

Summary
Invasive carcinoma cells form actin-rich matrix-degrading protrusions called invadopodia. These structures resemble podosomes produced by some normal cells and play a crucial role in extracellular matrix remodeling. In cancer, formation of invadopodia is strongly associated with invasive potential. Although deregulated signals from the receptor tyrosine kinase Met (also known as hepatocyte growth factor are linked to cancer metastasis and poor prognosis, its role in invadopodia formation is not known. Here we show that stimulation of breast cancer cells with the ligand for Met, hepatocyte growth factor, promotes invadopodia formation, and in aggressive gastric tumor cells where Met is amplified, invadopodia formation is dependent on Met activity. Using both GRB2-associated-binding protein 1 (Gab1)-null fibroblasts and specific knockdown of Gab1 in tumor cells we show that Met-mediated invadopodia formation and cell invasion requires the scaffold protein Gab1. By a structure–function approach, we demonstrate that two proline-rich motifs (P4/5) within Gab1 are essential for invadopodia formation. We identify the actin regulatory protein, cortactin, as a direct interaction partner for Gab1 and show that a Gab1–cortactin interaction is dependent on the SH3 domain of cortactin and the integrity of the P4/5 region of Gab1. Both cortactin and Gab1 localize to invadopodia rosettes in Met-transformed cells and the specific uncoupling of cortactin from Gab1 abrogates invadopodia biogenesis and cell invasion downstream from the Met receptor tyrosine kinase. Met localizes to invadopodia along with cortactin and promotes phosphorylation of cortactin. These findings provide insights into the molecular mechanisms of invadopodia formation and identify Gab1 as a scaffold protein involved in this process.
Key words: Invadopodia, Met RTK, Gab1, Cortactin, Matrix remodeling, Cell invasion
Introduction
Metastasis is the major cause of cancer-related mortality. During the initial steps of metastatic dissemination, some cancer cells acquire the ability to remodel extracellular matrix (ECM), invade surrounding tissue locally, intravasate into lymphatic and blood microvasculature by breaking basement membranes (BM) of the vessels and extravasate at distant sites (Chaffer and Weinberg, 2011). Enhanced invasive capacity of many such cancer cells, in particular carcinomas, is linked to their ability to form invadopodia, specialized actin-rich membrane protrusions that penetrate and remodel the ECM (Buccione et al., 2009; Gimona, 2008), and that are much like podosomes formed in macrophages and osteoclasts (Linder, 2007). Consequently, these invasive cancer cells can use invadopodia as functional structures to perforate the basement membranes and guide the cell body into blood vessels (Schoumacher et al., 2010).
Molecular mechanisms leading to invadopodia biogenesis are only beginning to emerge. Invadopodia-like cellular structures with the capacity to degrade ECM were originally identified in chicken embryonic fibroblasts transformed by Rous sarcoma virus (Chen, 1989), and were linked with constitutive activation of the v-Src oncogene (Hauck et al., 2002). Since then, many studies have established a role for increased Src kinase activity in the formation of invadopodia in cancer cells and in invadopodia-like structures of transformed fibroblasts, which are often referred to as podosomes (Ayala et al., 2009; Bowden et al., 2006; Webb et al., 2007; Oikawa et al., 2008; Balzer et al., 2010; Kelley et al., 2010; Mader et al., 2011).
In addition to Src, other non-receptor tyrosine kinases, Abl and Arg, localize to invadopodia, and are involved in biogenesis of these cellular structures in MDA-MB-231 breast carcinoma cells (Mader et al., 2011; Smith-Pearson et al., 2010). Activation of the epidermal growth factor (EGF) as well as platelet derived growth factor (PDGF) receptor tyrosine kinases (RTK) also promotes invadopodia biogenesis (Eckert et al., 2011; Mader et al., 2011). These findings raise the possibility that multiple receptor tyrosine kinases, when deregulated in cancer, converge signals to promote invadopodia biogenesis contributing to metastatic progression.
The inner structure of invadopodia consists of an actin-rich core, the formation of which is regulated by actin regulatory proteins, protein kinases, as well as regulators of lipid metabolism (Murphy and Courtneidge, 2011). During invadopodia formation in response to EGF, actin polymerization is promoted following cortactin tyrosine phosphorylation and localized release of the actin severing protein, cofilin (Oser et al., 2009). Src kinase also promotes tyrosine phosphorylation of cortactin (Bowden et al., 2006), as well as the scaffold protein Tks5 (Blouw et al., 2008; Seals et al., 2005). Tks5 recruits the adaptor protein, Nck, to form a trimeric complex (Stylli et al., 2009), which activates Wiscott–Aldrich syndrome protein (N-WASP) allowing recruitment of Arp2/3 to promote branched actin nucleation (Yamaguchi et al., 2005). Downstream from Src, a Tks5 protein complex is recruited to phosphatidylinositol 3,4-bisphosphate [PtdIns(3,4)P2]-rich membrane regions through its phox homology (PX) domain and initiates invadopodia biogenesis (Oikawa et al., 2008).
The Met RTK [also known as hepatocyte growth factor (HGF) receptor] is a proto-oncogene often implicated in cancer (Birchmeier et al., 2003). In normal tissues, Met and its ligand, HGF, activate signals that induce epithelial cell dispersal, epithelial remodeling and invasive growth, which are important during development (Birchmeier et al., 2003). Met exerts an invasive morphogenic program primarily through the scaffold protein GRB2-associated-binding protein 1 (Gab1). Gab1 contains a pleckstrin homology (PH) domain, which tethers Gab1 to membranes through interactions with PtdIns(3,4,5)P3 (Maroun et al., 1999) and is recruited to and phosphorylated on multiple tyrosine residues by an activated Met receptor (Birchmeier et al., 2003; Peschard et al., 2007). Upon phosphorylation, these residues serve as docking sites for numerous SH2-domain-containing adaptor and signaling proteins, including Crk, Nck, P85 subunit of PI3K, Shp2 and PLCγ (Abella et al., 2010; Schaeper et al., 2000; Garcia-Guzman et al., 1999; Gual et al., 2000; Lamorte et al., 2000; Maroun et al., 1999; Maroun et al., 2000; Cunnick et al., 2001). Gab1 contains six proline rich motifs, two of which (proline-rich motifs four and five) are implicated in the constitutive association with the adaptor protein growth factor receptor-bound protein 2 (Grb2) through its C-terminal SH3 domain (Lock et al., 2002). Once recruited to a Met–Gab1 complex, these proteins trigger activation of multiple signaling cascades, including PI3K–Akt (Maroun et al., 1999), Ras–MAPK (Maroun et al., 2000; Schaeper et al., 2000), Rac and Rap1 (Lamorte et al., 2000) and Nck/N-WASP (Abella et al., 2010) that promote cell survival, actin cytoskeleton remodeling, as well as increased migration and invasion (Benvenuti and Comoglio, 2007; Birchmeier et al., 2003; Lai et al., 2009; Peschard and Park, 2007).
Although aberrant activation of Met is linked to increased cancer cell invasion and is a hallmark of aggressive tumors with poor prognosis, the ability of Met to coordinate invadopodia formation has not been addressed (Camp et al., 1999; Cruz et al., 2003; Kammula et al., 2007; Lengyel et al., 2005; Okuda et al., 2008; Sawada et al., 2007; Tuynman et al., 2008; Wu et al., 1998; Ponzo et al., 2010). Here we demonstrate that an activated Met RTK promotes invadopodia in carcinoma cells and that fibroblasts transformed with the constitutively active oncogenic variant of the Met receptor, Tpr-Met, form invadopodia-like structures capable of remodeling ECM. We show that invadopodia induced by Met activity are dependent on recruitment of Gab1 that localizes to these structures and interacts directly with cortactin, a key regulator of actin dynamics within invadopodia. We demonstrate that Met colocalizes with cortactin to invadopodia, and that Met activity contributes to increased tyrosine phosphorylation of cortactin independent of Src kinase. By structure–function analysis, we have established that a Gab1–cortactin interaction is required for assembly of functional invadopodia, in response to oncogenic Met signals.
Results
Tpr-Met induces formation of invadopodia rosettes in fibroblasts
Recent studies have suggested that invasive and metastatic potential of cancer cells and malignantly transformed fibroblasts is tightly linked with the ability of these cells to produce invadopodia (Gimona, 2008; Buccione et al., 2009; Schoumacher et al., 2010). Therefore it is possible that malignant phenotypes, tumorigenicity and metastatic potential of cells transformed by oncogenic variants of the Met receptor are at least in part due to the acquired ability of these cells to produce invadopodia or similar actin-rich proteolytically active membrane protrusions that enable remodeling of ECM. To investigate this possibility, we used Fischer rat 3T3 (FR3T3) fibroblasts transformed with the oncogenic variant of the Met receptor, Tpr-Met. Upon Tpr-Met-mediated transformation, FR3T3 fibroblasts acquire many features of malignantly transformed cancer cells, including the ability to invade through the ECM, as well as to develop tumors and metastases in nude mice (Fixman et al., 1996; Fixman et al., 1997; Saucier et al., 2002). In line with our previous findings, control FR3T3 cells used in this study spread and formed a contact-inhibited monolayer in culture, whereas FR3T3 fibroblasts transformed with Tpr-Met developed a distinct elongated cell morphology, formed foci, lost contact inhibition and acquired increased migratory and invasive capacity (Fig. 1) (Fixman et al., 1995; Saucier et al., 2002).
Fig. 1.

