Mohammed S. (2010) Evaluation of metalloendopeptidase lys-n protease performance under different sample handling conditions. J. Proteome Res. 9, 4282–4288 [DOI] [PubMed] [Google Scholar]
- 30. Aguiar M., Haas W., Beausoleil S. A., Rush J., Gygi S. P. (2010) Gas-phase rearrangements do not affect site localization reliability in phosphoproteomics data sets. J. Proteome Res. 9, 3103–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen Y., Tolić N., Xie F., Zhao R., Purvine S. O., Schepmoes A. A., Moore R. J., Anderson G. A., Smith R. D. (2011) Effectiveness of CID, HCD, and ETD with FT MS/MS for degradomic-peptidomic analysis: comparison of peptide identification methods. J. Proteome Res. 10, 3929–3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jedrychowski M. P., Huttlin E. L., Haas W., Sowa M. E., Rad R., Gygi. S. P. (2011) Evaluation of hcd- and cid-type fragmentation within their respective detection platforms for murine phosphoproteomics. Mol. Cell. Proteomics (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frese C. K., Altelaar A. F., Hennrich M. L., Nolting D., Zeller M., Griep-Raming J., Heck A. J., Mohammed S. (2011) Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. J. Proteome Res. 10, 2377–2388 [DOI] [PubMed] [Google Scholar]
- 34. Swaney D. L., McAlister G. C., Coon J. J. (2008) Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat. Methods 5, 959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Swaney D. L., Wenger C. D., Thomson J. A., Coon J. J. (2009) Human embryonic stem cell phosphoproteome revealed by electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 106, 995–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim S., Mischerikow N., Bandeira N., Navarro J. D., Wich L., Mohammed S., Heck A. J., Pevzner P. A. (2010) The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteomics 9, 2840–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hogan J. M., Pitteri S. J., Chrisman P. A., McLuckey S. A. (2005) Complementary structural information from a tryptic N-linked glycopeptide via electron transfer ion/ion reactions and collision-induced dissociation. J. Proteome Res. 4, 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 39. Gupta N., Bandeira N., Keich U., Pevzner P. A. (2011) Target-decoy approach and false discovery rate: When things may go wrong. J. Am. Soc. Mass Spectrom. 22, 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Molina H., Matthiesen R., Kandasamy K., Pandey A. (2008) Comprehensive comparison of collision induced dissociation and electron transfer dissociation. Anal. Chem. 80, 4825–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chi H., Sun R. X., Yang B., Song C. Q., Wang L. H., Liu C., Fu Y., Yuan Z. F., Wang H. P., He S. M., Dong M. Q. (2010) pNovo: de novo peptide sequencing and identification using HCD spectra. J. Proteome Res. 9, 2713–2724 [DOI] [PubMed] [Google Scholar]
- 42. Hart S. R., Lau K. W., Gaskell S. J., Hubbard S. J. (2011) Distributions of ion series in ETD and CID spectra: making a comparison. Methods Mol. Biol. 696, 327–337 [DOI] [PubMed] [Google Scholar]
- 43. Compton P. D., Strukl J. V., Bai D. L., Shabanowitz J., Hunt. D. F., (2012) Optimization of electron transfer dissociation via informed selection of reagents and operating parameters. Anal. Chem. page Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim S., Gupta N., Bandeira N., Pevzner P. A. (2009) Spectral dictionaries: Integrating de novo peptide sequencing with database search of tandem mass spectra. Mol. Cell. Proteomics 8, 53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim M. S., Zhong J., Kandasamy K., Delanghe B., Pandey A. (2011) Systematic evaluation of alternating CID and ETD fragmentation for phosphorylated peptides. Proteomics 11, 2568–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin T., Glish G. L. (1998) C-terminal peptide sequencing via multistage mass spectrometry. Anal. Chem. 70, 5162–5165 [DOI] [PubMed] [Google Scholar]
- 47. Zhang Z., McElvain J. S. (2000) De novo peptide sequencing by two-dimensional fragment correlation mass spectrometry. Anal. Chem. 72, 2337–2350 [DOI] [PubMed] [Google Scholar]
- 48. Bandeira N., Olsen J. V., Mann M., Pevzner P.A. (2008) Multi-spectra peptide sequencing and its applications to multistage mass spectrometry. Bioinformatics 24, i416–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horn D. M., Zubarev R. A., McLafferty F. W. (2000) Automated de novo sequencing of proteins by tandem high-resolution mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 97, 10313–10317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Savitski M. M., Nielsen M. L., Kjeldsen F., Zubarev R. A. (2005) Proteomics-grade de novo sequencing approach. J. Proteome Res. 4, 2348–2354 [DOI] [PubMed] [Google Scholar]
