

Abstract
No consensus has been reached on the proper time to add stable-isotope labeled (SIL) peptides in protein cleavage isotope dilution mass spectrometry workflows. While quantifying 24 monolignol pathway enzymes in the xylem tissue of Populus trichocarpa, we compared the protein concentrations obtained when adding the SIL standard peptides concurrently with the enzyme or after quenching of the digestion (i.e. postdigestion) and observed discrepancies for nearly all tryptic peptides investigated. In some cases, greater than 30-fold differences were observed. To explain these differences and potentially correct for them, we developed a mathematical model based on pseudo-first-order kinetics to account for the dynamic production and decay (e.g. degradation and precipitation) of the native peptide targets in conjunction with the decay of the SIL peptide standards. A time course study of the digests confirmed the results predicted by the proposed model and revealed that the discrepancy between concurrent and postdigestion introduction of the SIL standards was related to differential decay experienced by the SIL peptide and the native peptide in each method. Given these results, we propose concurrent introduction of the SIL peptide is most appropriate, though not free from bias. Mathematical modeling of this method reveals that overestimation of protein quantities would still result when rapid peptide decay occurs and that this bias would be further exaggerated by slow proteolysis. We derive a simple equation to estimate the bias for each peptide based on the relative rates of production and decay. According to this equation, nearly half of the peptides evaluated here were estimated to have quantitative errors greater than 10% and in a few cases over 100%. We conclude that the instability of peptides can often significantly bias the protein quantities measured in protein cleavage isotope dilution mass spectrometry-based assays and suggest peptide stability be made a priority when selecting peptides to use for quantification.
In 1991, Desiderio and coworkers were the first to couple protein cleavage and isotope dilution mass spectrometry (PC-IDMS)1 when quantifying β-endorphin from human pituitary glands using a full-length (31 residue) deuterated analog (1). Since this seminal work, PC-IDMS has evolved and now describes three distinct methods applying bottom-up methodologies (i.e. proteolysis) in conjunction with stable isotope-labeled (SIL) standards to carry out absolute quantification of proteins. These three methods are commonly called AQUA (2–8), QconCAT (9, 10), and PSAQ (11), and are differentiated by the origin of the SIL peptides used as internal standards. The first method, demonstrated by Barr et al. (2) and later coined “AQUA” by Gygi and coworkers (4), employs SIL peptides produced via chemical synthesis, which offers the advantage of incorporating modifications within the standard peptide's sequence to quantify post-translationally modified proteins. To circumvent the limitations of chemical synthesis and the high cost associated with producing multiple peptides for large scale studies, Beynon and coworkers developed the QconCAT methodology in which all SIL peptides are first produced as a recombinant “concatemer,” which upon digestion produces the individual SIL peptide standards (9, 10). The last of the three methods, yet perhaps the most straight-forward, uses SIL protein standards to perform absolute quantification (i.e. PSAQ) and was shown by Brun and coworkers to provide more accurate quantification relative to QconCAT and AQUA (11).
Although each of these three methods have their own practical strengths and weaknesses, it is logical to assume the PSAQ approach could provide superior quantitative accuracy because a protein standard is being used to quantify a protein. If a SIL protein standard adopts the same confirmation as the native protein, the standard would account for nearly all potential biases associated with a PC-IDMS assay (digestion efficiency, etc.), thereby allowing for protein-level quantification despite the use of peptide-level detection. Nevertheless, the PSAQ approach has seen limited use because of the high cost associated with producing high purity SIL protein standards. Conversely, the AQUA and QconCAT approaches are significantly less expensive, but are limited to peptide-level quantification through their use of SIL peptide standards. Consequently, there is potential bias in using these techniques to infer protein-level quantities.
In the AQUA strategy, the main factors generally believed to contribute to inaccuracies are the digestion efficiency and labile amino acid residues within the target peptide sequence (12–14). Consequently, it has become routine to optimize the digestion protocol to ensure complete proteolysis and to avoid quantifying peptides containing commonly modified amino acids or sites of known (or highly probable) post-translational modification. To help ensure complete digestion, sites having high probabilities of missed cleavages are likewise avoided when selecting target peptides. More recently, Henrion and co-workers were the first to propose that the rate of proteolysis (not just the completeness) can be a potential source of bias in AQUA strategies (15). In their work, the authors showed slower digestion conditions resulted in an overestimation of two proteins' quantities compared with their rapid digestion conditions.
One point often taken for granted regarding the quantitative accuracy of the AQUA methodology is the timing of the SIL peptides' introduction to the sample. In various early reports using the AQUA method, the SIL peptides were added immediately before reduction and alkylation of the protein sample (i.e. predigestion) (3, 7, 8), whereas in others the standards were added concurrently with the digesting enzyme (2, 4), or following digestion (5, 6). Various articles highlighting the AQUA workflow indicate predigestion (or concurrent) and postdigest addition are equally acceptable approaches (14), whereas others have emphasized only one as appropriate (13, 16) or failed to specify an appropriate time (17). No distinction has ever been provided in the literature for “predigestion” or “concurrent” introduction of the SIL peptide to the best of our knowledge. Instead, the ambiguous phrase “prior to analysis” has often been used to describe when the SIL peptide was introduced into the sample. In short, no specific procedure has been agreed upon, or demonstrated to be best.
Given that lignin serves as a major hindrance to the production of pulp and the extraction of plant cell wall polysaccharides as a biofuel feedstock, we have been working toward providing a more comprehensive and quantitative understanding of monolignol biosynthesis at the metabolomic, proteomic, and genomic levels (18, 19). As part of this systems biology study we have been developing an AQUA-based assay to quantify 24 enzymes related to the biosynthesis of monolignols and their subsequent formation of lignin in the stem differentiating xylem (SDX) tissue of the model woody plant, Populus trichocarpa. During this work we compared adding the SIL peptides concurrently with the trypsin to adding the standards after quenching the digestions and observed several large discrepancies in the specific protein quantities determined by each method. Here, we seek to explain these discrepancies using mathematical models describing the digestion process. We find the observed differences are associated with variable rates of peptide production and decay during proteolysis, and demonstrate the protein quantities estimated by each method of SIL peptide addition (predigest, concurrent, and postdigest) are uniquely biased by these rates. Moreover, we use digestion time-course data to demonstrate that these models are sufficiently accurate and confirm that adding the SIL peptide standards concurrently with the proteolytic enzyme is the most appropriate (i.e. least biased) approach. Given these findings, we also propose that peptide stability be considered when selecting peptides for quantification to minimize the bias associated with peptide decay during proteolysis.