Tpr-Met-transformed FR3T3 fibroblasts form invadopodia. (A) FR3T3 cells or FR3T3 cells stably overexpressing Tpr-Met (Tpr-Met 3 and Tpr-Met 4) were cultured for 24 hours on glass coverslips coated with Oregon-Green-conjugated gelatin (gelatin matrix). Cells were stained with phalloidin and confocal images were taken at the ventral plane of the cells. Representative images are shown. (B) Quantification of the ability of FR3T3 cells to form actin rosettes or active invadopodia in response to Tpr-Met. Values are the means of three independent experiments. Active invadopodia are defined as actin-rich rosettes overlaying matrix remodeling. (C) SDS-PAGE was performed on cell lysates from FR3T3 cells or FR3T3 cells stably overexpressing Tpr-Met and probed for Met-P (pMet), Met and actin. (D) Confocal Z-sections were collected, deconvolved using IMARIS software and volume rendered to reconstruct the 3D image. View of invadopodia from above (arrows) and below (arrowheads) the gelatin matrix. (E) FR3T3 cells expressing Tpr-Met were plated on glass coverslips coated with unlabeled collagen for 24 hours and immunostained for invadopodia markers Tks5 and cortactin to identify invadopodia rosettes. The boxed region in the third image is shown at higher magnification in the fourth panel. Scale bars: 10 μm.
To establish whether introduction of the Tpr-Met oncogene could induce the formation of invadopodia and remodel the extracellular matrix, we examined the ability of control and Tpr-Met-transformed FR3T3 fibroblasts to produce ventral actin-rich protrusions when plated on fluorescent gelatin. In this assay, we observed that control FR3T3 fibroblasts formed extensive actin stress fibers (as defined by phalloidin staining), and were unable to remodel gelatin matrix (Fig. 1A). At the same time, FR3T3 fibroblasts transformed with Tpr-Met formed few stress fibers, but produced prominent ventral rosettes of actin filaments associated with underlying areas of degraded fluorescent gelatin matrix, typical of invadopodia (Fig. 1A,D; supplementary material Movie 1). To confirm that the observed structures were indeed invadopodia, we stained these cells with invadopodia markers Tks5 and cortactin and showed their colocalization with the actin rosettes (Fig. 1E; supplementary material Fig. S1). These cellular structures, which we call invadopodia rosettes, were found in 50% of the cells (usually one rosette per cell), and penetrated through the gelatin matrix in 35% of cells at steady state (Fig. 1B). To exclude the possibility that the observed formation of invadopodia rosettes by Tpr-Met-transformed FR3T3 fibroblasts was due to clonal effects, we repeated these experiments using independently derived clones of Tpr-Met-transformed FR3T3 fibroblasts and obtained similar results (Fig. 1A–C). Hence, our findings demonstrate that upon transformation with the Tpr-Met oncogene, FR3T3 fibroblasts acquire the ability to produce invadopodia rosettes, similar to those produced by cancer cells and fibroblasts transformed with the Src oncogene (Murphy and Courtneidge, 2011).
Met activity is required for invadopodia formation in cancer cells
Increased activity of receptor tyrosine kinases, including Met, is often observed in malignancies and is associated with invasive capacity of cancer cells (Birchmeier et al., 2003; Corso et al., 2005; Lai et al., 2009). The roles of different RTKs in biogenesis of invadopodia and similar proteolytically active protrusive cellular structures, of invasive cancer cells, are poorly understood. Given our observation that a constitutively active oncogenic form of Met, Tpr-Met, could induce formation of invadopodia rosettes in fibroblasts, and a number of previous reports that aberrant signaling downstream from Met can promote cancer invasion and metastasis (Lai et al., 2009), we sought to verify whether activation of Met in cancer cells could promote invadopodia biogenesis. For this purpose, we used two different cancer cell lines: an invasive basal breast carcinoma cell line MDA-MB-231, in which Met activation is HGF dependent and a gastric carcinoma cell line MKN45, in which Met is amplified and is constitutively active (Fushida et al., 1993). MDA-MB-231 cells increase invadopodia formation in response to activation of the epidermal growth factor receptor (EGFR) tyrosine kinase (Nam et al., 2007; Pichot et al., 2010) but also express the Met receptor (Fig. 2B). Stimulation of MDA-MB-231 cells plated on fluorescent gelatin in the presence of HGF, led to enhanced activation of Met receptor as visualized using a phosphorylation-specific antibody that recognizes the active receptor (Fig. 2B), but also increased the number of invadopodia by approximately twofold, compared with non-stimulated cells (Fig. 2A,C,D). Approximately 30% of MKN45 cells form invadopodia in the absence of HGF stimulation (Fig. 2E–H). Strikingly, specific inhibition of the Met RTK using a small molecule inhibitor, PHA665752 (Christensen et al., 2003), resulted in a profound change in cell morphology, and abrogated the ability of MKN45 cells to form invadopodia and remodel the gelatin matrix (Fig. 2E–H). To confirm that this is dependent on Met activity, we employed knockdown of Met expression using a specific siRNA and observed the same phenotype: the ability of MKN45 cells to form invadopodia upon inactivation of Met signal is decreased by half (Fig. 2E–H). Together, these data demonstrate that in invasive cancer cells, activation of Met, both dependent and independent of stimulation with HGF, elicits signals that result in increased invadopodia biogenesis and extracellular matrix remodeling.
Fig. 2.

Invasive breast cancer cells, MDA-MB-231, and gastric cancer cells, MKN45, form invadopodia in response to Met RTK signaling. (A) MDA-MB-231 cells were cultured on gelatin matrix for 3 hours and stimulated with 0.5 nM HGF for an additional 3 hours. Cells were stained for the invadopodia markers actin (phalloidin) or cortactin. (B) SDS-PAGE was performed on cell lysates of MDA-MB-231 cells stimulated with 0.5 nM HGF and non-stimulated cells. (C,D) The ability of MDA-MB-231 cells to form invadopodia in response to HGF stimulation was quantified. Values are the means of three independent experiments. (E) MKN45 cells were cultured on gelatin matrix in the presence of 0.1 µM Met inhibitor PHA665752 or DMSO for 24 hours. MKN45 cells were treated with 50 nM siRNA targeting Met or control siRNA. Cells were trypsinized 48 hours after treatment and plated on gelatin matrix and cultured for an additional 24 hours. Cells were stained for markers of invadopodia, actin (phalloidin) or cortactin and confocal images were acquired at the ventral plane of the cells. DIC images of cells stained with actin (red) and DAPI (blue) taken at a lower magnification (63×) are shown on the right. Representative images are shown. (F,G) The loss of invadopodia formation in MKN45 cells in response to treatment with Met inhibitor PHA665752 or siRNA-mediated knockdown of Met. (H) SDS-PAGE was performed on cell lysates of MKN45 cells treated with 0.1 µM Met inhibitor or 50 nM siRNA to Met or the respective vehicles (DMSO) or control siRNA and probed for Met-P (pMet), Met and tubulin. Scale bars: 10 μm.
Ability of fibroblasts to form functional invadopodia rosettes is determined by an intact multiprotein docking site of Tpr-Met
Constitutive activation of the Tpr-Met oncogene is accompanied by auto-phosphorylation at two key tyrosine residues of the C-terminal region (Y482 and Y489, corresponding to Y1349 and Y1356 residues of the wild-type Met receptor). These residues create a multiprotein docking site for phosphorylation-dependent recruitment of the adaptor proteins, Grb2 and Shc, and the scaffold protein Gab1 (Fixman et al., 1997; Fournier et al., 1996; Nguyen et al., 1997; Ponzetto et al., 1994) and are essential for the biological and transforming activity of the Tpr-Met oncogene and Met RTK (Fixman et al., 1995; Fixman et al., 1997; Lock et al., 2003; Ponzetto et al., 1996). Whereas interaction between Grb2 and Tpr-Met requires direct binding of the Grb2 SH2 domain to a phosphorylated Y1356 residue of Tpr-Met (Nguyen et al., 1997), Gab1 can be recruited to Tpr-Met both directly, through interaction of the Gab1 Met-binding domain (MBD) to a phosphorylated Y1349 residue (Weidner et al., 1996), and indirectly, through Grb2 (Lock et al., 2003; Lock et al., 2000; Nguyen et al., 1997).
To assess whether signals from the multiprotein docking site of Tpr-Met also determine the ability of FR3T3 fibroblasts to form functional invadopodia rosettes, we established stable cell lines expressing mutants of the Tpr-Met oncogene, carrying Y1349F and/or Y1356F substitutions, and quantified their ability to produce proteolytically active invadopodia rosettes on fluorescent gelatin. In comparison with Tpr-Met-transformed FR3T3 fibroblasts, substitution of Y1349 or Y1356 residues of Tpr-Met with phenylalanine led to slight decreases in the number of actin rosettes (∼20%) as well as proteolytically active invadopodia rosettes (∼30%; Fig. 3A,B). However, FR3T3 cells expressing Tpr-Met mutants with both Y1349 and Y1356 replaced by phenylalanine [Tpr-Met Y1349, Y1356F], produced considerably fewer actin rosettes (∼20% that of cells expressing WT Tpr-Met) and were unable to remodel the gelatin matrix (∼5% that of cells expressing WT Tpr-Met; Fig. 3A,B), even though this mutant Tpr-Met is catalytically active (Fixman et al., 1997; Nguyen et al., 1997). In a similar manner to parental FR3T3 fibroblasts, these cells also formed enhanced actin stress fibers (Fig. 3A,B), indicative of the reversal of the transformed phenotype.
Fig. 3.