- 51. Datta R., Bern M. (2009) Spectrum fusion: using multiple mass spectra for de novo peptide sequencing. J. Comput. Biol. 16, 1169–1182 [DOI] [PubMed] [Google Scholar]
- 52. Bertsch A., Leinenbach A., Pervukhin A., Lubeck M., Hartmer R., Baessmann C., Elnakady Y. A., Müller R., Böcker S., Huber C. G., Kohlbacher O. (2009) De novo peptide sequencing by tandem MS using complementary CID and electron transfer dissociation. Electrophoresis 30, 3736–3747 [DOI] [PubMed] [Google Scholar]
- 53. Medzihradszky K. F., Bohlen C. J. (2012) Partial De Novo Sequencing and Unusual CID Fragmentation of a 7 kDa, Disulfide-Bridged Toxin. J. Am. Soc. Mass Spectrom. Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 55. Craig R., Beavis R. C. (2004) Tandem: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 56. Bern M., Cai Y., Goldberg D. (2007) Lookup peaks: a hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal. Chem. 79, 1393–1400 [DOI] [PubMed] [Google Scholar]
- 57. Tabb D. L., Fernando C. G., Chambers M. C. (2007) Myrimatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang L. H., Li D. Q., Fu Y., Wang H. P., Zhang J. F., Yuan Z. F., Sun R. X., Zeng R., He S. M., Gao W. (2007) pFind 2.0: a software package for peptide and protein identification via tandem mass spectrometry. Rapid Commun. Mass Spectrom. 21, 2985–2991 [DOI] [PubMed] [Google Scholar]
- 59. Spivak M., Weston J., Bottou L., Käll L., Noble W. S. (2009) Improvements to the percolator algorithm for peptide identification from shotgun proteomics data sets. J. Proteome Res. 8, 3737–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 61. Baker P. R., Medzihradszky K. F., Chalkley R. J. (2010) Improving software performance for peptide electron transfer dissociation data analysis by implementation of charge state- and sequence-dependent scoring. Mol. Cell. Proteomics 9, 1795–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu X., Shan B., Xin L., Ma B. (2010) Better score function for peptide identification with ETD MS/MS spectra. BMC Bioinformatics 11, S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Deutsch E. W., Shteynberg D., Lam H., Sun Z., Eng J. K., Carapito C., von Haller P. D., Tasman N., Mendoza L., Farrah T., Aebersold R. (2010) Trans-proteomic pipeline supports and improves analysis of electron transfer dissociation data sets. Proteomics 10, 1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alves G., Ogurtsov A. Y., Wu W. W., Wang G., Shen R. F., Yu Y. K. (2007) Calibrating e-values for ms2 database search methods. Biol. Direct 2, 26–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Alves G., Ogurtsov A. Y., Yu Y. K. (2010) Raid_aps: Ms/ms analysis with multiple scoring functions and spectrum-specific statistics. PLoS One 5, e15438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alves G., Wu W. W., Wang G., Shen R. F., Yu Y. K. (2008) Enhancing peptide identification confidence by combining search methods. J. Proteome Res. 7, 3102–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Searle B. C., Turner M., Nesvizhskii A. I. (2008) Improving sensitivity by probabilistically combining results from multiple MS/MS search methodologies. J. Proteome Res. 7, 245–253 [DOI] [PubMed] [Google Scholar]
- 68. Shteynberg D., Deutsch E. W., Lam H., Eng J. K., Sun Z., Tasman N., Mendoza L., Moritz R. L., Aebersold R., Nesvizhskii A. I. (2011) iProphet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 10.1074/mcp.M111.007690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Olsen J. V., Mann M. (2004) Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc. Natl. Acad. Sci. U.S.A. 101, 13417–13422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ulintz P. J., Bodenmiller B., Andrews P. C., Aebersold R., Nesvizhskii A. I. (2008) Investigating MS2-MS3 matching statistics: A model for coupling consecutive stage mass spectrometry data for increased peptide identification confidence. Mol. Cell. Proteomics 7, 71–87 [DOI] [PubMed] [Google Scholar]
- 71. Zubarev R. A. (2003) Reactions of polypeptide ions with electrons in the gas phase. Mass Spectrom. Rev. 22, 57–77 [DOI] [PubMed] [Google Scholar]
- 72. Mikesh L. M., Ueberheide B., Chi A., Coon J. J., Syka J. E., Shabanowitz J., Hunt D. F. (2006) The utility of etd mass spectrometry in proteomic analysis. Biochim. Biophys. Acta 1764, 1811–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villén J., Li J., Cohn M. A., Cantley L. C., Gygi. S. P. (2004) Large-scale characterization of hela cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Palumbo A. M., Smith S. A., Kalcic C. L., Dantus M., Stemmer P. M., Reid. G. E. (2011) Tandem mass spectrometry strategies for phosphoproteome analysis. Mass Spectrom. Rev. 30, 600–625 [DOI] [PubMed] [Google Scholar]