EXPERIMENTAL PROCEDURES
Reagents
All solvents used here were of HPLC-grade and were purchased from Honeywell Burdick & Jackson (Muskegon, MI). All other chemicals and reagents were from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Synthetic Peptide Standards
Stable isotope-labeled synthetic peptides were obtained from the Mayo Clinic Proteomics Research Center (Rochester, MN). Stock solutions of each SIL peptide were quantified according to the Scope's method (20) and, subsequently, combined to produce the standard mixture (SIL mixture) used for this experiment. All peptides in the SIL mixture were present at 200 nm with the exception of CCoAOMT1.182–206 and CCoAOMT2.182–206, which were both present at 10 μm. Upon addition of the SIL mixture to each sample (10 μl per digest), this resulted in addition of 100 pmol of CCoAOMT1.182–206 and CCoAOMT2.182–206 and 2 pmol of all other SIL peptides. Likewise, a 200 nm solution of the double-isotope labeled peptide for HCT1.338–354 was produced and added to each sample separately.
SDX Protein Extracts
Stem differentiating xylem was collected from three six-month-old Populus trichocarpa (genotype Nisqually-1), which were grown in a greenhouse as previously described (18). Protein extracts were prepared from each tree individually, by grinding 3 g of SDX in liquid nitrogen then homogenizing the cells (2 min, on ice) in 15 ml of extraction buffer containing: 50 mm Bis-Tris (pH 8.0), 20 mm sodium ascorbate, 0.4 m sucrose, 100 mm NaCl, 5 mm dithiothreitol, and 10% (w/w) polyvinylpolypyrrolidone. After removing the cell debris by centrifugation (3000 × g, 4 °C, 15 min, 2 times), the three protein extracts were pooled and the protein concentration measured using a Coomassie Plus Bradford assay (Thermo Scientific, Rockford, IL) prior to storage at −80 °C.
Filter-aided Sample Preparation
Filter-aided sample preparation (FASP) (21) was performed using an abbreviated protocol optimized to ensure complete proteolysis of all proteins targeted here. Briefly, several 200 μl aliquots of pooled SDX protein extract were reduced for 30 min at 56 °C with an equal volume of 100 mm dithiothreitol and, subsequently, alkylated for 60 min at 37 °C after the addition of 100 μl of 1 m iodoacetamide. Both reagents were prepared in 8 m urea containing 50 mm Tris-HCl (pH 8). To ensure a uniform reduced and alkylated sample, the various aliquots were then pooled and, from this pool, an appropriate volume containing 200 μg of protein was added to individual 10 kDa Amicon Ultra-0.5 ml MWCO-filters (Millipore, Billerica, MA). After an initial concentration step (15 min at 14,000 × g), each sample was exchanged three times with 400 μl digestion buffer. Digestion was then initiated by adding 40 μg of bovine trypsin in 90 μl of the digestion buffer containing 2 m urea, 10 mm CaCl2, and 50 mm Tris-HCl (pH 8.0). After the specified time (0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, or 16 h), digestion was quenched by the addition of 100 μl of 1% formic acid containing 0.001% Zwittergent 3–16 (Calbiochem, La Jolla, CA). Following elution of the peptides, the remaining contents of each MWCO-filter were diluted with 400 μl of the quenching solution, which was passed through the filter-unit and combined with the first eluent. Lastly, each sample was diluted to a final volume of 1 ml with quenching solution and the total protein concentration measured by UV-Vis (ε280 = 1 ml mg−1 cm−1) prior to LC-SRM analysis to confirm good peptide recovery from the MWCO-filter units. Within this FASP workflow, SIL peptide standards (10 μl) were added either concurrently with the trypsin or after the quenching step (i.e. postdigestion) in order to perform quantification (see below).
LC-SRM Data Acquisition and Analysis
Each sample was analyzed in triplicate by liquid chromatography-selected reaction monitoring (LC-SRM), using an Eksigent 2D-nanoLC system (Eksigent, Dublin, CA) interfaced to a TSQ Vantage triple-stage quadrupole mass spectrometry (Thermo Scientific, San Jose, CA). The nanoLC system was equipped with an AS1 autosampler and a cHiPLC-nanoflex system, which was operated in Trap-Elute mode. For each run, 5 μl of sample was loaded onto a Nano cHiPLC trap column (200 μm × 0.5 mm) at 1.5 μl/min by way of a 9000 nL metered injection using 100% mobile phase A. The trap column was then placed in-line with the Nano cHiPLC analytical column (75 μm × 15 cm) and the 400 nL/min gradient elution was initiated. The gradient program consisted of an initial 22 min ramp from 5 to 38.5% B, followed by a 0.5 min ramp to 95% B where it was held for 8 min before reinstating initial conditions. Mobile phases A and B consisted of water/acetonitrile/formic acid (98/2/0.2 and 2/98/02, respectively) and ChromXP C18-CL (3 μm, 120 Å) was the packing material in both columns.
The column eluent was subjected to electrospray ionization using a 10 μm SilicaTip emitter (Woburn, MA) and ESI potential of 1400 V. Scheduled SRM detection was performed for all NAT and SIL peptide pairs using a scan time of 1.5 s, Q1 peak width of 0.7 (FWHM), and chrom filter setting of 30 s. A list of each peptide's SRM transitions can be found in the supplemental data (supplemental Table S1). SRM transition development and peak detection/integration was performed using Skyline v1.1.0.2905 (22), and the resulting peak area data was exported into Microsoft Excel 2010 (Redmond, WA) for further analysis. In particular, nonlinear least-square regression analysis was performed using the Solver add-in for Excel. Calculation of the protein quantities in each run was performed using the ratio of the NAT and SIL peptide peak areas, which was calculated for each peptide species as the sum of all peak areas measured by each transition.