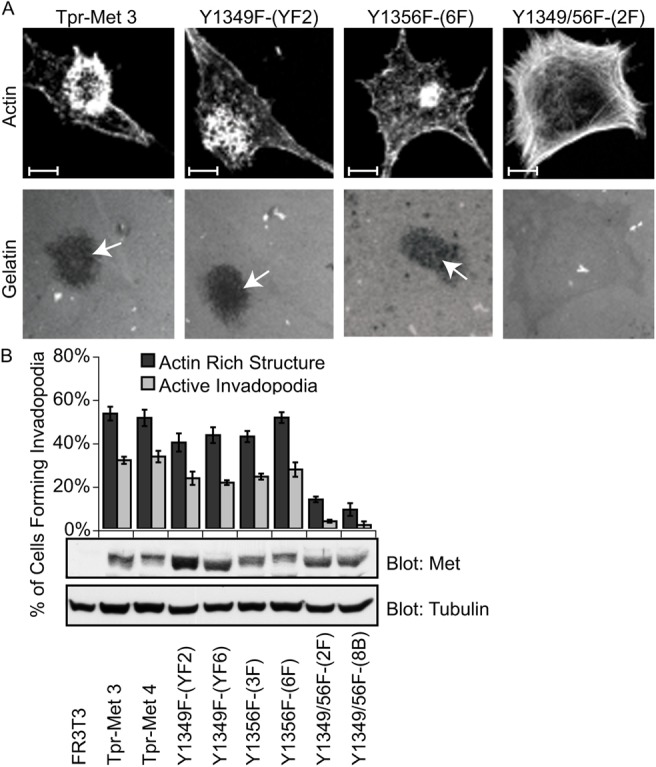
A multi-substrate docking site on Tpr-Met is required for functional invadopodia formation. (A) FR3T3 cells stably overexpressing various mutants of Tpr-Met with phenylalanine substituted for tyrosine, were cultured on gelatin-matrix-coated coverslips for 24 hours. Cells were stained with phalloidin and confocal images were acquired. Arrows indicate areas of matrix remodeling. Representative images are shown. (B) The ability of Tpr-Met mutants to promote formation of actin rosettes or active invadopodia was quantified. Values are the means of three independent experiments. SDS-PAGE was performed on lysates from cells expressing Tpr-Met mutants and probed for Met and tubulin. Scale bars: 10 μm.
Gab1 is required for invadopodia formation induced by Met in fibroblasts and cancer cells
Because both tyrosine resides, 1349 and 1356, of Tpr-Met are required for recruitment of the Gab1 scaffold (Fixman et al., 1997; Nguyen et al., 1997), we tested the requirement for Gab1 in invadopodia formation. Tpr-Met was stably expressed in immortalized mouse embryonic fibroblasts with knockout of Gab1 (Gab1−/− MEFs) (Holgado-Madruga and Wong, 2003) and the ability of these cells to form invadopodia rosettes when cultured on fluorescent gelatin matrix was examined. In the absence of Gab1, expression of Tpr-Met failed to initiate the formation of actin rosettes or matrix remodeling (Fig. 4A–C; supplementary material Movie 2). However, when expression of Gab1 was rescued by introduction of a GFP-fused variant of this scaffold protein, Gab1−/− MEFs expressing Tpr-Met acquired the ability to form proteolytically active actin rosettes, in a manner similar to Tpr-Met-transformed FR3T3 fibroblasts (Fig. 4A–C; supplementary material Movie 3, Fig. S2C,D). In addition, upon rescue of Gab1 expression in Gab1−/− MEFs, Tpr-Met signals lead to increased peripheral actin ruffles and reduced actin stress fibers (Fig. 4A).
Fig. 4.

Gab1 is required for Met-induced invadopodia formation. (A) Gab1 null cells stably overexpressing Tpr-Met (Gab1 null Tpr-Met) and rescued with GFP–Gab1 were cultured on gelatin-matrix-coated coverslips for 24 hours. Cells were stained with phalloidin and confocal images were acquired. (B) Proteins from cell extracts of Gab1 null Tpr-Met cells as well as GFP–Gab1-rescued cells were resolved by SDS-PAGE and immunoblotted for GFP, Met-P (pMet), Met and actin. (C) Gab1 null Tpr-Met cells or cells rescued with GFP–Gab1 were transiently transfected with RFP–actin and subjected to time-lapse video microscopy. One frame from each video is shown. (D) MKN45 cells were treated with 100 nM control siRNA or smartpool siRNA against Gab1 for 48 hours. Cells were trypsinized and plated on gelatin matrix for 24 hours, stained with phalloidin and confocal images were acquired. (E,F) The ability of MKN45 cells to form invadopodia in response to treatment with siRNA against Gab1 or control siRNA was quantified. An immunoblot showing the knockdown of Gab1 in MKN45 cells treated with siRNA against Gab1 but not in control siRNA-treated cells is shown. Scale bars: 10 μm.
To establish whether Gab1 expression is also crucial for Met-dependent invadopodia formation in cancer cells, we performed siRNA-mediated knockdown of Gab1 expression in MKN45 gastric cancer cells. Decreased Gab1 expression, led to a fourfold decrease in invadopodia formation (Fig. 4D–F). Together these data support that Gab1 plays an essential role in the formation of invadopodia, both in Tpr-Met transformed fibroblasts and in Met-dependent gastric cancer cells.
Proline-rich motifs of Gab1 are required for formation of functional invadopodia rosettes and invasion of Tpr-Met-transformed fibroblasts
Upon binding to a phosphorylated Met receptor, or Tpr-Met oncogene, Gab1 recruits many proteins that trigger diverse downstream signaling cascades (Birchmeier et al., 2003; Lai et al., 2009). Having established that Gab1 is crucial for biogenesis of invadopodia rosettes in Tpr-Met-transformed fibroblasts, and invadopodia in gastric carcinoma MKN45 cells, we sought to identify downstream interaction partners of Gab1 implicated in these events using a structure–function approach. For this, we rescued Gab1 expression in Gab1−/− MEFs stably expressing Tpr-Met (2B-Tpr-Met-1) using various deletion or point mutants of the Gab1 scaffold protein. In contrast to wild-type (WT) Gab1, two of the Gab1 deletion mutants, Gab1ΔMBD, lacking the Met binding domain, and Gab1ΔP4/5, missing the fourth and fifth proline-rich motifs of Gab1, failed to appreciably rescue the formation of actin rosettes in Gab1−/− MEFs expressing Tpr-Met (Fig. 5A,B,D). Instead, cells expressing Gab1ΔMBD and Gab1ΔP4/5 mutants to similar levels as WT Gab1, formed prominent actin stress fibers, and their ability to form actin rosettes were decreased by 75%, compared with cells expressing WT Gab1 (Fig. 5B,D,F).
Fig. 5.

Met-RTK-driven invadopodia biogenesis is dependent on two PxxP motifs in Gab1. (A) Schematic diagram of Gab1, indicating Met binding domain (MBD) and a proline-rich region 4/5 (P4/5) and sites of recruitment for downstream signaling proteins. PH; pleckstrin homology domain. (B) Gab1 null Tpr-Met cells rescued with either WT Gab1, Gab1ΔMBD or Gab1ΔP4/5 were cultured on gelatin matrix for 24 hours, fixed, stained with phalloidin and confocal images acquired. Arrows indicate rosettes and arrowheads indicate matrix remodeling. (C) Cells rescued with either WT Gab1 or the indicated Gab1 mutants were subjected to Boyden chamber invasion assays. Representative images are shown. (D,E) Quantification of invadopodia response (D) and Boyden chamber invasion response (E) is shown for three clones for each mutant and WT Gab1-rescued cells. Values are the means of three independent experiments. (F) SDS-PAGE was performed on lysates from the corresponding rescue cells and probed for GFP, Met-P (pMet), Met and tubulin. Scale bars: 10 μm.
Although Gab1−/− MEFs expressing Tpr-Met (2B-Tpr-Met-1) could invade reconstituted extracellular matrix (Matrigel) at a basal level, rescue of WT Gab1 expression increased the invasive capacity of these cells by fourfold (Fig. 5C,E). Furthermore, consistent with the reduced capacity of Gab1−/− Tpr-Met MEFs rescued using Gab1ΔMBD or Gab1ΔP4/5 deletion mutants to form actin rosettes, there was a more than 50% reduction in their invasive capacity, when compared with cells expressing WT Gab1 (Fig. 5C,E).
The MBD domain of Gab1 binds directly to a tyrosine-phosphorylated Met receptor, and Gab1 proline-rich motifs 4 and 5 are implicated in indirect recruitment to Met, mediated through the Grb2 adaptor protein (Lock et al., 2003). To determine whether the failure of the Gab1ΔP4/5 or Gab1ΔMBD deletion mutants to support formation of actin rosettes and rescue invasiveness of Gab1−/− Tpr-Met MEFs is due to their inability to bind Tpr-Met, we performed reciprocal immunoprecipitations of Tpr-Met or Gab1 mutants in HEK 293 cells, and examined co-immunoprecipitates for the presence of Gab1 and Tpr-Met, respectively. Whereas Gab1ΔMBD failed to bind Tpr-Met, Gab1ΔP4/5 was recruited, although its tyrosine phosphorylation was reduced by 20% (Fig. 6A–E). Notably, FR3T3 fibroblasts, transformed by a Tpr-Met mutant (N1358H), specifically uncoupled from Grb2, formed actin rosettes to a similar level as cells expressing WT Tpr-Met, indicating that Grb2-dependent recruitment of Gab1 to the Met receptor is dispensable for actin rosette formation (Fig. 6H–J).
Fig. 6.

Gab1ΔP4/5 is recruited to Tpr-Met and localizes to invadopodia but fails to initiate actin rosette formation. (A,D) HEK 293 cells were transiently transfected with the indicated constructs; proteins from lysates were immunoprecipitated with Met 147 antibody (A) or HA antibody (D) and probed as indicated using the Odessey detection system (Li-Cor). (B,E) Densitometric analysis of western blots was performed using Odessey software and tyrosine phosphorylation of Gab1 mutants. Their ability to be recruited to Met is depicted as a percentage of WT Gab1. Values are the means of three independent experiments. (C) Proteins from lysates of Gab-null Tpr-Met cells rescued with GFP–Gab1 (WT) or GFP–Gab1ΔP4/5 were immunoprecipitated with GFP antibody and probed as indicated. (F) FR3T3 Tpr-Met cells were transiently transfected with RFP-actin and GFP-Gab1ΔP4/5 and subjected to time-lapse video microscopy. One frame depicting Gab1ΔP4/5 localization to an actin rosette is shown. (G) FR3T3 Tpr-Met cells were transiently transfected with GFP-Gab1ΔP4/5, plated on gelatin matrix for 24 hours and stained for cortactin and GFP–Gab1. (H) FR3T3 cells stably overexpressing a Tpr-Met mutant that is specifically uncoupled from Grb2 (N1358H) were plated on gelatin matrix for 24 hours and stained for invadopodia markers actin (phalloidin) and cortactin. (I) Ability of Tpr-Met N1358H mutants to induce actin rosette was assessed. Values are the means of two independent clones (FR3T3 Tpr-Met-N1358H-1 and FR3T3 Tpr-Met-N1358H-2). Levels of Tpr-Met were assessed by western blotting. (J) Tpr-Met was immunoprecipitated from lysates prepared from FR3T3 Tpr-Met-N1358H-1 and FR3T3 Tpr-Met-N1358H-2 clones as well as from FR3T3 Tpr-Met 3 cells using Met 147 antibody. SDS-PAGE was performed and proteins immunoblotted as indicated. Scale bars: 10 μm.
