

Abstract
Necroptosis is a regulated caspase-independent cell death pathway with morphological features resembling passive non-regulated necrosis. Several diverse structure classes of necroptosis inhibitors have been reported to date, including a series of 3,3a,4,5-tetrahydro-2H-benz[g]indazoles (referred to as the Nec-3 series) displaying potent activity in cellular assays. However, evaluation of the tricyclic necroptosis inhibitor’s stability in mouse liver microsomes indicated that they were rapidly degraded. A structure-activity relationship (SAR) study of this compound series revealed that increased liver microsomal stability could be accomplished by modification of the pendent phenyl ring and by introduction of a hydrophilic substituent (i.e. α-hydroxyl) to the acetamide at the 2-position of the tricyclic ring without significantly compromising necroptosis inhibitory activity. Further increases in microsomal stability could be achieved by utilizing the 5,5-dioxo-3-phenyl-2,3,3a,4-tetrahydro-[1]benzothiopyrano[4,3-c]pyrazoles. However, in this case necroptosis inhibitory activity was not maintained. Overall, these results provide a strategy for generating potent and metabolically stable tricyclic necrostatin analogs (e.g. 33, LDN-193191) potentially suitable for in vivo studies.
Necroptosis is a regulated caspase-independent cell death pathway with morphological features resembling passive non-regulated necrosis.1, 2 This type of cell death can be initiated with various stimuli (e.g. TNF-α and Fas ligand) and in a variety of cell types (e.g. monocytes, fibroblasts, lymphocytes, macrophages, epithelial cells and neurons). Furthermore, necroptosis may represent a significant contributor to and in some cases predominant mode of cellular demise under pathological conditions involving excessive cell stress, rapid energy loss and massive oxidative species generation, not conducive for highly energy-dependent processes, such as apoptosis. Regulated necrotic cell death mechanisms, such as necroptosis, raises the possibility of novel therapeutic intervention strategies for the treatment of conditions where necrosis is known to play a prominent role, such as organ ischemia (i.e. stroke3 and myocardial infarction4), trauma and possibly some forms of neurodegeneration.5
To date several diverse structure classes of necroptosis inhibitors have been reported, including hydantoin containing indole derivatives (i.e. 1),6 rel-(3R,3aR)-3-phenyl-3,3a,4,5-tetrahydro-2H-benz[g]indazoles (i.e. 2),7 substituted 3H-thieno[2,3-d]pyrimidin-4-ones (i.e. 3),8 [1,2,3]thiadiazole benzylamides (i.e. 4)9 and pyrrole benzylamides (i.e. 5)10 (Figure 1). Many of these compounds block necroptosis through inhibition of receptor interacting protein 1 (RIP1) kinase.11 In addition, (±)-1 has demonstrated in vivo activity in the temporary and permanent middle cerebral artery occlusion (MCAO) model of cerebral ischemia1, in a mouse model of ischemia/reperfusion heart injury,12 in the controlled cortical impact (CCI) model of traumatic brain injury (TBI),13 a retinal ischemia-reperfusion injury model,14 a systemic inflammatory response syndrome (SIRS) model,15 and a Huntington’s disease model.16
Figure 1.

Necrostatins. The numbering convention of 2 is also shown.
In order to evaluate the in vivo pharmacology of other necroptosis inhibitors via preferred administration routes (i.e. oral, intravenous, intraperitoneal or subcutaneous) they must possess adequate metabolic stability, in addition to in vitro potency. One efficient and cost effective method of assessing a compound’s metabolic stability is to measure its resistance to metabolism over time in the presence of liver microsomes.17 Utilizing this technique with mouse liver microsomes, compound 2, which inhibits necroptosis induced with TNF-α in FADD-deficient variant of human Jurkat T cells with an EC50 value of 0.29 μM, demonstrated poor metabolic stability with a half-life (t1/2) of 8.2 min and intrinsic clearance (CLint) of 169 ± 2.0 μL/min/mg protein. Herein, we describe the results of a SAR study to optimize the in vitro liver microsomal stability of the tricyclic (Nec-3) class of necroptosis inhibitors.