RESULTS
Theoretical Implications of Peptide Decay
PC-IDMS is based on the principle the measured concentration ratio between the native, surrogate peptide (i.e. the proteotypic/signature peptide produced enzymatically for quantification) and its SIL analog accurately reflect the molar ratio between the intact protein and the SIL internal standard in the interrogated sample. For the AQUA strategy, this ideal relationship is described mathematically as:
 |
Herein [PNAT] and [PSIL] are the concentrations of the native and SIL peptide following proteolysis, respectively; [SNAT]0 is the target protein (i.e. substrate) concentration prior to proteolysis; and [PSIL]0 is the known concentration of SIL internal standard peptide added to the interrogated sample. Thus, if the measured concentration ratio, [PNAT]/[PSIL], is approximated by the corresponding ratio of their analytical responses, ANAT/ASIL, the protein concentration can be calculated by the equality:
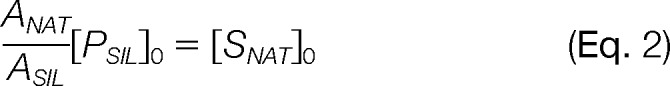 |
where the magnitude of the analytical responses (e.g. peak areas) are represented by the values, ANAT and ASIL. However, these equalities work under the assumptions that the protein substrate has been fully converted into its peptide form prior to the analytical measurement and the native and SIL peptides do not decay prior to or during proteolysis, where decay refers to any chemical or physical process altering the solubilized concentrations of the targeted molecular species. If no such assumptions are made, this situation is more appropriately defined as a quotient of the pseudo-first-order, integrated rate equations describing the concentrations of PNAT and PSIL as a function of time, t, and the rate constants for peptide production (i.e. proteolysis), kp, and peptide decay, kd (Supplemental Data, Fig. S1 and Eq. S1, S2, and S3). In its simplified form this quotient is:
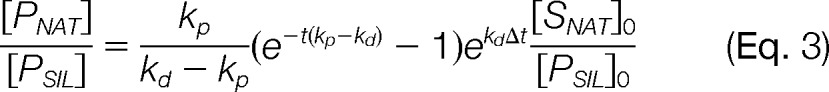 |
In the presence of peptide decay, another factor impacting the measured ratio in the AQUA strategy is the timing of the SIL peptide's introduction into the sample. This is because of the fact that the SIL internal standard will undergo decay differently than the endogenous peptide sequence until its production via proteolysis. As such, this relationship also incorporates the time difference, Δt, between the start of the digestion (t0 = 0) and the introduction of the SIL peptide standard (ti) (Fig. 1A).
Fig. 1.

Theoretical models for peptide production and decay during AQUA workflows. A, Shown is the sample preparation timeline for AQUA-workflows. Here, t0 is the time point when digestion begins and is set to 0 h in this frame of reference such that the length of the digestion period is defined by t. The time point at which the SIL peptide is introduced to the sample is called ti and the time difference, Δt, between the start of the digestion (t0) and the introduction of the SIL peptide (ti) is defined by the equation shown. B, Shown is the model for the ideal situation, where no decay of the NAT peptide (PNAT) or SIL peptide (PSIL) occurs. C, Depicted are the modeled results when the SIL is introduced into the sample predigestion (Δt = 2 h), concurrently with the enzyme (Δt = 0), or postdigestion (Δt = –t). The blue and red lines in each plot indicate [PNAT] and [PSIL], respectively. The light purple line indicates the correct ratio of [PNAT] and [PSIL] under ideal conditions, while the dark purple line shows the “measured” ratio. These plots were created using the following criteria: [SNAT]0 = [PSIL]0 (to give an expected ratio of 1), kp = 0.750 h−1, and kd = 0.075 h−1.
In AQUA workflows, there are three practical time points to introduce the SIL peptide into the sample: prior to all chemical and enzymatic treatments (ti < t0; i.e. Δt > 0), concurrently with the enzyme (ti = t0; i.e. Δt = 0), or following completion of the digestion (ti = t; i.e. Δt = –t). Herein, these three time points of introduction are referred to as predigest, concurrent, and postdigest, respectively. When the SIL peptide is added predigestion, the relationship follows Eq. 3. If the standard is added concurrently, the equation simplifies to:
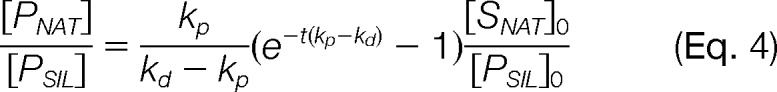 |
Finally, if the SIL standard peptide is added post-digestion then the SIL peptide essentially undergoes no degradation, in which case Eq. 3 becomes:
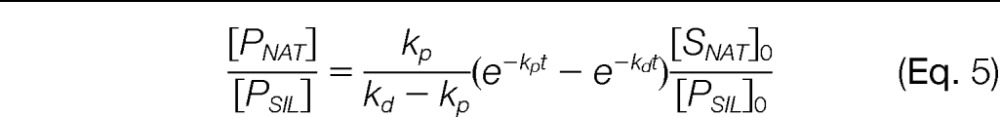 |
To illustrate the quantitative differences between these three points of SIL peptide introduction and the implications of peptide decay, we used these mathematical models to produce theoretical curves depicting how the peptide concentrations and their measured ratio progresses over the course of the digestion (Fig. 1). In the ideal situation, when no peptide decay occurs, Eq. 3 simplifies to an increasing form of an exponential decay function:
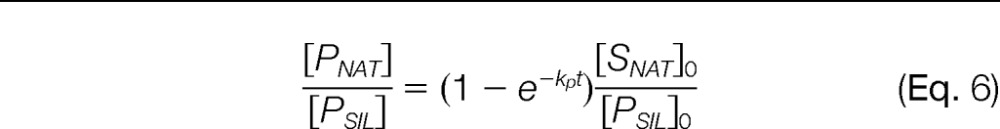 |
Once digestion reaches completion, the measured ratio between the native peptide and its SIL analog will exactly match the expected ratio, regardless of when the SIL peptide is added (Fig. 1B). However, when the peptides do undergo decay, different biases are expected depending on when the SIL peptide is introduced to the sample (Fig. 1C). If it is added predigestion or concurrently, the SIL peptide will decay more than the native peptide over the course of the digestion and will result in a measured ratio ([PNAT]/[PSIL]) greater the expected. In contrast, when the SIL peptide is added postdigestion, the measured ratio will be less than the expected ratio because of the degradation of the native peptide and lack of degradation of the SIL peptide. In the latter situation, the model also predicts the bias will increase over time as the native, surrogate peptide continues to degrade. Comparison of all three models reveals the bias between the measured and expected ratio will always be less when the SIL peptide is added concurrently because the decay time for the SIL peptide will be closer to that of the native, surrogate peptide.