Given that Gab1ΔP4/5 recruitment to Met is not impaired, we examined the ability of the Gab1ΔP4/5 mutant to localize to invadopodia. For this, we performed both confocal analysis on fixed cells and time-lapse video microscopy analysis of FR3T3-Tpr-Met cells that form actin rosettes and examined the ability GFP-Gab1 WT or GFP-Gab1ΔP4/5 to localize to these structures. We observed that GFP-Gab1ΔP4/5 localized to the actin rosettes in a similar manner to GFP–Gab1WT (Fig. 6F,G; supplementary material Movies 4, 5). Together, these results substantiate that the formation of actin rosettes in Tpr-Met-transformed fibroblasts is dependent on assembly of multiprotein complexes that involve Tpr-Met, Gab1, and possibly other protein(s) recruited through the fourth and/or fifth proline-rich motifs of Gab1.
Proline-rich motifs of Gab1 are involved in interaction with cortactin
Previous studies have identified the adaptor protein cortactin as a key component of invadopodia, both in cancer cells and oncogenically transformed fibroblasts (Bowden et al., 1999; Bowden et al., 2006; Clark and Weaver, 2008; Clark et al., 2007; Cortesio et al., 2008; Cosen-Binker and Kapus, 2006; DesMarais et al., 2009; Oser et al., 2009). Cortactin is thought to promote invadopodia formation by association with actin regulatory proteins Nck, N-WASP and Arp2/3, which initiate actin polymerization (DesMarais et al., 2009; Murphy and Courtneidge, 2011; Yamaguchi et al., 2005).
The fourth and fifth proline-rich motifs of Gab1, which are crucial for formation of actin rosettes in Tpr-Met transformed fibroblasts, are similar to xPPxPxKPx consensus sequences recognized by the SH3 domain of cortactin (Rubini et al., 2010; Sparks et al., 1996). This raises the possibility that Gab1 serves as a scaffold to recruit cortactin to sites of emerging actin rosette or vice versa. To test this hypothesis, we performed reciprocal immunoprecipitation of Gab1 and cortactin and detected co-immunoprecipitation between the two proteins when either transiently expressed in HEK 293 cells (Fig. 7A,B) or between endogenous Gab1 and cortactin in BT549 cells (Fig. 7H).
Fig. 7.

Cortactin interacts with Gab1 through its SH3 domain with proline-rich regions on Gab1. HEK 293 cells were transiently transfected with the indicated constructs; proteins from lysates were immunoprecipitated with HA antibody (A,D,E) or cortactin antibody (B) and immunoblotted as indicated. (C) Schematic diagram depicting a potential interaction of the cortactin SH3 domain and Gab1 proline-rich consensus motifs. (F,G) FR3T3 cells expressing Tpr-Met were transiently transfected with GFP-Gab1 and then plated on gelatin matrix for 24 hours. Cells were stained for GFP, cortactin and phalloidin. (F) X–Z and Y–Z projection of a 40 Z-stack showing the localization of Gab1 and cortactin to membrane protrusions (arrows). (H) Endogenous Gab1 was immunoprecipitated from BT549 cells and probed for cortactin. (I) GST–cortactin SH3 or GST–cortactin SH3-W525K mutant fusion proteins were coupled to GST beads and used to pull down proteins from lysates of HEK 293 cells transiently expressing HA–Gab1. (J) Proteins from HEK 293 cells, transfected with GFP-Gab1 or HA-dynamin, were immunoprecipitated with either HA or GFP antibody. The immune complex was separated by SDS-PAGE and transferred to a nitrocellulose membrane. Nitrocellulose membranes were incubated with fusion proteins of either cortactin GST–SH3 domain or GST–SH3-W525K and immunoblotted as indicated. Scale bars: 10 μm.
By undertaking a structure–function analysis in HEK 293 cells, we established that Gab1-cortactin interaction is dependent on the SH3 domain of cortactin (Fig. 7E,C). Notably, WT cortactin was efficiently co-immunoprecipitated with Gab1, whereas both a cortactin mutant lacking the SH3 domain (cortactinΔSH3) and cortactin with a point mutation rendering the SH3 domain unable to bind consensus proline-rich sequences (cortactin W525K), failed to bind Gab1 (Fig. 7E,C). Additionally, in reciprocal experiments, cortactin co-immunoprecipitated efficiently with WT Gab1, and could interact, although to a lesser extent, with either Gab1ΔP4 or Gab1ΔP5 mutants alone, but failed to interact with the Gab1ΔP4/5 mutant (Fig. 7D). Gab1 was efficiently pulled down from cell lysates by a GST fusion protein containing the SH3 domain of cortactin, but not by a fusion protein expressing the cortactin SH3 W525K mutant (Fig. 7I), indicating that an intact SH3 domain of cortactin is both necessary and sufficient for interaction with Gab1. Although the interaction between cortactin and Gab1 requires the SH3 domain of cortactin and the proline-rich motifs of Gab1, it does not exclude the possibility that this interaction is mediated by an intermediate. To confirm that this interaction was indeed direct, we performed far-western blotting of Gab1, and a known cortactin SH3 binding partner dynamin as positive control, with the GST-fused cortactin SH3 or SH3 W525K mutant. As expected dynamin interacted with GST–cortactin SH3 domain fusion protein but failed to interact with GST–cortactin SH3-W525K mutant fusion protein (Fig. 7J). In a similar manner, Gab1 bound to the intact SH3 domain of cortactin but failed to interact with the SH3 W525K mutant, indicating that the interaction between the proline-rich motifs of Gab1 and SH3 domain of cortactin is direct (Fig. 7C,J).
Cortactin colocalizes with Gab1 in Tpr-Met-transformed fibroblasts and is required for formation of invadopodia rosettes
Recently we demonstrated that in the process of circular dorsal ruffle formation in response to Met activation, Gab1 served as a scaffold protein linking activated Met to nucleation of branched actin filaments, by recruiting the adaptor protein Nck and actin nucleation promoting factor, N-WASP (Abella et al., 2010). In order to understand whether a Gab1–cortactin complex could play a similar role within invadopodia rosettes, we examined the colocalization of Gab1 and cortactin in Tpr-Met-transformed fibroblasts. Both Gab1 and cortactin were found to colocalize at actin rosettes, representative of proteolytically active invadopodia rosettes (Fig. 7F).
The ability of Tpr-Met-transformed fibroblasts to produce proteolytically active invadopodia rosettes is strongly linked with the potential to produce ventral actin rosettes (Fig. 1A,B). Hence both proteolytically active and non-degrading actin rosettes probably represent a similar cellular structure at different stages of formation. Using this advantage, we further validated the importance of cortactin for assembly of Tpr-Met-dependent invadopodia rosettes. Knockdown of cortactin expression in Tpr-Met-transformed FR3T3 cells, using two different siRNA duplexes, led to more prominent actin stress fibers and a 50% decrease in the formation of actin rosettes, as compared with control, Tpr-Met-transformed FR3T3 cells transfected with non-targeting siRNA duplexes (supplementary material Fig. S3A,B). Furthermore, in each case, the remaining actin rosettes observed in Tpr-Met-transformed FR3T3 fibroblasts were only partially formed, compared with cells treated with non-targeting siRNAs (supplementary material Fig. S3A). In summary, these experiments substantiate Gab1 and cortactin as components of a protein complex that determines the capacity of Tpr-Met-transformed fibroblasts to form invadopodia rosettes and remodel ECM.
Met RTK localizes to invadopodia and promotes tyrosine phosphorylation of cortactin
So far, only non-receptor tyrosine kinases, Src, Arg and Abl, have been shown to localize to invadopodia. Given that Met was found at other structurally related actin-rich structures, such as circular dorsal ruffles, we sought to explore the possibility that Met could localize to invadopodia. Indeed, in MKN45 cells, which show constitutive Met activity and produce cortactin-positive actin-rich protrusions that remodel the underlying matrix, Met localized to actively degrading invadopodia protrusions, as defined by colocalization with cortactin and degraded fluorescent gelatin areas (Fig. 8A). Given the observed colocalization of Met receptor and cortactin at invadopodia and the role of tyrosine phosphorylation of cortactin in regulation of invadopodia dynamics, we examined whether Met could also promote phosphorylation of cortactin on tyrosine residues. When expressed in HEK 293 cells without additional stimuli, cortactin was not detectably tyrosine phosphorylated; however, co-expression with Tpr-Met was sufficient to trigger its strong tyrosine phosphorylation of cortactin (Fig. 8B). In a similar fashion, in MKN45 cells, where Met is amplified and constitutively active, cortactin was tyrosine phosphorylated (Fig. 8C), which was abolished upon treatment of the cells with the Met inhibitor PHA665752, suggesting that tyrosine phosphorylation of cortactin in these cancer cells is dependent on Met kinase activity (Fig. 8C). Intriguingly, it has been shown previously that cortactin is a substrate for the non-receptor tyrosine kinase Src, and that cortactin phosphorylation on tyrosine is triggered by Src activity. However, in MKN45 cells, inhibition of Met RTK does not detectably alter Src activity (Fig. 8C). Moreover, treatment of MKN45 cells with Src inhibitors PP2 or SU6656 or the Abl inhibitor Imatinib, or in combinations, had little to no effect on cortactin tyrosine phosphorylation (Fig. 8C) indicating that in MKN45 gastric cancer cells Src or Abl kinase activity is not essential to trigger tyrosine phosphorylation of cortactin, whereas Met activation is required. Additionally, tyrosine phosphorylation of cortactin in MKN45 cells treated with inhibitors of Src, Abl or Met, correlate with the ability of treated cells to form invadopodia, with Met inhibition exerting the strongest impact (Fig. 8D–F). Taken together, these data support the proposal that Met can promote tyrosine phosphorylation of cortactin and invadopodia biogenesis independently of Src.