Many of the tricyclic derivatives evaluated herein were prepared according to the procedure outlined in Scheme 1.7 1-Tetralones, 4-chromanones and 4-thiochromanones, 6, were treated with benzaldehydes or phenylacetaldehyde under basic or acid conditions to give 7. The chalcones were allowed to react with hydrazine hydrate utilizing various acids (R3CO2H) as solvent to give a mixture of two diastereomers, the (3R,3aR)-rel-isomers 8a – 11a and the (3R,3aS)-rel-isomers 8b – 11b. The diastereomers were readily separated by column chromatography on silica gel and the stereochemical assignments were made using 1H-NMR. Removal of the benzyl group in 9a was accomplished by hydrogenation in the presence of 10% Pd/C to give alcohol 12. Oxidation of 10a and 11a with m-chloroperoxybenzoic acid (m-CPBA) gave sulfones 13 and 14, respectively. Sulfone 14 was subsequently converted to alcohol 15 by hydrogenation.
Scheme 1.

(a) Ar(CH2)nCHO, 8N NaOH, EtOH, rt, 2 h or Ar(CH2)nCHO, conc HCl, MeOH, Δ, 4 h (60 – 75%); (b) R3CO2H, NH2NH2·xH2O, 120 °C, 15 h (70 – 80% when n = 0; 20% when n = 1); (c) H2 (1 atm), 10% Pd/C, EtOH (47–75%); (d) MCPBA, DCM, rt, 16 h (90–96%).
Derivatives containing a methyl group at the 3a-position were prepared according to the procedure outlined in Scheme 2. Initial attempts to prepare these derivatives from an α-methyl-1-tetralone derivative utilizing the same synthetic strategy employed for the preparation of the 3a-H derivatives were unsuccessful. Instead, 7-methoxy-1-tetralone, 16, was deprotonated with NaN(TMS)2 and then allowed to react with 4-methyoxybenzoyl chloride to give the 1,3-diketone 17. This compound was again subjected to the same process, except that the anion was quenched with iodomethane to give 1,3-diketone 18. Condensation of 18 with hydrazine hydrate in the presence of 4Å molecular sieves gave 19.18 Treatment of 19 at −78 °C with acetyl chloride followed by reduction of the in situ generated acyl iminium gave a mixture of diastereomers 20a and 20b in a ratio of 1:1.7, favoring isomer 20b where the hydride anion approaches the acyl iminium intermediate distal to the 3a-Me. The 3a-Me of 20b, which is syn to the pendent phenyl, was shielded and appeared at δ 0.69 in the 1H NMR spectra.19a The 3a-Me of 20a appeared further downfield at δ 1.59.19b The structure of 20b was confirmed by single crystal x-ray analysis and reaffirmed the regioselectivity of the acylation reaction and the stereochemical assignments of the diastereomers (Figure 2).20
Scheme 2.

(a) NaN(TMS)2, THF, 0 °C, then 4-MeO-PhC(O)Cl, 0 °C to rt (80%); (b) NaN(TMS)2, THF, 0 °C, then MeI, 0 °C to rt (91%); (c) NH2NH2·xH2O, DCM, 4Å molecular sieves (52%); (d) MeC(O)Cl, DCM, −78 °C, then NaBH(OAc)3, −78 °C to rt (95%).
Figure 2.

Thermal ellipsoidal drawing of 20b as determined by single crystal x-ray analysis.
In vitro microsomal stability was determined in pooled mouse liver microsomes. Test compounds (Table 1) were incubated in the presence and absence of NADPH for 0 – 60 min and the amount of remaining compound was quantified.21 Necroptosis inhibitor (±)-1 demonstrated good metabolic stability in this assay with a t1/2 = 59.1 min and CLint = 23.5 ± 2.1 μL/min/mg protein. Evaluation of necroptosis inhibitory activity was performed using a FADD-deficient variant of human Jurkat T cells treated with TNF-α as previously described.1, 7 Utilizing these conditions the cells efficiently underwent necroptosis, which was completely and selectively inhibited by (±)-1 (EC50 = 0.21 μM). For EC50 value determinations, cells were treated with 10 ng/mL of human TNF-α in the presence of increasing concentration of test compounds (eleven doses between 30 nM to 100 μM) for 24 h followed by ATP-based viability assessment.
Table 1.
Compounds prepared for microsomal stability studies and EC50 determinations for necroptosis inhibition in FADD-deficient Jurkat T cells treated with TNF-α.