Experimental Modeling of Quantitative Digestion
To determine the veracity of the mathematical models, two parallel sets of digestions were carried out in which the SIL peptides (SIL mixture) were added concurrently with trypsin or postdigestion (Fig. 2). Utilization of a FASP workflow precluded evaluating predigest introduction of the SIL mixture. Digestions were quenched and analyzed at successive time points over the course of 16 h to observe the progress of the quantitative measurements over time. Quantification in each sample was performed as is typically done using Eq. 2 and nonlinear least squares regression was used to fit the two data sets to their corresponding models (supplemental Data, Eq. S4 and Eq. S5). In doing this, the rate constants, kd and kp, and the native protein concentration, [SNAT]0, was determined for each peptide (Table I). The endogenous surrogate peptides for three proteins targeted (PO2.213–230, PO3.300–310, and PAL3.239–252) were only detected in two or fewer samples, which prevented regression analysis. Three other peptides (CCoAOMT3.104–115, CCoAOMT3.217–232, and PAL1.238–251) were only detectable during the earlier time points and, consequently, regression was performed using only the time points up to and including 4 h. In nearly all cases were regression was feasible, the resulting data matched the mathematical models well, as evidenced by the high coefficients of determination (R2). Those few peptides that showed the poorest correlation (R2 < 0.85) were also observed to decay most rapidly (kd > 0.3 h−1) and, consequently, had very poor signal that led to low precision measurements.
Fig. 2.
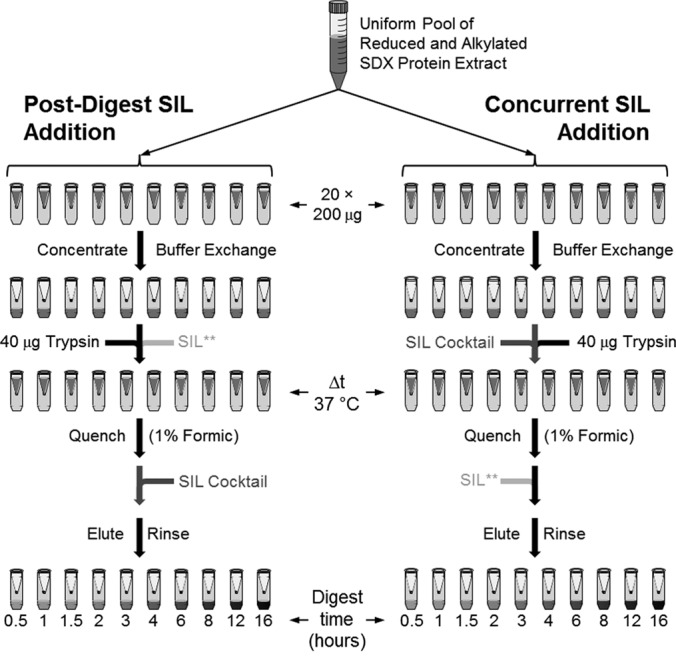
Experimental outline for temporal comparison of SIL peptide introduction into a FASP workflow. Twenty 200 μl aliquots of SDX protein extract were reduced and alkylated then pooled to create a uniform, bulk sample that was aliquoted into 20, 10-kDa MWCO-filters for FASP-digestions. Two sets of digests were performed in parallel. In the first set, the SIL mixture was added immediately following the digest quenching step (postdigest). In the second set, the SIL mixture was added concurrently with the trypsin (concurrent). During both sets of digests, the double-labeled peptide for HCT1.338–354 (indicated as SIL**) was added at the opposite time of the SIL mixture containing the corresponding single-labeled peptide. Note, predigest introduction of the SIL mixture could not be tested within this FASP workflow as the SIL peptides would be removed by the various rinse steps.
Table I. Nonlinear regression results. Shown are the experimental results obtained from fitting the temporal digest data to their mathematical models.
| Peptide | Sequence | kpb | kdb | [SNAT]0c | R2 | kp/kd |
|---|---|---|---|---|---|---|
| 4CL3.262–273 | FDIGTLLGLIEK | 0.344 | 0.140 | 61.1 | 0.9224 | 2.45 |
| 4CL5.262–273 | FEIGSLLGLIEK | 0.466 | 0.179 | 3.33 | 0.9152 | 2.61 |
| C3H3.125–134 | VCTLELFSPK | 0.762 | 0.075 | 16.3 | 0.9605 | 10.1 |
| C4H1.255–261 | DYFVDER | 1.436 | 0.003 | 16.8 | 0.9632 | 427 |
| C4H2.255–261 | DYFVEER | 1.822 | 0.002 | 5.34 | 0.9451 | 847 |
| CAD1.184–198 | GGILGLGGVGHMGVK | 1.011 | 0.184 | 35.0 | 0.8903 | 5.49 |
| CAld5H1.426–435 | FLEPGVPDFK | 1.670 | 0.016 | 15.3 | 0.9628 | 102 |
| CAld5H2.427–436 | FMKPGVPDFK | 1.976 | 0.046 | 4.37 | 0.9490 | 43.3 |
| CAld5H2.L.427–436 | FLKPGVPDFK | 1.881 | 0.020 | 11.0 | 0.9748 | 91.9 |
| CCoAOMT1.182–206 | VGGLIGYDNTLWNGSVVAPPDAPMR | 1.647 | 0.330 | 108 | 0.6355 | 5.00 |
| CCoAOMT2.182–206 | VGGLIGYDNTLWNGSVVAPADAPMR | 1.631 | 0.682 | 85.9 | 0.7946 | 2.39 |
| CCoAOMT3.104–115a | EAYEIGLPFIQK | 1.031 | 0.571 | 36.6 | 0.5861 | 1.81 |
| CCoAOMT3.217–232a | VEISQISIGDGVTLCR | 2.505 | 0.963 | 66.2 | 0.6535 | 2.60 |
| CCR2.299–308 | DLGFEFTPVK | 2.486 | 0.043 | 12.4 | 0.9438 | 57.2 |
| COMT2.51–69 | AGPGAFLSTSEIASHLPTK | 0.880 | 0.071 | 1208 | 0.9734 | 12.4 |
| HCT1.338–354 | SALDFLELQPDLSALVR | 1.264 | 0.103 | 20.1 | 0.9387 | 12.2 |
| HCT6.338–354 | SALDYLELQPDLSALVR | 1.328 | 0.091 | 11.5 | 0.9517 | 14.7 |
| PAL1.238–251a | AAGIDSGFFELQPK | 0.982 | 0.553 | 21.0 | 0.8248 | 1.78 |
| PAL1.664–675 | EELGTGLLTGEK | 2.399 | 0.014 | 5.75 | 0.9763 | 172 |
| PAL2.661–672 | EELGTILLTGEK | 1.147 | 0.109 | 21.8 | 0.9693 | 10.6 |
| PAL3.665–676 | EELGTVLLTGEK | 1.089 | 0.138 | 5.34 | 0.9473 | 7.92 |
| PAL4 5.614–622 | IGSFEEELK | 2.335 | 0.005 | 31.2 | 0.9819 | 430 |
| PO1.136–149 | DGIVSLGGPHIPLK | 1.470 | 0.024 | 0.549 | 0.8513 | 61.3 |
| PO8.113–121 | AFEIIEDLR | 0.701 | 0.019 | 17.3 | 0.9455 | 36.4 |
a Regression performed using digests times ≤4 hours owing to rapid decay of native peptide.
b Units are hr−1.
c Units are fmoles per microgram total protein.
Overall, the protein concentration measured by each peptide was different between the concurrent and postdigest workflows (Fig. 3). As the model predicts, the measured values were always greater when the SIL peptides were added concurrently. For over half of the detectable peptides (13 of 24) the discrepancy between the two methods was greater than twofold and, in two cases, the difference was more than 30-fold. The peptides showing the smallest difference between the two strategies were those with slowest decay rates whereas those with the greatest differences had the largest decay rates (Table I). Lending further weight to the validity of the mathematical models, the measured difference between the two sets of digests was compared with the expected difference calculated by the regression models and showed good correlation (supplemental Data, Fig. S2).
Fig. 3.
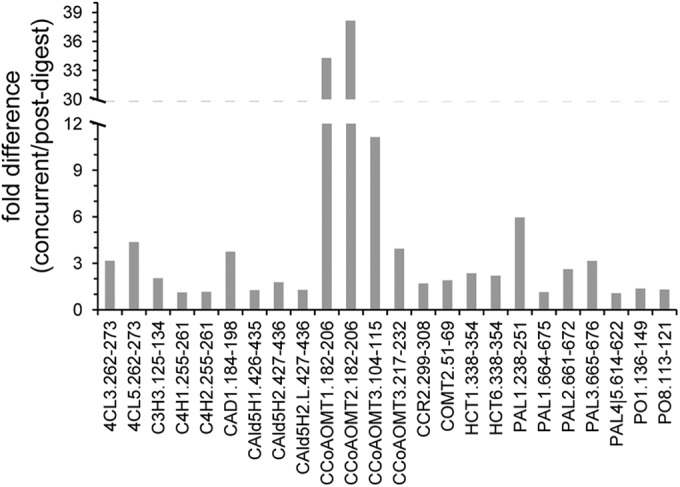
Differences observed in the measured concentrations between the concurrent and post-digest SIL additions at t = 8 h. The protein concentrations were calculated using Equation 2. The 8 h time point was selected given it was the earliest time point in which all targeted peptides were fully produced by proteolysis. The fold difference between the two methods was calculated as the concurrent/postdigest ratio. The protein concentrations measured were always greater when the SIL peptide was added concurrently, which explains why all of the calculated fold differences are greater than 1.
To show the four general combinations of peptide production and decay, the resulting data from four such peptides are depicted (Fig. 4). In the first example, a near ideal situation was observed in which PAL4 5.614–622 was produced rapidly and decayed slowly (Fig. 4A). This was exhibited by the peptide's large rate constant for production (kp = 2.335 h−1) and comparatively small rate constant for decay (kd = 0.005 h−1). This resulted in both digests' curves being practically identical and, once the digests reached completion, the protein concentrations measured nearly matched the value, [SNAT]0, determined by the nonlinear regression. In comparison, protein concentrations measured in the postdigestion workflow were observed to decrease significantly overtime when using peptides such as CCR2.299–308 (Fig. 4B) or PO8.113–121 (Fig. 4C), which decayed more rapidly (kd = 0.043 h−1) or were produced more slowly (kp = 0.701 h−1), respectively. In these cases, the measured protein quantities from the postdigest addition of the SIL peptide were much lower than those determined by regression. However, when the same peptides were added concurrently, the measured protein concentration eventually plateaued and matched well with the regression value, [SNAT]0. These observations are explained by the fact that the SIL peptide, when added concurrently, also undergoes decay and, therefore, can better account for the decay experienced by the native peptide. However, as the decay rates increase relative to the production rates, such as in the case of C3H3.125–135 (kp = 0.762 h−1, kd = 0.075 h−1), the SIL peptide introduced concurrently undergoes decay to a greater extent than the native peptide due to slow production of the native peptide (Fig. 4D). In situations such as these, the protein concentrations measured by the “concurrent” workflow overestimated the true protein concentration determined by the regression. In comparison, the values measured by the “postdigest” workflow drastically underestimated the regression value in these situations because of large degradation of the native peptide prior to addition of the SIL standard.
Fig. 4.

Digestion curves obtained for four distinct peptides showing different combinations of peptide production and decay rates. The results for these peptides are categorized by the production-decay rate combinations (A) rapid-slow, (B) rapid-rapid, (C) slow-slow, and (D) slow-rapid. The circles show the quantitative results obtained when the SIL mixture was added concurrently with the enzyme and the diamonds show the results when it was added post-digestion. Each data point is the mean value calculated with equation 2 from three replicate injections and the error bars represent the 95% confidence interval. Using the proposed mathematical models for each type of SIL introduction strategy, the curves for each peptide were obtained by performing non-linear least square regression analysis on both sets of experimental data simultaneously. The rate constants (kp and kd) and coefficients of determination are shown. The dotted line shows the target protein concentration, [S]0, determined by the regression analysis.
In order to demonstrate the observed differences between the two sets of digests were not the result of a systematic bias, a double-labeled SIL peptide for HCT1.338–354 (denoted as SIL**) was added at the opposite time point of the SIL mixture (Fig. 2). More specifically, the SIL** peptide was added postdigest when the SIL mixture was added concurrently and vice versa when then the SIL mixture was added postdigest. Given that the SIL mixture contained the corresponding single-labeled peptide (denoted as SIL), this provided an unbiased comparison of the two introduction strategies in each sample. For instance, when the SIL peptide was added concurrently and the SIL** peptide added postdigest, the amount of protein calculated was markedly greater at every time point when using the SIL peptide as the internal standard (Fig. 5). It could be argued this observation was simply because of a systematic bias in absolute quantities of the SIL and SIL** standards; however, this was ruled out during the opposite experiment when calculations using the SIL** standard gave the larger protein quantities across all time points (data not shown). This unique experimental design also provided the ability to absolutely quantify the progression of the SIL peptide added concurrently to the sample. Doing this confirmed the standard also underwent degradation throughout the digest, which in turn strongly supports that concurrent addition of SIL peptides provides more accurate quantification.