Fig. 8.

Met colocalizes with cortactin to invadopodia, and cortactin tyrosine phosphorylation is highly dependent on Met kinase activity. (A) MKN45 cells were plated on gelatin matrix for 24 hours and stained for cortactin and Met. The boxed regions are shown enlarged in the insets. (B) HEK 293 cells were transfected with the indicated constructs and the cell extracts were immunoprecipitated with cortactin antibody, 4F11. Immune complexes were separated by SDS-PAGE and probed as indicated. (C) MKN45 cells were treated with 10 µM PP2, 10 µM SU6656, 10 µM Imatinib, 0.1 µM PHA665752 or vehicle (DMSO). Cell extracts were separated by SDS-PAGE and probed as indicated or immunoprecipitated using anti-cortactin (4F11) or anti-Crk antibodies. Immune complexes were separated by SDS-PAGE and probed as indicated. (D) MKN45 cells were plated on gelatin matrix in the presence of 10 µM PP2, 10 µM SU6656, 10 µM Imatinib, 0.1 µM PHA665752 or vehicle (DMSO) for 24 hours and stained with phalloidin. Representative images are shown. (E,F) The ability of MKN45 cells to form invadopodia in the presence of PP2, SU6656, Imatinib or PHA665752 was quantified. Values are the mean of three independent experiments. Scale bars: 10 μm.
Discussion
It is becoming increasingly evident that the invasive capacity of cancer cells is often determined by their ability to assemble invadopodia, actin-rich protrusive cellular structures that mediate focal ECM remodeling (Gimona, 2008; Buccione et al., 2009; Murphy and Courtneidge, 2011). Invadopodia are produced predominantly by cancer cells, and with some exceptions, are absent in normal cells, and hence might serve as a feasible specific target for first-line anti-metastatic therapies. Despite this attractive therapeutic possibility, molecular mechanisms that lead to invadopodia biogenesis in cancer remain largely undefined.
Research performed so far has attributed induction of invadopodia and invadopodia-like structures mostly to oncogenic activation of the non-receptor tyrosine kinase Src in fibroblasts (Hauck et al., 2002; Bowden et al., 2006; Webb et al., 2007; Oikawa et al., 2008; Balzer et al., 2010; Kelley et al., 2010; Murphy and Courtneidge, 2011), as well as Abl and Arg kinases (Smith-Pearson et al., 2010; Mader et al., 2011), and growth factor receptor tyrosine kinases, including EGFR (Kimura et al., 2010; Mader et al., 2011) and PDGFRα (Eckert et al., 2011).
Here we report for the first time that signals from an activated Met RTK, which is implicated in cancer invasiveness (Birchmeier et al., 2003; Peschard and Park, 2007), can induce invadopodia rosettes in fibroblasts, and increase invadopodia biogenesis in human cancer cells. We demonstrate that upon transformation with a constitutively active oncogenic variant of the Met receptor, Tpr-Met, fibroblasts acquire the ability to form ventral proteolytically active actin rosettes (Fig. 1). In accordance with a recently proposed nomenclature (Murphy and Courtneidge, 2011), we refer to this structure as an ‘invadopodia rosette’. Importantly, we also show that activation of Met, either through engagement with the ligand, HGF, or as a result of genomic amplification of the MET locus, increases invadopodia biogenesis in basal-like breast cancer and gastric carcinoma cells (Fig. 2). These observations are physiologically relevant. Increase in plasma HGF levels, as well as elevated levels of Met expression and activity, including constitutive activation resulting from gene amplification, are linked with increased cancer invasiveness and metastasis, and correlate with poor prognosis in many types of cancer, including basal-like breast cancers (Camp et al., 1999; Lengyel et al., 2005; Ponzo et al., 2009) and gastric carcinomas (Kammula et al., 2007; Tuynman et al., 2008; Wu et al., 1998).
Invadopodia observed in basal breast cancer MDA-MB-231 cells upon HGF stimulation (Fig. 2) morphologically resemble those formed in response to epidermal growth factor (EGF) activation of the EGFR (Oser et al., 2009; Mader et al., 2011), suggesting that a similar molecular machinery promoting invadopodia formation is be driven by multiple upstream signals. By contrast, in gastric carcinoma MKN45 cells, which carry genomic amplification of MET, formation of invadopodia is abolished by Met inhibition or substantially decreased following Met knockdown (Fig. 2) indicating that in Met-addicted cancer cells, the Met signal is the major upstream driver of invadopodia biogenesis.
Sequential signals are involved in assembly of functional invadopodia (Oikawa et al., 2008). These involve initial signals that trigger the establishment of precursor actin-rich membrane protrusions, followed by signals that regulate targeted secretion of metalloproteases for ECM remodeling known as invadopodia maturation (Artym et al., 2006; Murphy and Courtneidge, 2011). In many cell types the formation of invadopodia in response to Met signaling is dependent on the scaffold protein Gab1 (Fig. 4). In Gab1−/− fibroblasts, Tpr-Met fails to induce formation of invadopodia rosettes and this phenotype is rescued by expression of Gab1 (Fig. 4; supplementary material Movies 2,3). SiRNA-mediated knockdown of Gab1 also leads to decreased Met-dependent invadopodia biogenesis in MKN45 gastric carcinoma cells (Fig. 4). Following Gab1 knockdown in MKN45 cells or in fibroblasts expressing a Tpr-Met mutant unable to recruit Gab1 Y1349F/Y1356FY, the inability to produce invadopodia and/or actin rosettes (Figs 3, 4) is due to decreased assembly of actin-rich core structures (supplementary material Movies 2,3), consistent with a role for Met-Gab1 signaling in the regulation of an early step in invadopodia formation. This supports previous reports that Gab1 plays a role in Met-dependent regulation of other actin-rich cellular structures, such as lamellipodia (Frigault et al., 2008) and circular dorsal ruffles (Abella et al., 2010). Hence, depending on the cellular context, Gab1 provides a common link between Met signals and assembly of actin-rich structures at the cell periphery.
Using a structure–function approach we have identified the requirement of two proline-rich motifs of Gab1 (P4/5) for actin rosette formation and have shown that these provide direct binding sites for the SH3 domain of cortactin, a protein involved in actin dynamics (Figs 5, 7). The P4/5 motifs also bind the Grb2 adaptor protein, which indirectly recruits Gab1 to Met (Lock et al., 2002). Given that direct recruitment of Grb2 to Met is dispensable for actin rosette formation (Fig. 6), whereas the Gab1ΔP4/5 mutant itself is still recruited to and phosphorylated by Tpr-Met (Fig. 6), but fails to rescue assembly of actin rosettes in Gab1−/− fibroblasts, we propose that a Gab1–cortactin interaction mediated through proline-rich motifs of Gab1 is required for Met-dependent invadopodia formation (Fig. 5).
Cortactin recruitment is essential for invadopodia formation downstream from multiple signals. In one model, invadopodia assembly in response to EGF is initiated at early precursors that contain cortactin, N-WASP and Arp2/3 (Oser et al., 2009; Artym et al., 2006). Recruitment of cortactin to the early precursors is thought to be mediated through the Arp2/3 complex (Uruno et al., 2001) and subsequent phosphorylation of cortactin on tyrosine residues promotes release and activation of cofilin, resulting in actin severing and increased barbed end formation (Oser et al., 2009). A second model proposes that the podosome, in Src transformed fibroblasts, is initiated by the accumulation of PtdIns(3,4)P2 in the vicinity of existing focal adhesions, and by subsequent recruitment of Tks5 scaffold protein (Oikawa et al., 2008; Murphy and Courtneidge, 2011). Clustering of N-WASP on Tks5 (Oikawa et al., 2008; Oikawa et al., 2009) and recruitment of cortactin (Crimaldi et al., 2009) is responsible for actin nucleation during initiation of actin rosettes. Interestingly, Gab1 interacts with N-WASP and cortactin and localizes to actin-rich circular dorsal ruffles (Abella et al., 2010) as well as invadopodia in response to Met activation and might thus function in a similar manner to Tks5 to promote the assembly of actin rosette.
Met promotes robust tyrosine phosphorylation of cortactin (Fig. 8) (Crostella et al., 2001). Hence one possible role for a Gab1–cortactin complex at invadopodia precursors is to promote localized Met–dependent tyrosine phosphorylation of cortactin, leading to increased branched actin nucleation and actin rosette assembly. In support of this, Gab1 and Met localize to cortactin-positive matrix remodeling invadopodia (Fig. 8). To date Met is the only RTK shown to localize to invadopodia. Hence Met could promote actin nucleation, not only through phosphorylation-dependent regulation of cortactin, but also by influencing activity of other invadopodia-associated proteins. In support of this, Src activity is not essential for invadopodia formation in Tpr-Met-transformed fibroblasts or Met-addicted MKN45 carcinoma cells (supplementary material Fig. S4; Fig. 8). Interestingly, overexpression of cortactin in human non-small cell lung cancer (HNSCLC) cells is associated with acquired resistance to treatment with EGFR inhibitors, a phenomenon that is often observed in HNSCLC and other cancer cells as a result of genomic amplification of MET and increased Met activation (Kosaka et al., 2011; Lai et al., 2009). Overexpression of cortactin in HNSCLC cells has also been linked to attenuated Met receptor downregulation and augmented Met-mediated biological responses (Timpson et al., 2007), further supporting a physiological role for Met–cortactin functional interaction.