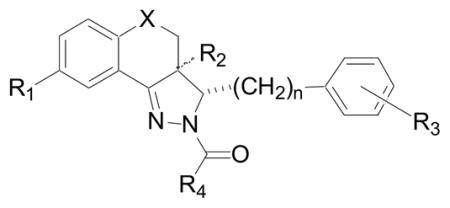
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | X | n |
| 2 | OMe | β-Ha | 4-OMe | Me | CH2 | 0 |
| 20a | OMe | β-Me | 4-OMe | Me | CH2 | 0 |
| 20b | OMe | α-Me | 4-OMe | Me | CH2 | 0 |
| 21 | OMe | β-H | 4-OMe | CF3 | CH2 | 0 |
| 22 | OMe | β-H | 4-OMe | Me | O | 0 |
| 23 | F | β-H | 4-OMe | Me | O | 0 |
| 24 | OMe | β-H | 4-OMe | Me | S | 0 |
| 25 | OMe | β-H | 4-OMe | Me | SO2 | 0 |
| 26 | OMe | β-H | H | Me | CH2 | 1 |
| 27 | OMe | β-H | 4-OMe | CH2OH | CH2 | 0 |
| 28 | OMe | β-H | 3-F, 4-OMe | Me | CH2 | 0 |
| 29 | OMe | β-H | 3-F, 4-OMe | CH2OH | CH2 | 0 |
| 30 | OMe | β-H | 4-OCF3 | Me | CH2 | 0 |
| 31 | OMe | β-H | 4-OCF3 | CH2OH | CH2 | 0 |
| 32 | OMe | β-H | 4-OCF3 | CH2OH | SO2 | 0 |
| 33 | OMe | β-H | 3-F, 4-OCF3 | CH2OH | CH2 | 0 |
α-H refers to the (3R,3aS)-rel-diastereomer; β-H refers to the (3R,3aR)-rel-diastereomer.
Several regions of the tricyclic necroptosis inhibitor 2 were considered potential liability sites responsible for the compound’s poor metabolic stability in mouse liver microsomes. These sites included the methoxy groups at both the 8-position of the tricyclic ring and the 4-position of the pendent phenyl, the dihydropyrazole ring, the benzylic carbon at the 5-positions and the amide at the 2-position.
Introduction of a methyl group at the 3a-position of the tricyclic ring (20a and 20b), which was envisioned to block potential oxidation of the dihydropyrazole, did not result in improved stability. Interestingly, necroptosis inhibitory actively was dramatically decreased for both diastereomers. Replacement of the benzylic methylene at the 5-position with oxygen (22) did not result in an increase in metabolic stability. When the methoxy group at the 8-position of the tricyclic ring was further replaced with a fluorine (23) metabolic stability remained poor. Substitution of the benzylic methylene with sulfur (24) similarly did not improve stability. However, replacement with a sulfone (25) did result in a significant increase in metabolic stability (t1/2 = 40 min and CLint = 34.6 ± 2.6 μL/min/mg protein) with a slight decrease in necroptosis inhibitory activity. Introduction of a methylene group between the 3-position of the tricyclic ring and the pendent phenyl ring (26) similarly did not increase stability, but did eliminate necroptosis inhibitory activity. Addition of a hydroxyl group on the α-position of the amide (27 vs. 2 and 28 vs. 29) or introduction of a trifluoromethoxy in place of a methoxy at the 4-position of the pendent phenyl ring (30 vs. 2) resulted in increased metabolic stability. A combination of these changes (31) yielded a further increase in stability (t1/2 = 54 min and CLint = 25.5 ± 3.8 μL/min/mg protein). A similar result was also found with a sulfone derivative (25 vs. 32), albeit necroptosis inhibitory activity was compromised. Although introduction of fluorine at the 3-position of the pendent phenyl ring did not increase stability in one case (2 vs. 28), in another instance this change in combination with a hydroxyl group on the α-position of the amide and a trifluoromethoxy at the 4-position of the pendent phenyl (2 vs. 33) resulted in a significant (18-times) stability increase (t1/2 = 148 min and CLint = 9.38 ± 1.4 μL/min/mg protein) with only a modest (2-fold) decrease in necroptosis inhibitory activity.