Fig. 5.
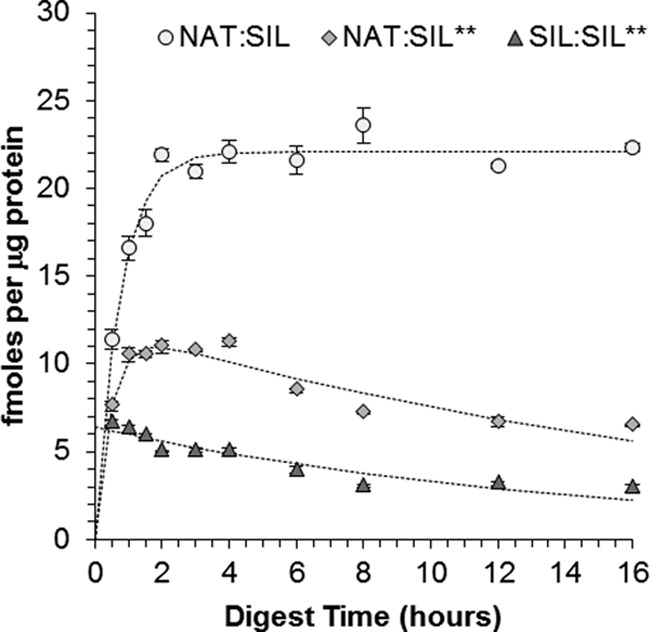
Concentrations of HCT1.338–354 measured over the course of the SDX digestion. The data shown here was produced when the SIL Mixture was added concurrent to the trypsin and the double-labeled peptide of HCT1.338–354 (SIL**) was added after the quenching step. The circles show the concentration of endogenous HCT1 measured using the single-labeled standard peptide (SIL) added concurrently and the diamonds show the concentration measured using the double-labeled standard peptide added postdigestion. Similarly, the triangles show the concentration decay of the single-labeled peptide over the course of the digestion as it was measured using the double-labeled peptide.
Determination of Quantitative Bias
Although it is obvious from these results and the mathematical models that concurrent addition of the SIL peptide provides the most accurate (i.e. least biased) protein quantification, it is important to note this method will overestimate the true protein concentration if the surrogate peptide undergoes decay. Given this, it is essential to determine the extent to which the quantities obtained via this strategy are biased. To accomplish this, Eq. 4 was used to derive the following equation (Supplemental Data, Eq. S6, Eq. S7, and Eq. S8), which shows the error in the amount of protein quantified is related to the relative rates of peptide production and decay:
 |
This equation also infers concurrent introduction of the SIL peptide will always overestimate the true protein concentration as the error will always be positive. In addition, this equation reveals the difference between the true protein quantity and the measured protein quantity will decrease as the rate of peptide production becomes much larger than the rate of peptide decay. For example, the rate of peptide production was ∼10-fold greater than the decay rate (kp/kd = 10.1) for C3H3.125–134 (Fig. 4D) and, consequently, the relative error in the quantitative measurement is expected to be 11% according to Eq. 7. In contrast, the relative error is expected to be only 1.8% for CCR2.299–308 (Fig. 4B), whose peptide production occurs at a rate much faster than the rate of decay (kp/kd = 57.2). Calculating the anticipated relative errors for all peptides analyzed here shows many will yield low quantitative biases, whereas others will produce much greater biases (Fig. 6). In fact, just over half of the detectable peptides (13 of 24) yielded kp/kd ratios less than 11; which, according to Eq. 7, is the threshold required to achieve quantitative errors less than 10%.
Fig. 6.
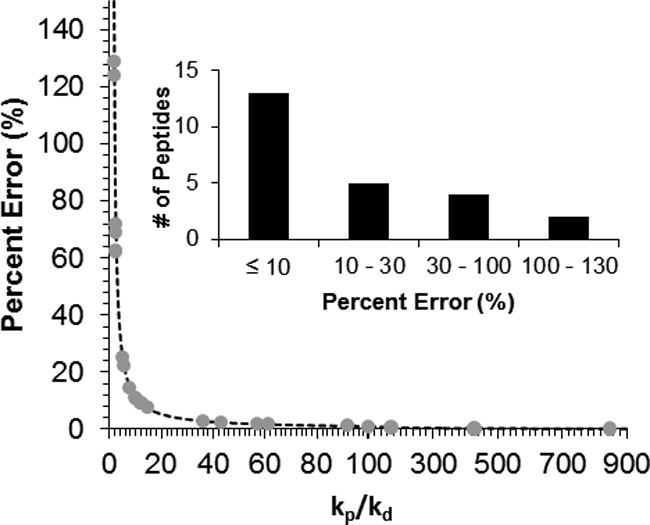
Plot showing the expected quantitative error during concurrent SIL addition as a function of the relative peptide production and decay rates. The dotted line shows the theoretical function when the rate constant for peptide production, kp, is greater than that for peptide decay, kd. The circles show the expected error calculated for all peptides whose rate constants could be determined. Note, the scale of the x axis changes at kp/kd = 100 for better visualization of the data. The inset shows the distribution of the expected relative errors for the 24 detectable surrogate peptides.
Interestingly, peptides derived from the same protein were determined to have drastically different relative errors. For example, the peptides PAL1.238–251 and PAL1.664–675 had calculated relative errors of 129% and 0.6%, respectively, indicating that latter peptide is more likely to accurately reflect the true protein concentration. As expected, these peptides also showed large differences in the protein concentrations measured in the concurrent digest workflow (36.4 verses 6.22 fmoles/μg total protein, respectively); however, the difference in the calculated protein quantities was reduced somewhat after correction based on their expected relative errors (15.9 and 6.18 fmoles/μg total protein).
DISCUSSION
Until now, peptide decay has largely been ignored with regards to its implications on the quantitative accuracy of PC-IDMS. When taking it into consideration for the AQUA strategy, a mathematical model (Eq. 3) developed using principles of pseudo-first-order kinetics revealed all such quantitative measurements are inherently biased by the differential decay between the native peptide and its SIL peptide standard. This model also indicates introduction of the SIL peptide at different points in the AQUA workflow will result in different amounts of protein being quantified. Temporal quantification of 24 enzymes in SDX tissue of P. trichocarpa confirmed the results anticipated by the model and demonstrated the most appropriate time to add the SIL internal standards is concurrently with the proteolytic enzyme. This is not to say adding the SIL peptide concurrent to the digest will provide entirely accurate quantification; on the contrary, the accuracy is still dependent upon the relative rates of peptide production and decay. According to these findings, true absolute quantification will only be achieved when the surrogate peptide is produced much faster than the rate at which it decays. These findings have significant implications on previous studies and could explain why different peptides from the same protein frequently result in different measured protein concentrations.