In summary, in this study we have identified a Met signaling axis as an important determinant of invadopodia biogenesis. We have demonstrated that Gab1, one of the main scaffold proteins recruited to active Met, is crucial for the formation of Met-dependent invadopodia, by regulating assembly of their actin rosettes. Our study highlights Met–Gab1 signaling as an alternative target for therapeutic disruption of these structures in invasive cancer cells.
Materials and Methods
Cell culture and cDNA transfections
FR3T3, HEK 293, MEFs and MDA231 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). MKN45 cells were maintained in RPMI containing 10% FBS. The generation of FR3T3 cell lines stably overexpressing the WT Tpr-Met and Tpr-Met mutants has been previously described by Saucier et al. and Fixman et al. GFP-tagged WT and mutant Gab1 were generated as described in Abella et al. (Saucier et al., 2002; Fixman et al., 1997; Abella et al., 2010). Tpr-Met was cloned into pLXSH vector and stably expressed in MEFs by retroviral infection. Cell lines were selected in 0.5 mg/ml hygromysin or 2 µg/ml of puromysin. pCDNA-cortactin WT and the mutants were described previously (Stuible et al., 2008). DsRed–cortactin was a kind gift from Mark A. McNiven (Mayo Clinic, Rochester, MN). For transient transfection assays, HEK 293 cells were transfected with Lipofectamine Plus reagent and MEFs and FR3T3 cells were transfected with Lipo2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). Cells were used for biochemical assays 24 hours post-transfection.
siRNA knockdown
Human Gab1- and Met-specific siRNAs were purchased from Dhamacon Inc. (Lafayette, CO) as a smartpool containing four different oligonucleotides: siGENOME, SMARTpool M-003553, Human Gab1 and siGENOME SMARTpool L-003156, Human Met; siRNAs were transfected using Hiperfect transfection reagent at 100 nM and 50 nM concentrations, respectively. After 48 hours, cells were trypsinized and plated for biological assays or for western blotting for an additional 24 hours. Rat cortactin-specific siRNA sequences, S102732177 and S100169386, were purchased from Qiagen (Valencia, CA) and transfected at 50 nM concentration using Hiperfect transfection reagent. After 48 hours, cells were trypsinized and plated for biological assays or for western blot analysis for an additional 24 hours.
Antibodies and reagents
Antibody 147 was raised against a C-terminal peptide of the human Met protein (Maroun et al., 1999; Rodrigues et al., 1991). Tks5 antibody (1736) was a kind gift from Sara Courtneidge (Sanford-Burnham Medical Research Institute, CA). Commercial antibodies: Gab1 and cortactin 4F11 were from Upstate Biotechnology (Lake Placid, NY), pan phospho tyrosine (pTYR-100) and pMet1234/35 (phosphorylated Met;Met-p) were from Cell Signaling Technologies (Danvers, MA), actin was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), Tubulin was from Sigma (St Louis, MO), GFP antibody, Phalloidin Alexa Fluor 488, 546 and 647, and Alexa-Fluor-488-, 555- and 647-conjugated secondary antibodies were from Molecular Probes (Eugene, OR), HA.11 monoclonal antibody was from Covance (Berkeley, CA). Met inhibitor PHA665752 was a kind gift from Pfizer Inc. (New York, NY). Src inhibitors PP2 and Su6656 were purchased from EMD Chemicals (Gibbstown, NJ) and Sigma-Aldrich, respectively. HGF was a generous gift from Genentech (San Francisco, USA).
Immunoprecipitation and western blotting
Cells were harvested in T&D lysis buffer (150 mM NaCl, 20 nM Tris-HCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1% deoxycholate, pH 7.4). All lysis buffers were supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM sodium vanadate, 1 mM sodium fluoride, 10 µg/ml aprotinin and 10 µg/ml leupeptin. For immunoprecipitation, lysates (1 mg) were incubated with the indicated antibodies for 2 hours at 4°C with gentle rotation. To collect immune complexes, 25 µl of 50% slurry of either protein-A– or protein-G–Sepharose was added for an additional hour. The immune complex was washed three times in the lysis buffer and resolved by SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blocked in 3% BSA in TBST (10 mM Tris pH 8.0, 150 mM NaCl, 2.5 mM EDTA, 0.1% Tween 20) for 1 hour, incubated with primary and secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) in TBST for 2 hours and 1 hour, respectively. After four washes with TBST, bound proteins were visualized with an ECL detection kit (Amersham Biosciences) or Odessey detection system (Li-Cor, Lincoln, NE). Densitometric analysis of western blots was performed using Odyssey software.
Fluorescent gelatin degradation assay
Coverslips were coated with poly-D lysine for 20 minutes, washed three times with PBS, incubated for 20 minutes in 0.4% glutaraldehyde (Sigma-Aldrich) and washed three times with PBS. Oregan-Green-conjugated gelatin (Invitrogen) was diluted to 20 µg/ml in 0.1% unconjugated gelatin (Stem Cell Technologies, Vancouver, BC, Canada) and incubated on the coverslips at 37°C for 1 hour, washed three times with PBS and quenched with 70% ethanol for 20 minutes. Finally Oregan-Green-conjugated gelatin-coated coverslips were washed with DMEM, and 50,000 cells were plated and incubated at 37°C in 5% CO2 for 24 hours unless specified otherwise. Cells were fixed with 4% paraformaldehyde and continued with regular immunofluorescence protocol, as described previously (Abella et al., 2010). Samples were mounted using Immunomount from Thermo Scientific (Pittsburgh, PA) and images were acquired using a 100× objective on a confocal microscope, unless mentioned otherwise.
Live cell imaging
Cells grown on gelatin-coated glass coverslips (35 mm) were positioned on the motorized stage on the Axiovert 200 M (Carl Zeiss, Inc.) inverted microscope, equipped with 100× plan Apochromat NA 1.4 objective and an AxioCam HRM digital camera, and equipped with a small transparent environmental chamber, Climabox (Carl Zeiss, Inc.) with 5% (v/v) CO2 in air at 37°C. The microscope was driven by AxioVision LE software (Carl Zeiss, Inc.). The motorized stage advanced to pre-programmed locations and photographs were collected for 30 minutes.
Far-western blotting
HEK 293 cells were transiently transfected with the indicated constructs, immunoprecipitated and separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with either cortactin GST–SH3 or GST–SH3–W525K fusion proteins in lysis buffer A (20 mM Hepes pH 7.5, 120 mM NaCl, 2 mM EDTA, 10% glycerol, 1 mM PMSF, 10 µg/ml aprotinin and 10 µg/ml leupeptin) and bound GST-fused proteins were detected using anti-GST antibodies.
Supplementary Material
Acknowledgments
We thank members of the Park lab for their helpful comments on the manuscript. We thank Claire Brown and Aleksandrs Spurmanis for their help with deconvolution of confocal Z-stacks using IMARIS software. We thank Ken McDonald for his help with FACS. We would like to thank Genentech Inc. for HGF and Marina Holgado-Madruga for Gab1-null cells.
Footnotes
Funding
This work was supported by a fellowship from Canadian Institutes of Health Research [grant number CGD-96470 to C.V.R.]; the US Department of Defense Breast Cancer Research Initiative [grant number XWH-09-1-00 to R.V.]; Canadian Institutes of Health Research/Fonds de la Recherche en Santé du Québec training grant in cancer research from the McGill Integrated Cancer Research Training Program [grant number FRN53888 to S.H. and K.Z.]; and by an operating grant from the Canadian Institutes of Health Research [grant number MOP-106635 to M.P.]. M.P. holds the Diane and Sal Guerrera Chair in Cancer Genetics. Deposited in PMC for immediate release.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.100834/-/DC1
References
- Abella J. V., Vaillancourt R., Frigault M. M., Ponzo M. G., Zuo D., Sangwan V., Larose L., Park M. (2010). The Gab1 scaffold regulates RTK-dependent dorsal ruffle formation through the adaptor Nck. J. Cell Sci. 123, 1306–1319 10.1242/jcs.062570 [DOI] [PubMed] [Google Scholar]
- Artym V. V., Zhang Y., Seillier–Moiseiwitsch F., Yamada K. M., Mueller S. C. (2006). Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 66, 3034–3043 10.1158/0008-5472.CAN-05-2177 [DOI] [PubMed] [Google Scholar]
- Ayala I., Giacchetti G., Caldieri G., Attanasio F., Mariggiò S., Tetè S., Polishchuk R., Castronovo V., Buccione R. (2009). Faciogenital dysplasia protein Fgd1 regulates invadopodia biogenesis and extracellular matrix degradation and is up-regulated in prostate and breast cancer. Cancer Res. 69, 747–752 10.1158/0008-5472.CAN-08-1980 [DOI] [PubMed] [Google Scholar]
- Balzer E. M., Whipple R. A., Thompson K., Boggs A. E., Slovic J., Cho E. H., Matrone M. A., Yoneda T., Mueller S. C., Martin S. S. (2010). c-Src differentially regulates the functions of microtentacles and invadopodia. Oncogene 29, 6402–6408 10.1038/onc.2010.360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuti S., Comoglio P. M. (2007). The MET receptor tyrosine kinase in invasion and metastasis. J. Cell. Physiol. 213, 316–325 10.1002/jcp.21183 [DOI] [PubMed] [Google Scholar]
- Birchmeier C., Birchmeier W., Gherardi E., Vande Woude G. F. (2003). Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 4, 915–925 10.1038/nrm1261 [DOI] [PubMed] [Google Scholar]