- 75. Chen R., Wang F., Tan Y., Sun Z., Song C., Ye M., Wang H., Zou H. (2012) Development of a combined chemical and enzymatic approach for the mass spectrometric identification and quantification of aberrant N-glycosylation. J. Proteomics Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 76. Hanisch F. G. (2012) O-glycoproteomics: site-specific O-glycoprotein analysis by CID/ETD electrospray ionization tandem mass spectrometry and top-down glycoprotein sequencing by in-source decay MALDI mass spectrometry. Methods Mol. Biol. Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 77. Nagaraj N., D'Souza R. C., Cox J., Olsen J. V., Mann M. (2010) Feasibility of large-scale phosphoproteomics with higher energy collisional dissociation fragmentation. J. Proteome Res. 9, 6786–6794 [DOI] [PubMed] [Google Scholar]
- 78. Chalkley R. J., Clauser K. R. (2012) Modification site localization scoring: Strategies and performance. Mol. Cell. Proteomics 11, 10.1074/mcp.R111.015305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 80. Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphors. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 81. Kelstrup C. D., Hekmat O., Francavilla C., Olsen J. V. (2011) Pinpointing phosphorylation sites: Quantitative filtering and a novel site-specific x-ion fragment. J. Proteome Res. 10, 2937–2948 [DOI] [PubMed] [Google Scholar]
- 82. Chowdhury S. M., Du X., Tolić N., Wu S., Moore R. J., Mayer M. U., Smith R. D., Adkins J. N. (2009) Identification of cross-linked peptides after click-based enrichment using sequential collision-induced dissociation and electron transfer dissociation tandem mass spectrometry. Anal. Chem. 81, 5524–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jufvas A., Strålfors P., Vener A. V. (2011) Histone variants and their post-translational modifications in primary human fat cells. PLoS One 6, e15960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lu Y., Zhou X., Stemmer P., Reid G. (2011) Sulfonium ion derivatization, isobaric stable isotope labeling and data dependent CID- and ETD-MS/MS for enhanced phosphopeptide quantitation, identification and phosphorylation site characterization. J. Am. Soc. Mass Spectrom. 23, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Duncan M. W., Aebersold R., Caprioli R. M. (2010) The pros and cons of peptide-centric proteomics. Nat. Biotechnol. 28, 659–664 [DOI] [PubMed] [Google Scholar]
- 86. Depontieu F. R., Qian J., Zarling A. L., McMiller T. L., Salay T. M., Norris A., English A. M., Shabanowitz J., Engelhard V. H., Hunt D. F., Topalian S. L. (2009) Identification of tumor-associated, MHC class II-restricted phosphopeptides as targets for immunotherapy. Proc. Natl. Acad. Sci. U.S.A. 106, 12073–12078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Walsh G. (2010) Post-translational modifications of protein biopharmaceuticals. Drug Discov. Today 15, 773–780 [DOI] [PubMed] [Google Scholar]
- 88. Robertson C. R., Flynn S. P., White H. S., Bulaj G. (2011) Anticonvulsant neuropeptides as drug leads for neurological diseases. Nat. Prod. Rep. 28, 741–762 [DOI] [PubMed] [Google Scholar]
- 89. King G. F. (2011) Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin. Biol. Ther. 11, 1469–1484 [DOI] [PubMed] [Google Scholar]
- 90. Burkard M. E., Maciejowski J., Rodriguez-Bravo V., Repka M., Lowery D. M., Clauser K. R., Zhang C., Shokat K. M., Carr S. A., Yaffe M. B., Jallepalli P. V. (2009) Plk1 self-organization and priming phosphorylation of hscyk-4 at the spindle midzone regulate the onset of division in human cells. PLoS Biol. 7, e1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Savitski M. M., Lemeer S., Boesche M., Lang M., Mathieson T., Bantscheff M., Kuster B. (2011) Confident phosphorylation site localization using the mascot delta score. Mol. Cell. Proteomics (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Falkner J. A., Andrews P. C. (2007) P6-T Tranche: secure decentralized data storage for the proteomics community. J. Biomol. Tech. 18, 3 [Google Scholar]
- 93. Desiere F., Deutsch E. W., King N. L., Nesvizhskii A. I., Mallick P., Eng J., Chen S., Eddes J., Loevenich S. N., Aebersold R. (2006) The PeptideAtlas project. Nucleic Acids Res. 34,655–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Savitski M., Kjeldsen F., Nielsen M. L., Zubarev. R. A. (2007) Hydrogen rearrangement to and from radical z fragments in electron capture dissociation of peptides. J. Am. Soc. Mass Spectrom. 18, 113–120 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.