External calibration curves are frequently employed for internal standard assays for two reasons: they demonstrate the linear dynamic range of the measurements and they account for differences in the response factors of the target analyte and the internal standard. When using heavy isotope analogs as internal standards, a special case forms in which the analyte and internal standard share the exact same physiochemical properties. In the case of LC-SRM analysis, this results in the NAT and SIL analogs having the same ionization and fragmentation efficiencies and, thus, the same instrumental response factor. Consequently, the relative responses of the NAT and SIL peptides will mirror their relative concentration at the time of LC-SRM analysis. If the relative responses of the two species are within an order of magnitude, then considerations such as isotopic overlap can generally be ignored and linearity can be assumed—thus negating the need for external calibration curves (23). For small molecules, fragmentation efficiency is not always conserved between NAT and SIL analogs because of the greater prevalence of isotope effects, in which case calibration curves are required to account for the different response factors. This is the same for traditional (i.e. nonisotope dilution) internal standard assays.
We bring up this point because it is common to generate external calibration curves with standard peptides and use these for quantitative calculations. In such cases, it is assumed the signal ratio observed in the sample can be compared with the ratio on the calibration curve to estimate the native peptide concentration and, thereby, the native protein concentration. This is somewhat redundant because the NAT and SIL peptide have identical response factors (if within their linear dynamic range). Moreover, our work suggests the concentration ratio of the NAT and SIL peptide at the time of LC-SRM analysis does not necessarily reflect the initial (pre-digestion) concentration ratio of the NAT protein and SIL peptide. To account for this by a calibration curve would require intact protein standards that, like the native proteins, would undergo proteolysis. If these were generated in a matched matrix, this would account for the differential decay of the NAT and SIL peptides and provide reliable quantification. For obvious reasons, generation of such calibration curves is impractical and, to the best of our knowledge, has not been performed.
Similar results to those obtained here were previously reported by Arsene et al. when they showed conditions favoring faster digestion provided more accurate quantification than slow digestion conditions (15). Although the authors attributed their results to the differential decay of the SIL peptide and native peptide, they did not fully explore the implications of this on of the SIL introduction strategy. Consequently, their comparison may have been unintentionally biased when they used predigestion and concurrent SIL peptide addition for their slow and rapid digest protocols, respectively. Introduction of the SIL peptides at different time points could explain why differences have been commonly observed across laboratories and across different assays. For example, Williams et al. compared quantification of C-reactive protein in human plasma using an AQUA-based assay to a CLIA-certified ELISA-based assay and reported the AQUA-based assay systematically gave protein quantities ∼12-fold greater than the ELISA-based assay (8). Although this result could possibly be explained solely by the different molecular specificities of the two assays, the potential overestimations of the AQUA-base assay could, in part, be caused by their predigest addition of the SIL peptide standard.
The implications of peptide decay may also apply to other PC-IDMS strategies as well. Beynon and coworkers recently showed that accurate quantification by the QconCAT method is best achieved when the SIL peptide is produced from the QconCAT standard at the same, rapid rate as the native peptide (24). The authors suggest this is because of complete production of the proper limit peptides via rapid proteolysis. However, the concepts introduced by our model suggest accurate quantification during the QconCAT method will be achieved, at least in part, by the identical production rates of the SIL and native peptides allowing both to decay to the same extent during the digestion (supplemental Data, Eq. S9 and Fig. S3). This also implies that overestimation or underestimation can occur in the QconCAT method when the SIL peptide is produced faster or slower, respectively, than the native peptide. Similar biases could potentially result in the PSAQ method (11) as well if the SIL protein standard is digested at a different rate because of an altered conformational or post-translationally modified state.
All peptides tested in this study were fully produced within a relatively small time frame, yet had a much broader dynamic range in terms of their stability (Supplemental Data, Fig. S4 and Table S2). This observation suggests the property most likely to negatively impact the quantitative accuracy of PC-IDMS is peptide stability. As with all assays, there were several potential sources for peptide decay here including peptide precipitation/aggregation, nonspecific proteolysis, chemical modification, absorption, etc. To minimize absorptive losses and improve peptide solubility we added a detergent, Zwittergent 3–16, when quenching the digest. The use of urea in our digestion buffer could have potentially contributed to the decay of the peptides trough carbamylation (25); however, we performed an abbreviated form of this study without urea and no appreciable decrease in decay was observed (data not shown). Given sources of peptide decay are likely to be unavoidable, we would suggest peptide stability be made a greater priority during selection of surrogate peptides. Having said that, a priori knowledge of a peptide's stability is often unavailable and no prediction algorithm exists that is capable of accurately assessing all properties affecting peptide stability, nor is it necessary that peptide decay rates will be conserved across different biological matrices. As such, prudent researchers should select multiple peptides and empirically validate their stability (i.e. decay rates) relative to their rate of production in order to determine which will provide the most accurate quantification.
In instances where options for surrogate peptides are inherently limited, such as when targeting a specific PTM site, nonlinear least square regression analysis like that performed here may provide a more accurate estimate of the protein concentration, [SNAT]0. Although this would likely be impractical for large scale studies, it could be performed once to estimate the relative error for all peptides (Eq. 7) and calculate correction factors that could be applied to all subsequent measurements. However, we would caution readers to avoid using correction factors or nonlinear regression to estimate absolute protein quantities because of the additional uncertainty they would create. Moreover, researchers intending to employ such unorthodox methods would need to verify their veracity, which we could not do here because we did not have knowledge of the “true” protein concentrations, nor an orthogonal method to determine them. Instead, we would suggest selecting “stable” surrogate peptides, which the model and experimental data presented here suggest have little or no quantitative bias and require no correction.
Alternatively, the development of cleavable SIL peptides could be an option to circumvent or mitigate the issue of peptide decay in the AQUA strategy. Cleavable SIL peptides are synthetic peptides with extensions at either or both the N- and C terminus, which allow the standard peptide to undergo proteolysis and mimic the digestion of their native counterparts. Although preliminary studies by our group showed little success in achieving accurate quantification with such peptides (26), this approach would provide more accurate quantification without sacrificing the benefits of having synthetic SIL standards if proteolysis of the SIL standard and native protein are validated to occur at the same rate.