- Blouw B., Seals D. F., Pass I., Diaz B., Courtneidge S. A. (2008). A role for the podosome/invadopodia scaffold protein Tks5 in tumor growth in vivo. Eur. J. Cell Biol. 87, 555–567 10.1016/j.ejcb.2008.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden E. T., Barth M., Thomas D., Glazer R. I., Mueller S. C. (1999). An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene 18, 4440–4449 10.1038/sj.onc.1202827 [DOI] [PubMed] [Google Scholar]
- Bowden E. T., Onikoyi E., Slack R., Myoui A., Yoneda T., Yamada K. M., Mueller S. C. (2006). Co-localization of cortactin and phosphotyrosine identifies active invadopodia in human breast cancer cells. Exp. Cell Res. 312, 1240–1253 10.1016/j.yexcr.2005.12.012 [DOI] [PubMed] [Google Scholar]
- Buccione R., Caldieri G., Ayala I. (2009). Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 28, 137–149 10.1007/s10555-008-9176-1 [DOI] [PubMed] [Google Scholar]
- Camp R. L., Rimm E. B., Rimm D. L. (1999). Met expression is associated with poor outcome in patients with axillary lymph node negative breast carcinoma. Cancer 86, 2259–2265 [DOI] [PubMed] [Google Scholar]
- Chaffer C. L., Weinberg R. A. (2011). A perspective on cancer cell metastasis. Science 331, 1559–1564 10.1126/science.1203543 [DOI] [PubMed] [Google Scholar]
- Chen W. T. (1989). Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J. Exp. Zool. 251, 167–185 10.1002/jez.1402510206 [DOI] [PubMed] [Google Scholar]
- Christensen J. G., Schreck R., Burrows J., Kuruganti P., Chan E., Le P., Chen J., Wang X., Ruslim L., Blake R., et al. (2003). A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 63, 7345–7355 [PubMed] [Google Scholar]
- Clark E. S., Weaver A. M. (2008). A new role for cortactin in invadopodia: regulation of protease secretion. Eur. J. Cell Biol. 87, 581–590 10.1016/j.ejcb.2008.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E. S., Whigham A. S., Yarbrough W. G., Weaver A. M. (2007). Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 67, 4227–4235 10.1158/0008-5472.CAN-06-3928 [DOI] [PubMed] [Google Scholar]
- Corso S., Comoglio P. M., Giordano S. (2005). Cancer therapy: can the challenge be MET? Trends Mol. Med. 11, 284–292 10.1016/j.molmed.2005.04.005 [DOI] [PubMed] [Google Scholar]
- Cortesio C. L., Chan K. T., Perrin B. J., Burton N. O., Zhang S., Zhang Z. Y., Huttenlocher A. (2008). Calpain 2 and PTP1B function in a novel pathway with Src to regulate invadopodia dynamics and breast cancer cell invasion. J. Cell Biol. 180, 957–971 10.1083/jcb.200708048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosen–Binker L. I., Kapus A. (2006). Cortactin: the gray eminence of the cytoskeleton. Physiology (Bethesda) 21, 352–361 10.1152/physiol.00012.2006 [DOI] [PubMed] [Google Scholar]
- Crimaldi L., Courtneidge S. A., Gimona M. (2009). Tks5 recruits AFAP-110, p190RhoGAP, and cortactin for podosome formation. Exp. Cell Res. 315, 2581–2592 10.1016/j.yexcr.2009.06.012 [DOI] [PubMed] [Google Scholar]
- Crostella L., Lidder S., Williams R., Skouteris G. G. (2001). Hepatocyte Growth Factor/scatter factor-induces phosphorylation of cortactin in A431 cells in a Src kinase-independent manner. Oncogene 20, 3735–3745 10.1038/sj.onc.1204474 [DOI] [PubMed] [Google Scholar]
- Cruz J., Reis–Filho J. S., Silva P., Lopes J. M. (2003). Expression of c-met tyrosine kinase receptor is biologically and prognostically relevant for primary cutaneous malignant melanomas. Oncology 65, 72–82 10.1159/000071207 [DOI] [PubMed] [Google Scholar]
- Cunnick J. M., Mei L., Doupnik C. A., Wu J.2001). Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J. Biol. Chem. 27624380–24387 10.1074/jbc.M010275200 [DOI] [PubMed] [Google Scholar]
- DesMarais V., Yamaguchi H., Oser M., Soon L., Mouneimne G., Sarmiento C., Eddy R., Condeelis J. (2009). N-WASP and cortactin are involved in invadopodium-dependent chemotaxis to EGF in breast tumor cells. Cell Motil. Cytoskeleton 66, 303–316 10.1002/cm.20361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert M. A., Lwin T. M., Chang A. T., Kim J., Danis E., Ohno–Machado L., Yang J. (2011). Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 19, 372–386 10.1016/j.ccr.2011.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fixman E. D., Naujokas M. A., Rodrigues G. A., Moran M. F., Park M. (1995). Efficient cell transformation by the Tpr-Met oncoprotein is dependent upon tyrosine 489 in the carboxy-terminus. Oncogene 10, 237–249 [PubMed] [Google Scholar]
- Fixman E. D., Fournier T. M., Kamikura D. M., Naujokas M. A., Park M. (1996). Pathways downstream of Shc and Grb2 are required for cell transformation by the tpr-Met oncoprotein. J. Biol. Chem. 271, 13116–13122 10.1074/jbc.271.22.13116 [DOI] [PubMed] [Google Scholar]
- Fixman E. D., Holgado–Madruga M., Nguyen L., Kamikura D. M., Fournier T. M., Wong A. J., Park M. (1997). Efficient cellular transformation by the Met oncoprotein requires a functional Grb2 binding site and correlates with phosphorylation of the Grb2-associated proteins, Cbl and Gab1. J. Biol. Chem. 272, 20167–20172 10.1074/jbc.272.32.20167 [DOI] [PubMed] [Google Scholar]
- Fournier T. M., Kamikura D., Teng K., Park M. (1996). Branching tubulogenesis but not scatter of madin-darby canine kidney cells requires a functional Grb2 binding site in the Met receptor tyrosine kinase. J. Biol. Chem. 271, 22211–22217 10.1074/jbc.271.36.22211 [DOI] [PubMed] [Google Scholar]
- Frigault M. M., Naujokas M. A., Park M. (2008). Gab2 requires membrane targeting and the Met binding motif to promote lamellipodia, cell scatter, and epithelial morphogenesis downstream from the Met receptor. J. Cell. Physiol. 214, 694–705 10.1002/jcp.21264 [DOI] [PubMed] [Google Scholar]
- Fushida S., Yonemura Y., Urano T., Yamaguchi A., Miyazaki I., Nakamura T., Shiku H. (1993). Expression of hepatocyte growth factor(hgf) and C-met gene in human gastric-cancer cell-lines. Int. J. Oncol. 3, 1067–1070 [DOI] [PubMed] [Google Scholar]
- Garcia–Guzman M., Dolfi F., Zeh K., Vuori K. (1999). Met-induced JNK activation is mediated by the adapter protein Crk and correlates with the Gab1-Crk signaling complex formation. Oncogene 18, 7775–7786 10.1038/sj.onc.1203198 [DOI] [PubMed] [Google Scholar]
- Gimona M. (2008). The microfilament system in the formation of invasive adhesions. Semin. Cancer Biol. 18, 23–34 10.1016/j.semcancer.2007.08.005 [DOI] [PubMed] [Google Scholar]
- Gual P., Giordano S., Williams T. A., Rocchi S., Van Obberghen E., Comoglio P. M. (2000). Sustained recruitment of phospholipase C-gamma to Gab1 is required for HGF-induced branching tubulogenesis. Oncogene 19, 1509–1518 10.1038/sj.onc.1203514 [DOI] [PubMed] [Google Scholar]
- Hauck C. R., Hsia D. A., Ilic D., Schlaepfer D. D. (2002). v-Src SH3-enhanced interaction with focal adhesion kinase at beta 1 integrin-containing invadopodia promotes cell invasion. J. Biol. Chem. 277, 12487–12490 10.1074/jbc.C100760200 [DOI] [PubMed] [Google Scholar]
- Holgado–Madruga M., Wong A. J.2003). Gab1 is an integrator of cell death versus cell survival signals in oxidative stress. Mol. Cell. Biol. 234471–4484 10.1128/MCB.23.13.4471-4484.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammula U. S., Kuntz E. J., Francone T. D., Zeng Z., Shia J., Landmann R. G., Paty P. B., Weiser M. R. (2007). Molecular co-expression of the c-Met oncogene and hepatocyte growth factor in primary colon cancer predicts tumor stage and clinical outcome. Cancer Lett. 248, 219–228 10.1016/j.canlet.2006.07.007 [DOI] [PubMed] [Google Scholar]
- Kelley L. C., Ammer A. G., Hayes K. E., Martin K. H., Machida K., Jia L., Mayer B. J., Weed S. A. (2010). Oncogenic Src requires a wild-type counterpart to regulate invadopodia maturation. J. Cell Sci. 123, 3923–3932 10.1242/jcs.075200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura F., Iwaya K., Kawaguchi T., Kaise H., Yamada K., Mukai K., Matsubara O., Ikeda N., Kohno N. (2010). Epidermal growth factor-dependent enhancement of invasiveness of squamous cell carcinoma of the breast. Cancer Sci. 101, 1133–1140 10.1111/j.1349-7006.2010.01527.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T., Yamaki E., Mogi A., Kuweno H. (2011). Mechanism of resistence to EGFR TKIs and development of a new generation of drugs in non-small-cell lung canacer. J. Biomed. Biotechnol. 12, 1–7 10.1155/2011/165214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. Z., Abella J. V., Park M. (2009). Crosstalk in Met receptor oncogenesis. Trends Cell Biol. 19, 542–551 10.1016/j.tcb.2009.07.002 [DOI] [PubMed] [Google Scholar]
- Lamorte L., Kamikura D. M., Park M. (2000). A switch from p130Cas/Crk to Gab1/Crk signaling correlates with anchorage independent growth and JNK activation in cells transformed by the Met receptor oncoprotein. Oncogene 19, 5973–5981 10.1038/sj.onc.1203977 [DOI] [PubMed] [Google Scholar]
- Lengyel E., Prechtel D., Resau J. H., Gauger K., Welk A., Lindemann K., Salanti G., Richter T., Knudsen B., Vande Woude G. F., et al. (2005). C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int. J. Cancer 113, 678–682 10.1002/ijc.20598 [DOI] [PubMed] [Google Scholar]
- Linder S. (2007). The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 17, 107–117 10.1016/j.tcb.2007.01.002 [DOI] [PubMed] [Google Scholar]