In conclusion, increased liver microsomal stability as well as improvement in activity were accomplished for the tricyclic (Nec-3) series of necroptosis inhibitors by modification of the pendent phenyl and by introduction of a hydrophilic substituent (i.e. α-hydroxyl) to the acetamide at the 2-position, resulting in inhibitor 33 (LDN-193191).19c The benzylic position of the tricyclic ring also appeared to influence metabolic stability. Although replacement of the methylene group with a sulfone increased metabolic stability, it significantly decreased necroptosis inhibitory activity. Additional optimization of tricyclic necroptosis inhibitors utilizing the information from this study may result in further increases in metabolic stability and provide a unique set of necroptosis inhibitors suitable for in vivo analysis of the pathologic role of necroptosis following acute and potentially chronic injury.
Table 2.
EC50 determinations for necroptosis inhibition in FADD-deficient Jurkat T cells treated with TNF-α and mouse microsomal stability values.
| Compound | EC50 (μM)a | t1/2 (min) | CLint (μL/min/mg protein) |
|---|---|---|---|
| 2 | 0.29 | 8.2 | 169 ± 2.0 |
| 20a | > 100 | 2.9 | 476 ± 53.9 |
| 20b | 15 | 7.2 | 194 ± 5.9 |
| 21 | 0.39 | 15 | 90.7 ±13.4 |
| 22 | 0.46 | 10 | 135 ± 9.3 |
| 23 | 12 | 11 | 120 ± 39.3 |
| 24 | 0.28 | 12 | 115 ± 18.5 |
| 25 | 0.75 | 40 | 34.6 ± 2.6 |
| 26 | > 100 | 3.0 | 463 ± 78.1 |
| 27 | 0.16 | 33 | 42.4 ± 5.5 |
| 28 | 0.090 | 8.6 | 162 ± 5.7 |
| 29 | 0.28 | 27 | 50.5 ± 3.6 |
| 30 | 0.33 | 20 | 68.7 ± 18.0 |
| 31 | 0.64 | 54 | 25.5 ± 3.8 |
| 32 | 27 | 115 | 12.1 ± 1.4 |
| 33 | 0.61 | 148 | 9.38 ± 1.4 |
Standard deviation < 10%.
Acknowledgments
SC and GDC thank the Harvard NeuroDiscovery Center (HNC) for financial support. AD and JY thank the National Institute on Aging, National Institute of General Medical Sciences and American Health Assistance Foundation for financial support. SC, GDC and JY thank the National Institute of Neurological Disorders and Stroke (NINDS) for financial support. AD is a recipient of NIH Mentored Scientist Development Award from the National Institute on Aging (NIA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison T, Moskowitz M, Yuan J. Nat Chem Biol. 2005;1:112. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 2.For review articles related to necroptosis see: Yuan J, Kroemer G. Genes Dev. 2010;24:2592. doi: 10.1101/gad.1984410.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Nat Rev Mol Cell Biol. 2010;11:700. doi: 10.1038/nrm2970.Christofferson DE, Yuan J. Curr Opin Cell Biol. 2010;22:263. doi: 10.1016/j.ceb.2009.12.003.Degterev A, Yuan J. Nat Rev Mol Cell Biol. 2008;9:378. doi: 10.1038/nrm2393.