Supplementary Material
Acknowledgments
We thank Dr. Quanzi Li for providing the SDX protein extracts used for these experiments and to Shan Randall for his help with this research.
Footnotes
* This work was supported by North Carolina State University, the W. M. Keck Foundation, and the National Science Foundation Plant Genome Research Program (Grant DBI-0922391).
 This article contains supplemental Tables S1 and S2 and Results.
This article contains supplemental Tables S1 and S2 and Results.
1 The abbreviations used are:
- PC-IDMS
- protein cleavage isotope dilution mass spectrometry
- AQUA
- absolute quantification of proteins via peptide standards
- PSAQ
- protein standards for absolute quantification
- QconCAT
- concatemer of quantitative peptide standards
- SIL
- stable isotope-labeled
- NAT
- natural/native
- SDX
- stem-differentiating xylem
- FASP
- filter-aided sample preparation
- SRM
- selected reaction monitoring.
REFERENCES
- 1. Dass C., Kusmierz J. J., Desiderio D. M. (1991) Mass Spectrometric Quantification of Endogenous beta-Endorphin. Biol. Mass Spectrom. 20, 130–138 [DOI] [PubMed] [Google Scholar]
- 2. Barr J. R., Maggio V. L., Patterson D. G., Jr, Cooper G. R., Henderson L. O., Turner W. E., Smith S. J., Hannon W. H., Needham L. L., Sampson E. J. (1996) Isotope dilution mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin. Chem. 42, 1676–1682 [PubMed] [Google Scholar]
- 3. Barnidge D. R., Dratz E. A., Martin T., Bonilla L. E., Moran L. B., Lindall A. (2003) Absolute quantification of the g protein-coupled receptor rhodopsin by LC/MS/MS using proteolysis product peptides and synthetic peptide standards. Anal. Chem. 75, 445–451 [DOI] [PubMed] [Google Scholar]
- 4. Gerber S. A., Rush J., Stemman O., Kirschner M. W., Gygi S. P. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. P. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuhn E., Wu J., Karl J., Liao H., Zolg W., Guild B. (2004) Quantification of C-Reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and C-13-labeled peptide standards. Proteomics 4, 1175–1186 [DOI] [PubMed] [Google Scholar]
- 6. Anderson N. L., Anderson N. G., Haines L. R., Hardie D. B., Olafson R. W., Pearson T. W. (2004) Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA). J. Proteome Res. 3, 235–244 [DOI] [PubMed] [Google Scholar]
- 7. Barnidge D. R., Goodmanson M. K., Klee G. G., Muddiman D. C. (2004) Absolute quantification of the model biomarker prostate-specific antigen in serum by LC-MS/MS using protein cleavage and isotope dilution mass spectrometry. J. Proteome Res. 3, 644–652 [DOI] [PubMed] [Google Scholar]
- 8. Williams D. K., Muddiman D. C. (2009) Absolute quantification of C-reactive protein in human plasma derived from patients with epithelial ovarian cancer utilizing protein cleavage isotope dilution mass spectrometry. J. Proteome Res. 8, 1085–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beynon R. J., Doherty M. K., Pratt J. M., Gaskell S. J. (2005) Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat. Methods 2, 587–589 [DOI] [PubMed] [Google Scholar]
- 10. Pratt J. M., Simpson D. M., Doherty M. K., Rivers J., Gaskell S. J., Beynon R. J. (2006) Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat. Protoc. 1, 1029–1043 [DOI] [PubMed] [Google Scholar]
- 11. Brun V., Dupuis A., Adrait A., Marcellin M., Thomas D., Court M., Vandenesch F., Garin J. (2007) Isotope-labeled protein standards. Mol. Cell. Proteomics 6, 2139–2149 [DOI] [PubMed] [Google Scholar]
- 12. Lange V., Picotti P., Domon B., Aebersold R. (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kettenbach A. N., Rush J., Gerber S. A. (2011) absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat. Protoc. 6, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kito K., Ito T. (2008) Mass spectrometry-based approaches toward absolute quantitative proteomics. Curr. Genomics 9, 263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arsene C. G., Ohlendorf R., Burkitt W., Pritchard C., Henrion A., O'Connor G., Bunk D. M., Güttler B. (2008) Protein quantification by isotope dilution mass spectrometry of proteolytic fragments: cleavage rate and accuracy. Anal. Chem. 80, 4154–4160 [DOI] [PubMed] [Google Scholar]
- 16. Brun V., Masselon C., Garin J., Dupuis A. (2009) Isotope dilution strategies for absolute quantitative proteomics. J. Proteomics 72, 740–749 [DOI] [PubMed] [Google Scholar]
- 17. Pan S., Aebersold R., Chen R., Rush J., Goodlett D. R., McIntosh M. W., Zhang J., Brentnall T. A. (2009) Mass spectrometry based targeted protein quantification: methods and applications. J. Proteome Res. 8, 787–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shi R., Sun Y. H., Li Q., Heber S., Sederoff R., Chiang V. L. (2010) Towards a systems approach for lignin biosynthesis in Populus trichocarpa: transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol. 51, 144–163 [DOI] [PubMed] [Google Scholar]
- 19. Chen H. C., Li Q., Shuford C. M., Liu J., Muddiman D. C., Sederoff R. R., Chiang V. L. (2011) membrane protein complexes catalyze both 4- and 3-hydroxylation of cinnamic acid derivatives in monolignol biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 21253–21258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scopes R. K. (1974) Measurement of protein by spectrophotometry at 205-nm. Anal. Biochem. 59, 277–282 [DOI] [PubMed] [Google Scholar]
- 21. Wiśniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 22. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuzyk M. A., Smith D., Yang J. C., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brownridge P., Beynon R. J. (2011) The importance of the digest: Proteolysis and absolute quantification in proteomics. Methods 54, 351–360 [DOI] [PubMed] [Google Scholar]
- 25. Stark G. R. (1965) Reactions of cyanate with functional groups of proteins. 3. Reactions with amino and carboxyl groups. Biochemistry 4, 1030–1036 [DOI] [PubMed] [Google Scholar]
- 26. Barnidge D. R., Hall G. D., Stocker J. L., Muddiman D. C. (2004) Evaluation of a cleavable stable isotope labeled synthetic peptide for absolute protein quantification using LC-MS/MS. J. Proteome Res. 3, 658–661 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.