- Lock L. S., Royal I., Naujokas M. A., Park M. (2000). Identification of an atypical Grb2 carboxyl-terminal SH3 domain binding site in Gab docking proteins reveals Grb2-dependent and -independent recruitment of Gab1 to receptor tyrosine kinases. J. Biol. Chem. 275, 31536–31545 10.1074/jbc.M003597200 [DOI] [PubMed] [Google Scholar]
- Lock L. S., Maroun C. R., Naujokas M. A., Park M. (2002). Distinct recruitment and function of Gab1 and Gab2 in Met receptor-mediated epithelial morphogenesis. Mol. Biol. Cell 13, 2132–2146 10.1091/mbc.02-02-0031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock L. S., Frigault M. M., Saucier C., Park M. (2003). Grb2-independent recruitment of Gab1 requires the C-terminal lobe and structural integrity of the Met receptor kinase domain. J. Biol. Chem. 278, 30083–30090 10.1074/jbc.M302675200 [DOI] [PubMed] [Google Scholar]
- Mader C. C., Oser M., Magalhaes M. A., Bravo–Cordero J. J., Condeelis J., Koleske A. J., Gil–Henn H. (2011). An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 71, 1730–1741 10.1158/0008-5472.CAN-10-1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroun C. R., Holgado–Madruga M., Royal I., Naujokas M. A., Fournier T. M., Wong A. J., Park M. (1999). The Gab1 PH domain is required for localization of Gab1 at sites of cell-cell contact and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell. Biol. 19, 1784–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroun C. R., Naujokas M. A., Holgado–Madruga M., Wong A. J., Park M. (2000). The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell. Biol. 20, 8513–8525 10.1128/MCB.20.22.8513-8525.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D. A., Courtneidge S. A. (2011). The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 12, 413–426 10.1038/nrm3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J. M., Onodera Y., Mazaki Y., Miyoshi H., Hashimoto S., Sabe H. (2007). CIN85, a Cbl-interacting protein, is a component of AMAP1-mediated breast cancer invasion machinery. EMBO J. 26, 647–656 10.1038/sj.emboj.7601534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L., Holgado–Madruga M., Maroun C., Fixman E. D., Kamikura D., Fournier T., Charest A., Tremblay M. L., Wong A. J., Park M. (1997). Association of the multisubstrate docking protein Gab1 with the hepatocyte growth factor receptor requires a functional Grb2 binding site involving tyrosine 1356. J. Biol. Chem. 272, 20811–20819 10.1074/jbc.272.33.20811 [DOI] [PubMed] [Google Scholar]
- Oikawa T., Itoh T., Takenawa T. (2008). Sequential signals toward podosome formation in NIH-src cells. J. Cell Biol. 182, 157–169 10.1083/jcb.200801042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K., Sasaki H., Yukiue H., Yano M., Fujii Y. (2008). Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 99, 2280–2285 10.1111/j.1349-7006.2008.00916.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser M., Yamaguchi H., Mader C. C., Bravo–Cordero J. J., Arias M., Chen X., DesMarais V., van Rheenen J., Koleske A. J., Condeelis J. (2009). Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J. Cell Biol. 186, 571–587 10.1083/jcb.200812176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschard P., Park M. (2007). From Tpr-Met to Met, tumorigenesis and tubes. Oncogene 26, 1276–1285 10.1038/sj.onc.1210201 [DOI] [PubMed] [Google Scholar]
- Pichot C. S., Arvanitis C., Hartig S. M., Jensen S. A., Bechill J., Marzouk S., Yu J., Frost J. A., Corey S. J. (2010). Cdc42-interacting protein 4 promotes breast cancer cell invasion and formation of invadopodia through activation of N-WASP. Cancer Res. 70, 8347–8356 10.1158/0008-5472.CAN-09-4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponzetto C., Bardelli A., Zhen Z., Maina F., dalla Zonca P., Giordano S., Graziani A., Panayotou G., Comoglio P. M. (1994). A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 77, 261–271 10.1016/0092-8674(94)90318-2 [DOI] [PubMed] [Google Scholar]
- Ponzetto C., Zhen Z., Audero E., Maina F., Bardelli A., Basile M. L., Giordano S., Narsimhan R., Comoglio P. (1996). Specific uncoupling of GRB2 from the Met receptor. Differential effects on transformation and motility. J. Biol. Chem. 271, 14119–14123 10.1074/jbc.271.24.14119 [DOI] [PubMed] [Google Scholar]
- Ponzo M. G., Lesurf R., Petkiewicz S., O'Malley F. P., Pinnaduwage D., Andrulis I. L., Bull S. B., Chughtai N., Zuo D., Souleimanova M., et al. (2009). Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc. Natl. Acad. Sci. USA 106, 12903–12908 10.1073/pnas.0810402106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues G. A., Naujokas M. A., Park M. (1991). Alternative splicing generates isoforms of the met receptor tyrosine kinase which undergo differential processing. Mol. Cell. Biol. 11, 2962–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubini C., Ruzza P., Spaller M. R., Siligardi G., Hussain R., Udugamasooriya D. G., Bellanda M., Mammi S., Borgogno A., Calderan A., et al. (2010). Recognition of lysine-rich peptide ligands by murine cortactin SH3 domain: CD, ITC, and NMR studies. Biopolymers 94, 298–306 10.1002/bip.21350 [DOI] [PubMed] [Google Scholar]
- Saucier C., Papavasiliou V., Palazzo A., Naujokas M. A., Kremer R., Park M. (2002). Use of signal specific receptor tyrosine kinase oncoproteins reveals that pathways downstream from Grb2 or Shc are sufficient for cell transformation and metastasis. Oncogene 21, 1800–1811 10.1038/sj.onc.1205261 [DOI] [PubMed] [Google Scholar]
- Sawada K., Radjabi A. R., Shinomiya N., Kistner E., Kenny H., Becker A. R., Turkyilmaz M. A., Salgia R., Yamada S. D., Vande Woude G. F., et al. (2007). c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 67, 1670–1679 10.1158/0008-5472.CAN-06-1147 [DOI] [PubMed] [Google Scholar]
- Schaeper U., Gehring N. H., Fuchs K. P., Sachs M., Kempkes B., Birchmeier W. (2000). Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J. Cell Biol. 149, 1419–1432 10.1083/jcb.149.7.1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoumacher M., Goldman R. D., Louvard D., Vignjevic D. M. (2010). Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 189, 541–556 10.1083/jcb.200909113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals D. F., Azucena E. F., Jr, Pass I., Tesfay L., Gordon R., Woodrow M., Resau J. H., Courtneidge S. A. (2005). The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 7, 155–165 10.1016/j.ccr.2005.01.006 [DOI] [PubMed] [Google Scholar]
- Smith–Pearson P. S., Greuber E. K., Yogalingam G., Pendergast A. M. (2010). Abl kinases are required for invadopodia formation and chemokine-induced invasion. J. Biol. Chem. 285, 40201–40211 10.1074/jbc.M110.147330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks A. B., Rider J. E., Hoffman N. G., Fowlkes D. M., Quillam L. A., Kay B. K. (1996). Distinct ligand preferences of Src homology 3 domains from Src, Yes, Abl, Cortactin, p53bp2, PLCgamma, Crk, and Grb2. Proc. Natl. Acad. Sci. USA 93, 1540–1544 10.1073/pnas.93.4.1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuible M., Dubé N., Tremblay M. L.2008). PTP1B regulates cortactin tyrosine phosphorylation by targeting Tyr446. J. Biol. Chem. 28315740–15746 10.1074/jbc.M110.115295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stylli S. S., Stacey T. T., Verhagen A. M., Xu S. S., Pass I., Courtneidge S. A., Lock P. (2009). Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J. Cell Sci. 122, 2727–2740 10.1242/jcs.046680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpson P., Wilson A. S., Lehrbach G. M., Sutherland R. L., Musgrove E. A., Daly R. J. (2007). Aberrant expression of cortactin in head and neck squamous cell carcinoma cells is associated with enhanced cell proliferation and resistance to the epidermal growth factor receptor inhibitor gefitinib. Cancer Res. 67, 9304–9314 10.1158/0008-5472.CAN-07-0798 [DOI] [PubMed] [Google Scholar]
- Tuynman J. B., Lagarde S. M., Ten Kate F. J., Richel D. J., van Lanschot J. J. (2008). Met expression is an independent prognostic risk factor in patients with oesophageal adenocarcinoma. Br. J. Cancer 98, 1102–1108 10.1038/sj.bjc.6604251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uruno T., Liu J., Zhang P., Fan Y., Egile C., Li R., Mueller S. C., Zhan X. (2001). Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 3, 259–266 10.1038/35060051 [DOI] [PubMed] [Google Scholar]
- Webb B. A., Jia L., Eves R., Mak A. S. (2007). Dissecting the functional domain requirements of cortactin in invadopodia formation. Eur. J. Cell Biol. 86, 189–206 10.1016/j.ejcb.2007.01.003 [DOI] [PubMed] [Google Scholar]
- Weidner K. M., Di Cesare S., Sachs M., Brinkmann V., Behrens J., Birchmeier W. (1996). Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 384, 173–176 10.1038/384173a0 [DOI] [PubMed] [Google Scholar]
- Wu C. W., Li A. F., Chi C. W., Chung W. W., Liu T. Y., Lui W. Y., P'eng F. K. (1998). Hepatocyte growth factor and Met/HGF receptors in patients with gastric adenocarcinoma. Oncol. Rep. 5, 817–822 [DOI] [PubMed] [Google Scholar]
- Yamaguchi H., Lorenz M., Kempiak S., Sarmiento C., Coniglio S., Symons M., Segall J., Eddy R., Miki H., Takenawa T., et al. (2005). Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 168, 441–452 10.1083/jcb.200407076 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.