- 3.(a) Mehta SL, Manhas N, Raghubir R. Brain Res Rev. 2007;54:34. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]; (b) Lo EH, Dalkara T, Moskowitz MA. Nat Rev Neurosci. 2003;4:399. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 4.(a) Whelan RS, Kaplinskiy V, Kitsis RN. Annu Rev Physiol. 2010;72:19. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S. Am J Physiol Heart Circ Physiol. 2004;286:H1923. doi: 10.1152/ajpheart.00935.2003. [DOI] [PubMed] [Google Scholar]
- 5.(a) Esposito E, Cuzzocrea S. Curr Med Chem. 2010;17:2764. doi: 10.2174/092986710791859324. [DOI] [PubMed] [Google Scholar]; (b) Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Brain Res Bull. 1998;46:281. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 6.Teng X, Degterev A, Jagtap P, Xing X, Choi S, Denu R, Yuan J, Cuny GD. Bioorg Med Chem Lett. 2005;15:5039. doi: 10.1016/j.bmcl.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 7.Jagtap PG, Degterev A, Choi S, Keys H, Yuan J, Cuny GD. J Med Chem. 2007;50:1886. doi: 10.1021/jm061016o. [DOI] [PubMed] [Google Scholar]
- 8.Wang K, Li J, Degterev A, Hsu E, Yuan J, Yuan C. Bioorg Med Chem Lett. 2007;17:1455. doi: 10.1016/j.bmcl.2006.11.056. [DOI] [PubMed] [Google Scholar]
- 9.Teng X, Keys H, Jeevanandam A, Porco JA, Jr, Degterev A, Yuan J, Cuny GD. Bioorg Med Chem Lett. 2007;17:6836. doi: 10.1016/j.bmcl.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teng X, Keys H, Yuan J, Degterev A, Cuny GD. Bioorg Med Chem Lett. 2008;18:3219. doi: 10.1016/j.bmcl.2008.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Degterev A, Hitomi J, Germsheid M, Ch’en I, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Nat Chem Biol. 2008;4:313. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith CCT, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Cardiovasc Drugs Ther. 2007;21:227. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 13.You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ. J Cereb Blood Flow Metab. 2008;28:1564. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. J Neurosci Res. 2010;88:1569. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. Immunity. 2011;35:908. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 16.Zhu S, Zhang Y, Bai G, Li H. Cell Death Dis. 2011;2:e115. doi: 10.1038/cddis.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baranczewski P, Stańczak A, Sundberg K, Svensson R, Wallin A, Jannson J, Garberg P, Postlind H. Pharmacol Rep. 2006;58:453. [PubMed] [Google Scholar]
- 18.Adam W, Librera CP. J Org Chem. 2002;67:576. doi: 10.1021/jo016224x. [DOI] [PubMed] [Google Scholar]
- 19.(a) 1H NMR of 20a (500 MHz, CDCl3): δ 1.59 (s, 3H), 1.78–1.82 (m, 2H), 2.35 (s, 3H), 2.35–2.45 (m, 2H), 3.78 (s, 3H), 3.85 (s, 3H), 5.39 (s, 1H), 6.74 (dd, J1 = 8.5 Hz, J2 = 3.0 Hz, 1H), 6.92–6.95 (m, 3H), 7.51 (d, J = 3.0 Hz, 1H), 7.70 (d, J = 8.5 Hz, 2H). (b) 1H NMR of 20b (500 MHz, CDCl3): δ 0.69 (s, 3H), 2.05–2.14 (m, 2H), 2.46 (s, 3H), 2.85–3.01 (m, 2H), 3.80 (s, 3H), 3.86 (s, 3H), 4.93 (s, 1H), 6.88 (d, J = 9.0 Hz, 1H), 6.92 (dd, J1 = 9.0 Hz, J2 = 2.5 Hz, 1H), 7.10–7.12 (m, 3H), 7.41 (d, J = 2.5 Hz, 1H). (c) 1H NMR of 33 (500 MHz, CDCl3): δ 1.01–1.10 (m, 1H), 1.79–1.84 (m, 1H), 2.81–2.88 (m, 2H), 3.58–3.64 (m, 1H), 3.88 (s, 3H), 4.59–4.74 (m, 2H), 5.69 (d, J = 11.0 Hz, 1H), 6.89–6.97 (m, 3H), 7.09 (d, J = 8.5 Hz, 1H), 7.24–7.28 (m, 1H), 7.48 (d, J = 2.5 Hz, 1H).
- 20.CCDC 875792 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
-
21.Microsomal stability was determined in pooled mouse liver microsomes. Test compound (3 μM final concentration) along with 0.5 mg/mL microsome protein and 1 mM NADPH was incubated for 0, 5, 15, 30 and 60 min. Incubation of test compound and microsomes in the absence of NADPH served as a negative control. The samples were quenched with methanol and centrifuged for 20 min at 2500 rpm to precipitate proteins. Sample supernatants were analyzed (N=3) by LC/MS. The ln peak area ratio (compound peak area/internal standard peak area) was plotted against time and the slope of the line determined to give the elimination rate constant [k = (−1)(slope)]. The half life (t1/2 in minutes), and the in vitro intrinsic clearance (CLint in μL/min/mg protein) were calculated according to the following equations, where V = incubation volume in μL/mg protein: