

Abstract
We recount several unexpected results observed in the course of our work toward the synthesis of welwitindolinone alkaloids. The surprising results provide an opportunity to refine one’s understanding of the interplay between chemical structure and reactivity.
Keywords: Welwitindolinones, Vilsmeier–Haack, O-acylation, Chlorination, Aromatic Cope Rearrangement
In the course of complex natural product synthesis, seemingly straightforward steps often yield unexpected results. Indeed, it is the exception rather than the norm that a multi-step synthesis proceeds precisely as planned; and it is this unpredictability that makes the field not only challenging, but also interesting. The unplanned results provide an opportunity to refine one’s understanding of the interplay between chemical structure and reactivity. In our ongoing efforts1 toward the synthesis of the welwitindolinone alkaloids (1–3, Figure 1),2,3,4 we have encountered more than a few hurdles along the way, and these have introduced new twists and turns in our synthetic strategy and have also provided some chemical surprises. We recount below several of the planned transformations and discuss the unforeseen outcomes. While some of the unexpected results have yet to be understood fully, others provide fresh insights on chemical reactivity.
Figure 1.

Representative Welwitindolinone Alkaloids
Regioselectivity and C vs. O Selectivity
We recently reported a concise synthesis of the welwitindolinone core via an oxidative arylation approach, which enabled the synthesis of diketone 5 from ketoester 4.1c The presence of the ester functionality was expected, whether through chelation or an electronic inductive effect, to enable regioselective functionalization of the desired α-carbon of the C13-ketone. Among the transformations that were examined to achieve this goal was a sequence involving a Vilsmeier–Haack reaction followed by deconjugative methylation (Scheme 1).5 The caged architecture of 6 renders only the convex face accessible to electrophiles, thus the methylation event was expected to occur with complete facial selectivity. Wittig methylenation of the tertiary aldehyde in 8 would then complete the C12 quaternary center.
Scheme 1.

Vilsmeier–Haack Deconjugative Methylation Approach
To put the plan into action, dione 5 was subjected to standard Vilsmeier–Haack conditions (POCl3, DMF, heat), and, while it produced a β-chloro-aldehyde product, it was solely the undesired regioisomer, 10 (Scheme 2). Varying the reaction conditions did not invert the inherent regioselectivity of this reaction.
Scheme 2.

Vilsmeier–Haack Chloroformylation of Dione 5
The regioselectivity of chloroformylation appeared inconsistent with the anticipated reactivity of the ketone, particularly with respect to the effect of the β-carboxylate group. To determine the inherent deprotonation selectivity, we investigated the conversion of the ketone to the corresponding silyl enol ether. Should that take place in the desired fashion, then that selectivity could be parlayed into the desired Vilsmeier–Haack reaction. To our surprise, silyl enol ether formation under soft-enolization conditions gave predominantly the undesired product (12, Scheme 3). The selectivity was reversed, however, under kinetic conditions. Indeed, deprotonation of dione 5 with KHMDS and quenching with an appropriate silyl chloride at −100 °C afforded the desired enol ether 11 with high regiocontrol. The presence of acidic by-products in the Vilsmeier–Haack reaction prompted us to favor bulky silyl groups like TBS and TIPS, which display higher stability to acidic conditions over the smaller and more labile TMS or TES groups. With the desired regioisomer of silyl enol ether (11) at hand we were poised to test the key Vilsmeier–Haack reaction. Treatment of 11 with the Vilsmeier–Haack reagent, generated in situ, once again yielded only the undesired regioisomer of the β-chloro-aldehyde (10). Investigation of the reaction revealed that the silyl enol ether (11) was undergoing isomerization under the reaction conditions to its regioisomer (12), which was responsible for the observed product (10). As the double bond isomerization is expected to be promoted by acid, the reaction was performed in the presence of various amine bases. Under these conditions, however, there was no reaction, and the starting silyl enol ether (11) was recovered in all cases.
Scheme 3.

Attempts to Functionalize the C12-Position of Dione 5
In a closely related strategy for installing the functionality at the neopentyl carbon (C12), we had hoped to capitalize on the successful kinetic deprotonation of dione 5 and functionalize the α-carbon. With that objective, the kinetic enolate was treated with Zayia’s reagent6 to afford the desired ketoaldehyde (13), albeit as the minor constituent of the reaction mixture. Disappointingly, this compound resisted our attempts at C-methylation. The O-methylated product was obtained as the predominant product, even under conditions known to favor C-methylation. The result highlighted for us the limitations that exist in the available methodology for a simple methylation reaction. The issue of C- vs. O-selectivity is one that we also confronted in a different route, as described below.
Unexpected Pd-Catalyzed Arylation Product
The inability to install the C12 quaternary center after assembling the core skeleton forced us to alter the strategy and perform the key C4–C11 bond forming step on a substrate that already had in place the requisite functionalities. To that end, we choose cyclohexanone 15,7 having the methyl and vinyl groups at C12, and converted it to compound 18 via a short sequence of steps (Scheme 4).8 With the indole-containing fragment attached, ketone 18 was treated with LDA followed by Mander’s reagent to deliver the expected carbomethoxylated product 19 (shown in the ketoester form), which was ready for the key Pd-enolate arylation step. Subjection of β-ketoester 19 to the palladium-mediated arylative-cyclization conditions (KOtBu, palladium(II) acetate, and PtBu3)1a afforded a tetracyclic product in modest yield. Curiously, the isolated product (20) lacked the C11 ester functionality, a surprising omission that was initially attributed to a hydrolytic-decarboxylation, a side reaction that was presumed to follow the palladium cyclization event. While plausible at first glance, this result was inconsistent with our earlier experiences using a model system, wherein a related ester was found to be highly resistant to basic hydrolysis.1a Moreover, the geometric constrains enforced by the bridged skeleton precludes the required stereoelectronic positioning of the carboxylate to the ketone. A careful examination of the overall reaction and the starting material provided the answer: there was an error in the structure assigned to the cyclization precursor (19). Acylation of the enolate of 18 with Mander’s reagent had delivered not the expected ketoester 19, but the enol carbonate 21. Evidently, the congested surroundings, specifically the quaternary centers at C12- and C16-positions, disfavor reaction of the electrophile at the carbon, allowing reaction only at the oxygen. After confirming the structure of the acylation product, it was then possible to provide a reasonable explanation for the formation of the decarboxylated product (20). Given the susceptibility of the enol carbonate to nucleophiles, it is likely that adventitious water, whether in the solvent or in the reagents, cleaves the enol carbonate 21. The resulting enolate (22) then undergoes a palladium-catalyzed intramolecular enolate arylation to provide tetracycle 20. Indeed, when the same reaction was performed under rigorously anhydrous conditions, none of the cyclized product (20) was observed. Various conditions and reagents were tried in order to functionalize the C11-position with a withdrawing group, but to no avail.9 The above difficulties forced us once again to modify the route and examine the possibility of installing the needed C11 electron-withdrawing group before coupling with the indole fragment.
Scheme 4.

Formation of the Decarboxylated Welwitindolinone
To circumvent the difficulties encountered with installation of the C11-withdrawing group, we decided to start with a more functionalized cyclohexanone fragment (23, Scheme 4). To our delight, this strategy proved successful and provided an efficient route to the fully functionalized welwitindolinone core (25), which ultimately culminated in the synthesis of N-methyl welwitindolinone D isonitrile (3).1b Tetracycle 25 was also expected to lay the path to several other welwitindolinones, especially toward welwitindolinone B (1), which required deoxygentative chlorination, indole oxidation, and conversion of the aldehyde to an isothiocyanate moiety.
Chlorination and Chloroselenation Surprises
In order to progress to welwitindolinone B (1), the C13 hydroxyl in 26 (Scheme 6) had to be transformed to the chloride, with inversion of configuration. Unfortunately, this simple transformation proved an insurmountable task and, even after considerable experimentation, we were unable to install the requisite halide at the C13-position of alcohol 26. Interestingly, when alcohol 26 was exposed to tri(2-furyl)phosphine and hexachloroacetone, it produced none of the desired chloride 27, but gave instead a fascinating rearrangement giving rise to cyclopropane 28 and diene 29.10 Interaction by the neighboring vinyl group evidently diverts the reaction from its normal course and produces compounds 28 and 29.
Scheme 6.

Unexpected Rearrangement during Deoxygenative Chlorination10
Another unexpected reaction was observed while searching for ways to protect the offending vinyl group, and thereby circumvent the homoallylic rearrangement. Attempted chloroselenation of TBS ether 25 with phenylselenium chloride11a,b or N-(phenylseleno)phthalimide (NPSP)11c gave pentacyclic selenide 31 rather than the expected addition product (30, Scheme 7). This anomalous result is consistent with the formation of an episelenonium ion (32) followed by an intramolecular Friedel–Crafts type reaction instead of the expected trapping by the halide counter ion. The result shows that the alkene is locked in close proximity to the indole-carbocycle. Significanlty, the constraints of the system and the stereoelectronic requirements are such that the Friedel-Crafts takes place at the primary carbon.
Scheme 7.

Unanticipated Intramolecular Reaction
Cross-Coupling Metamorphosis
The final and most remarkable result that we wish to discuss is a transformation that was encountered during our attempts to synthesize welwitindolinone C (2). The identifying feature of this natural product is the alkenyl chloride, a functional group that was expected to be available through the C13-ketone (33, Scheme 8), either directly or by way of the gem-dichloride. Several methods have been reported in the literature to effect this interconversion on ketones (e.g., WCl6,12 PCl5,13 MeSO3H/AcCl14) or their derivatives.15 Unfortunately, with a complex substrate such as ketoaldehyde 33 none of these conditions proved successful, and either no reaction or extensive decomposition of the starting material was observed. A less direct route to the alkenyl chloride is through a transition-metal catalyzed functionalization of the corresponding enol triflate, for example via the alkenyl stannane.16 In that regard, a recent communication by Hayashi and coworkers disclosed a ruthenium catalyzed transformation of enol triflates to alkenyl halides.17 A similar conversion, but promoted by a palladium catalyst, was reported by the Buchwald group shortly thereafter.18 In order to realize these opportunities we set forth to prepare the required alkenyl triflate. The reaction of the dione 33 with KHMDS in THF followed by addition of N-phenyl triflimide provided enol triflate 34 in 68% yield (Scheme 8). Upon subjection of 34 to Hayashi’s conditions, the starting material was transformed cleanly and in good yield into a new product. This substance was found, however, to not be the desired alkenyl chloride (35, X = Cl). Even a cursory examination of its proton NMR indicated that the skeleton of the product was different from that of the starting material. Extensive spectroscopic analysis, particularly HMBC, enabled the structural assignment of the product to be the rearranged compound 36. Compelling support for the assigned structure was obtained via X-ray crystallographic analysis (Figure 2). The same product was also obtained under palladium-catalyzed protocols.19 The dramatic reorganization of the carbon skeleton which is believed to arise from a Cope rearrangement19 between the vinyl group and the benzo-portion of indole, followed by C–C bond formation between the nucleophilic carbon (C11) of α-formylketone and the enol triflate carbon (C13).
Scheme 8.

Attempts to Synthesize the Alkenyl Chloride
Figure 2.
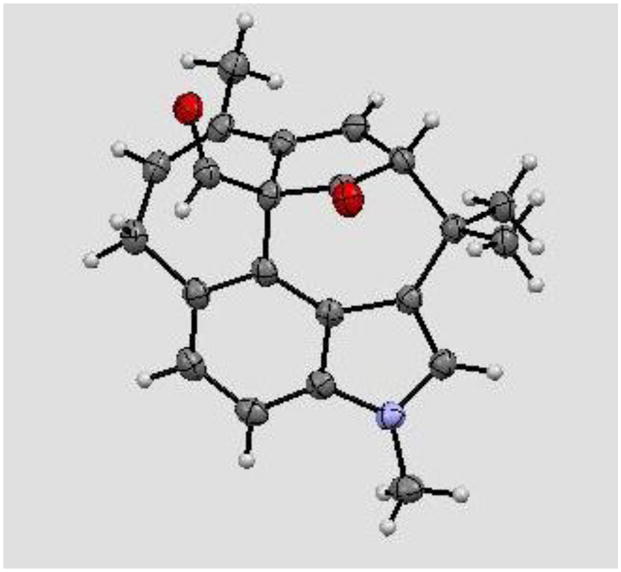
ORTEP Image of 36
The mechanistic underpinnings for the above transformation are interesting and informative for several reasons. First, the suggested [3,3]-sigmatropic rearrangement is an “aromatic Cope” rearrangement, of which there are relatively few examples.21,22 The loss of aromatic stabilization in the intermediate makes these rearrangements unfavorable unless driven by external factors, namely relief of ring strain, such as that of cyclopropane.20a The present example is especially noteworthy as the rearrangement takes place at just 60–80 °C. Second, Cope rearrangements typically give E- or Z-alkenes selectively, depending on whether the reaction is going through a chair or boat transition state. Third, it is unlikely that the enolate that forms after the rearrangement and proton loss can be alkenylated, since the enol triflate is incorrectly oriented for bond formation (38). It has E- rather than the Z-geometry required for the intramolecular alkenylation reaction. The alkene geometry must somehow be inverted to enable cyclization. Finally, it is unclear what role if any the metal plays in the [3,3]-rearrangement. With these considerations in mind, we propose the following rationale to explain the overall transformation (Scheme 9).23 Molecular models show, and the reactivity noted above for chloroselenation confirm, that the [4.3.1]-bridged bicyclic framework is strained—even more so with the enol triflate—and positions the vinyl group in close proximity to the aromatic ring. These strain and topological factors are expected to facilitate the Cope rearrangement. With regard to the E/Z-geometry of the product, the rigidity of the compound appears to favor the boat transition state over the chair form, such that the methyl-substituted alkene is expected to have E-geometry, opposite to what is observed in the final product. The resulting 10-membered ring intermediate with the alkenyl-Ru (or Pd) can undergo a stereospecific η1 to η1′ rearrangement, through an alkylidene-π-allyl intermediate, to produce the allenic intermediate 39.24 Some of the strain in 39 is relieved through a conformational change which gives rise to 40. A second η1 to η1′ rearrangement reforms the diene to yield 41. The net effect of the two rearrangements is to convert the E,E-diene (38) to the Z,Z-diene (41), having the alkenyl metal bond correctly oriented to enable the intramolecular alkenylation of the stabilized enolate via 42 to deliver pentacycle 36. It is worth noting that 34 is slowly consumed at 60 °C even in the absence of a catalyst, but gives rise to a mixture of products. Further studies on this fascinating rearrangement are currently underway, details of which will be reported in due course.
Scheme 9.

Plausible Mechanism for the Unusual Rearrangement23
The collection of unexpected results presented here serves to affirm what every synthetic organic chemist knows all too well: seemingly small perturbations in the structure of a molecule can significant alter its reactivity.25 The improved understanding of reactivity gained through this process has helped to define a successful path to several members of the welwitindolinone family.
Experimental
Unless stated otherwise, reactions were performed in flame or oven-dried glassware under an argon or nitrogen atmosphere using dry, deoxygenated solvents (distilled or passed over a column of activated alumina). Commercially available chemicals were used as received, unless otherwise stated. Ambient temperature refers to 22–26 °C. Higher than ambient reaction temperatures were controlled by IKAmag temperature modulators while for lower temperatures ice (0 °C), iPrOH/dry ice (−78 °C), and Et2O/liq N2 (−100 °C) baths were used. All reactions were monitored by thin-layer chromatography (TLC) which were performed using pre-coated Whatman K6F silica gel (250 μm, 60 Å) plates and Dynamic Adsorbents silica gel (250 μm, 60 Å) plates with F-254 indicator and visualized by UV fluorescence quenching, ceric ammonium molybdate, anisaldehyde, or potassium permanganate staining. Preparative-TLC was performed using Whatman Partisil PK6F pre-coated 60 Å silica gel plates (500 μm) with fluorescent indicator. Zeochem ZEOprep Flash ECO 40–63 Academic Grade silica gel was used for column chromatography. 1H and 13C NMR spectra were recorded on Bruker DRX-400, DRX-500 and DMX-500 and are reported relative to Me4Si (δ 0.0), unless otherwise stated. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (integration, multiplicity, coupling constant in Hz). Infrared spectra were recorded on Nicolet 6700 FT-IR spectrometer and are reported in frequency of absorption (cm−1). High resolution mass spectra (HRMS) were recorded at the Old Dominion University, Norfolk, and Hunter College, New York.
Chloro-aldehyde 10
A solution of dione 51c (15.0 mg, 0.04 mmol) in chloroform (0.3 mL) was cooled to 0 °C and DMF (25.0 μL) was added. POCl3 (13.0 μL) was added slowly to the reaction mixture. After 5 min, the reaction was placed in a 90 °C oil-bath. After 6h, the dark red reaction mixture cooled to ambient temperature and was quenched with 2 mL of 0.5 N HCl. The biphasic mixture was extracted with ether (3 × 5 mL). The combined organic layer was washed with water (3 × 5 mL), brine (10 mL) and dried (MgSO4). The crude product was purified by preparatory-TLC (plate size: 10 × 10 cm) with 4:1 hexanes:EtOAc as the eluent to obtain 9.8 mg (58%) of chloro-aldehyde 10. IR (Neat film, NaCl) 2951, 1740, 1716, 1691, 1607, 1476, 1452, 1417, 1371, 1342, 1261, 1200, 1092, 1054, 1037, 968, 910, 734 cm−1. 1H NMR (CDCl3) 10.00 (1H, s), 7.28-7.23 (2H, m), 6.75 (1H, d, J = 6.9 Hz), 3.95 (1H, dd, J = 18.8, 1.8 Hz), 3.84 (1H, d, J = 1.8 Hz), 3.80 (3H, s), 3.73 (3H, s), 3.23 (1H, d, J = 18.8 Hz), 1.77 (3H, s), 1.33 (3H, s). 13C NMR (CDCl3) δ 202.7, 191.0, 171.3, 169.1, 144.1, 135.9, 133.8, 128.9, 124.0, 122.5, 119.3, 113.9, 108.6, 67.8, 61.8, 53.0, 52.5, 38.7, 31.9, 31.2, 30.0. HRMS m/z calc’d for C21H19Cl2NO4Na+ [M+Na]+: 442.0583, found 442.0582.
Silyl enol ether 11
A solution of dione 5 (100 mg, 0.27 mmol) in 2.0 mL THF was cooled to −100 °C. KHMDS (1.33 mL, 0.5 M solution in toluene, 0.67 mmol) was added and the reaction was stirred for 40 min. TBSCl (81.4 mg, 0.54 mmol) in 0.4 mL THF was added and the reaction was allowed to warm up to −10 °C over a period of 1h. The reaction was quenched with 2 mL of aqueous saturated solution of NaHCO3 and extracted with 1:1 hexanes:ether mixture. Combined organic extracts were washed with water, brine and dried (MgSO4). The crude material was purified via column chromatography (4:1 hexanes:EtOAc) to afford 133.1 mg (~100%) and used immediately afterwards for the next step. The corresponding TIPS ether was prepared analogously. 1H NMR of the TBS ether (desired regioisomer, CDCl3) δ 7.18-7.13 (2H, m), 6.62 (1H, dd, J = 6.7, 1.7 Hz), 5.74 (1H, s), 3.72 (3H, s), 3.71 (3H, s), 2.83 (1H, ddd, J = 16.9, 7.0, 2.8 Hz), 2.74 (1H, dd, J = 9.0, 7.0 Hz), 2.34 (1H, ddd, J = 16.9, 9.0, 1.4 Hz), 1.65 (3H, s), 1.47 (3H, s), 0.84 (9H, s), 0.13 (3H, s), 0.04 (3H, s). 1H NMR of the TIPS ether (desired regioisomer, CDCl3) δ 7.16-7.11 (2H, m), 6.60 (1H, dd, J = 6.6, 1.7 Hz), 5.74 (1H, s), 3.71 (3H, s), 3.70 (3H, s), 2.86 (1H, ddd, J = 17.0, 7.2, 2.8 Hz), 2.74 (1H, dd, J = 9.1, 7.2 Hz), 2.39 (1H, dd, J = 17.0, 9.1 Hz), 1.66 (3H, s), 1.47 (3H, s), 1.13 (3H, sept, J = 7.5 Hz), 0.99 (9H, d, J = 7.4 Hz), 0.94 (9H, d, J = 7.4 Hz).
Vinylogous acid 14
A solution of dione 5 (33.0 mg, 0.088 mmol) in THF (0.66 mL) was cooled to −100 °C. KHMDS (0.5 M in toluene, 1.06 mL, 0.53 mmol) was added and the reaction mixture was stirred at −100 °C for 45 min. Neat TFEF (52.0 μL, 0.53 mmol) was added rapidly and the reaction mixture was allowed to warm to 10 °C over 3h. The reaction was quenched with 0.1 N HCl (2 mL) and was extracted with EtOAc (3 × 5 mL). The pH of the aqueous layer should be < 6 for complete extraction of the products into the organic layer. The combined organic layers were dried (MgSO4) and concentrated. The crude material was purified via preparatory-TLC (plate size 10 cm × 20 cm, eluent 4:1 hexanes:EtOAc) to afford acid 14 as a white solid (21.2 mg, 60%) and ~2 mg of 13 (~ 5%). Data for 14: IR (Neat film, NaCl) 2951, 2917, 1740, 1713, 1638, 1575, 1476, 1451, 1435, 1417, 1370, 1339, 1276, 1250, 1221, 1155, 1082, 1055, 1037, 744 cm−1. 1H NMR (CDCl3) δ 14.93 (1H, d, J = 4.7 Hz), 8.69 (1H, d, J = 4.7 Hz), 7.25-7.18 (2H, m), 6.73 (1H, d, J = 7.1 Hz), 3.80 (3H, s), 3.72 (3H, s), 3.66 (1H, d, J = 18.3 Hz), 3.53 (1H, s), 3.16 (1H, d, J = 18.3 Hz), 1.93 (3H, s), 1.33 (3H, s). 13C NMR (CDCl3) δ 204.1, 184.9, 183.8, 171.5, 136.0, 128.0, 124.4, 122.5, 120.3, 119.4, 113.9, 109.6, 108.6, 65.9, 61.6, 52.9, 49.0, 38.2, 30.8, 30.7, 30.0. HRMS m/z calc’d for C21H20ClNO5Na+ [M+Na]+: 424.0922, found 424.0924.
Bromoindole 17
To a solution of enol ether 437 (558.0 mg, 1.8 mmol) and indole 161a (881.0 mg, 2.2 mmol) in CH2Cl2 (40.0 mL) at −78 °C was added SnCl4 (0.42 mL, 3.6 mmol). The solution turned orange red and was stirred for 50 min. The reaction was quenched by the addition of saturated aqueous NaHCO3 (8 mL) and allowed to warm to room temperature. The mixture was filtered through Celite® and added to a separatory funnel. The organic layer was washed with saturated aqueous NaHCO3 (20 mL) and brine (20 mL), then dried (MgSO4) and concentrated to give a white foam that was a >10:1 diastereomeric ratio as determined by 1H NMR. The crude product was purified by flash chromatography using 100:1 toluene:ether as eluent to afford 537.0 mg of pure 17 (48%) as a white solid. IR (Neat film, NaCl, cm−1) 2969, 2931, 1726, 1715, 1456, 1406, 1369, 1280, 1176, 1149, 1090, 962. 1H NMR (C6D6 at 323K) δ 8.14 (1H, d, J = 8.1 Hz), 7.60 (1H, br s), 7.54 (2H, br d, J = 6.3 Hz), 7.25 (1H, d, J = 7.8 Hz), 6.69 (2H, d, J = 7.6 Hz), 6.66 (1H, t, J = 8.1 Hz), 5.48 (1H, dd, J = 17.6, 11.0 Hz), 5.14 (1H, d, J = 17.6 Hz), 4.95 (1H, d, J = 11.0 Hz), 4.88 (1H, br m), 4.13 (1H, dd, J = 13.1, 5.6 Hz), 2.48 (1H, d, J = 13.5 Hz), 2.28 (1H, d, J = 13.5 Hz), 1.92 (3H, br s), 1.85 (3H, s), 1.8-1.7 (2H, m), 1.38 (3H, m), 0.89 (9H, br s), 0.83 (3H, s). 13C NMR (C6D6 at 323K) δ 207.8, 176.8, 145.0, 142.7, 139.1, 135.5, 131.3, 130.3, 129.3, 126.8, 126.5, 125.1, 115.5, 114.2, 114.0, 73.7, 49.7, 49.5, 46.4, 38.8, 36.2, 30.8, 30.1, 27.5, 27.1, 24.9, 21.1. HRMS m/z calc’d for C32H38BrNO5SNa+ [M+Na]+: 650.1546, found 650.1547.
N-Methyl indole 18
To a solution of 17 (760.0 mg, 1.21 mmol) in EtOH (35.0 mL) was added crushed KOH (1.35 g, 24.0 mmol). The solution was heated at 60 °C for 40 min and monitored by TLC. The reaction was cooled to room temperature, quenched with saturated aqueous NH4Cl (20 mL) and extracted with EtOAc (3 × 20 mL). The organic layers were washed with brine (50 mL), dried (Na2SO4), and concentrated to give a brown solid. The crude product was dissolved in toluene (33 mL). Tetrabutylammonium hydroxide (40% aqueous solution, 2.5 mL, 3.8 mmol) and MeI (2.5 mL, 40.0 mmol) were added. The mixture was stirred at room temperature until the reaction was complete as judged by TLC (ca. 2h). After completion, 10% aqueous solution HCl (20 mL) was added along with saturated aqueous NH4Cl (20 mL), and the mixture was extracted with 1:1 hexanes:Et2O (3 × 50 mL). The organic layers were combined, washed with brine (80 mL), dried (Na2SO4), and concentrated to give a white foam. The crude product was purified by flash chromatography using 8:1 hexanes:EtOAc as eluent to provide 490.0 mg of 18 (83% over two steps). Pure material was obtained by crystallization from benzene:hexanes. IR (Neat film, NaCl) 2971, 2933, 2873, 1718, 1547, 1479, 1416, 1282, 1151, 1113, 911 cm−1. 1H NMR (C6D6 at 323K) δ 7.40 (1H, m), 6.8-6.7 (2H, m), 6.54 (1H, br s), 5.52 (1H, dd, J = 17.6, 11.2 Hz), 5.18 (1H, d, J = 17.6 Hz), 5.01 (1H, br s), 4.95 (1H, d, J = 11.2 Hz), 4.43 (1H, dd, J = 12.8, 5.6 Hz), 2.81 (3H, s), 2.62 (1H, d, J = 13.2 Hz), 2.35 (1H, d, J = 13.2 Hz), 2.16 (3H, br s), 1.90 (1H, dt, J = 13.6, 2.8 Hz), 1.72 (1H, br s), 1.55 (3H, s), 1.09 (9H, s), 0.88 (3H, s). 13C NMR (C6D6 at 323K) δ 208.3, 177.1, 143.0, 140.4, 128.5, 125.8, 125.7, 124.3, 122.0, 115.3, 114.3, 109.2, 74.2, 54.4, 51.0, 49.8, 46.5, 39.1, 36.2, 36.1, 32.1, 31.2, 27.4, 25.0. HRMS m/z calc’d for C26H34BrNO3Na+ [M+Na]+: 510.1614, found 510.1610.
Enol carbonate 21
To a solution of ketone 18 (63.0 mg, 0.13 mmol) in THF (0.5 mL) was cooled to −78 °C and a freshly prepared solution of LDA (0.16 mL, 0.16 mmol, 1M solution in THF) was added. The pale yellow solution was stirred for 30 min then treated with HMPA (27.0 μL, 0.16 mmol), followed by methylcyanoformate (13.0 μL, 0.17 mmol). The resulting solution was stirred for 5h then quenched with water (5 mL). The mixture was extracted with 1:1 ether:hexanes (3 × 10 mL) then the combined organic layers were washed with water (10 mL) and brine (10 mL). The solution was dried (Na2SO4) and concentrated to afford 60.0 mg (96%) of 21 as a foam that was sufficiently pure for the next reaction. IR (Neat film, NaCl) 2968, 2934, 2873, 1762, 1722, 1679, 1547, 1480, 1439, 1416, 1254, 1167, 1112, 1035, 998, 919 cm−1. 1H NMR (CDCl3) δ 7.37 (1H, d, J = 7.5 Hz), 7.24 (1H, d, J = 8.2 Hz), 7.02 (1H, br s), 6.98 (app t, J = 7.5 Hz), 5.82 (1H, dd, J = 16.1, 10.4 Hz), 5.15 (1H, d, J = 13.0 Hz), 5.09 (1H, d, J = 6.1 Hz), 4.95 (1H, br s), 4.33 (1H, br s), 3.74 (3H, s), 2.73 (3H, s), 2.27-2.24 (4H, m), 1.93-1.88 (1H, m), 1.62-1.58 (1H, m), 1.54 (3H, s), 1.37 (3H, s), 1.27 (9H, s), 1.05 (3H, s). 13C NMR (CDCl3) δ 178.4, 151.4 144.2, 139.5, 129.8, 128.5, 125.2, 121.6, 118.7, 115.7, 113.8 108.6, 73.3, 64.3, 54.0, 43.4, 39.1, 35.7, 32.9, 29.5, 27.4, 26.4, 24.8, 23.8, 19.1, 13.7. HRMS m/z calc’d for C28H36BrNO5Na+ [M+Na]+: 568.1669, found 568.1661.
Tetracycle 20
A flask was charged with Pd(OAc)2 (3.5 mg, 0.015 mmol) and KOtBu (5.0 mg, 0.044 mmol). The flask was purged with Ar. Tri-t-butylphosphine (138.0 μL, 0.031 mmol, 0.224 M solution in toluene) was added and the mixture was stirred at room temperature for 10 min. Next, a solution of substrate 21 (12.0 mg, 0.022 mmol) in toluene (1.0 mL) was added. The suspension was stirred for 5 min, then placed in a 70 °C oil bath and stirred for 4 h. The reaction was cooled to room temperature, filtered through a short plug of Celite with Et2O (5 mL) and concentrated. The crude product was purified by flash chromatography using 4:1 hexanes:EtOAc as the eluent to provide 3.0 mg (34%) of 20. IR (Neat film, NaCl) 2968, 2928, 2871, 1725, 1705, 1606, 1479, 1456, 1419, 1367, 1284, 1257, 1163, 1033 cm−1. 1H NMR (CDCl3) δ 7.19 (1H, d, J = 8.2 Hz), 7.11 (1H, dd, J = 8.1, 7.2 Hz), 6.98 (1H, s), 6.69 (1H, d, J = 7.2 Hz), 5.01-4.88 (3H, m), 4.86 (1H, dd, J = 11.5, 5.5 Hz), 3.75 (3H, s), 3.65 (1H, d, J = 1.6 Hz), 2.69 (1H, d, J = 8.5 Hz), 2.46 (1H, dd, J = 14.0, 5.5 Hz), 1.98 (1H, ddd, J = 14.0, 11.7, 8.5 Hz), 1.60 (3H, s), 1.29 (3H, s), 1.08 (3H, s), 1.02 (9H, s). 13C NMR (CDCl3) δ 211.3, 177.6, 142.9, 137.2, 126.6, 124.8, 124.7, 122.3, 121.7, 120.8, 113.6, 108.3, 70.0, 68.8, 59.7, 48.3, 38.6, 35.7, 35.6, 32.9, 27.9 27.6, 26.9, 17.3. HRMS m/z calc’d for C26H32NO3Na+ [M+Na]+: 430.2353, found 430.2350.
Pentacycle 31
To a solution of PhSeCl (6.0 mg, 0.031 mmol) in 0.5 mL DCM at 0 °C (ice-bath) was added tetracycle 251b (10.0 mg, 0.021 mmol) in 0.5 mL DCM and the reaction was stirred at 0 °C for 1h. After 1h, the ice bath was removed and the reaction was stirred for further 0.5h. The reaction was concentrated and the crude mixture was loaded on a preparatory-TLC plate (plate size 10 × 10 cm, eluent 4:1 hexanes:EtOAc) to afford 7.1 mg (53%) of pentacycle 31 as white solid. Better yield of 31 was obtained when N-phenylselenophthalimide (NPSP) and catalytic camphor sulfonic acid (CSA) was used in place of PhSeCl. IR (Neat film, NaCl): 2954, 2927, 2855, 1727, 1700, 1473, 1462, 1420, 1365, 1254, 1090, 1056 cm−1. 1H NMR (CDCl3) δ 9.58 (1H, s), 7.42 (2H, d, J = 7.1 Hz), 7.25-7.21 (4H, m), 7.09 (1H, d, J = 8.1 Hz), 6.93 (1H, s), 4.08 (1H, d, J = 4.6 Hz), 3.78 (3H, s), 3.73 (1H, br s), 3.31 (1H, dd, J = 14.7, 2.5 Hz), 3.11 (1H, dd, J = 14.9, 2.4 Hz), 2.72 (1H, dd, J = 11.3, 7.0 Hz), 2.58 (1H, dd, J = 13.7, 11.3 Hz), 2.02-1.91 (1H, m), 1.55 (3H, s), 1.50 (3H, s), 1.31 (3H, s), 0.87 (9H, s), −0.10 (3H, s), −0.23 (3H, s). 13C NMR (CDCl3) δ 212.8, 197.3, 136.3, 132.9, 132.8, 130.8, 129.3, 127.5, 127.1, 125.0, 123.4, 122.6, 120.4, 108.4, 74.1, 67.8, 59.2, 56.7, 55.1, 37.3, 36.3, 33.0, 32.97, 32.1, 32.0, 25.8, 23.4, 18.2, −4.6, −5.3. HRMS m/z calc’d for C34H44NO3SeSi [M+H]+: 622.2253, found 622.2251.
Enol triflate 34
A solution of dione 331b (23.0 mg, 0.066 mmol) in THF (3.5 mL) was cooled to −78 °C. KHMDS (0.5 M in toluene, 0.26 mL, 0.132 mmol) was added and the enolate solution was stirred at the same temperature for 15 min. PhNTf2 (47.2 mg, 0.132 mmol) in THF (1.5 mL) was then added and the reaction was followed by TLC. After 30 min, the reaction was quenched with 1 mL aqueous saturated solution of NaHCO3 and extracted with EtOAc (3 × 10 mL). Combined organic layers were dried (MgSO4) and concentrated. The crude material was purified via column chromatography (6:1 → 3:1 hexanes:EtOAc) to obtain 21.4 mg (68%) of enol triflate 34 as a yellow solid. IR (Neat film, NaCl): 2924, 2854, 1736, 1704, 1451, 1418, 1378, 1334, 1246, 1214, 1140, 1028, 1012 cm−1. 1H NMR (CDCl3) δ 9.66 (1H, s), 7.35 (1H, d, J = 8.0 Hz), 7.23 (1H, d, J = 8.0 Hz), 7.00 (1H, s), 5.87 (1H, d, J = 3.2 Hz), 5.52 (1H, dd, J = 17.6, 10.4 Hz), 5.42 (1H, d, J = 10.4 Hz), 5.41 (1H, d, J = 17.6 Hz), 3.80 (3H, s), 3.19 (1H, d, J = 3.2 Hz), 1.71 (3H, s), 1.64 (3H, s), 1.33 (3H, s). 13C NMR (CDCl3) δ 207.4, 194.1, 151.9, 137.9, 134.9, 126.6, 125.7, 123.5, 120.8, 120.6, 119.2, 118.7, 115.8, 110.3, 73.1, 60.1, 50.4, 36.7, 33.6, 33.1, 28.8, 21.0, 14.2. HRMS m/z calc’d for C23H22F3NO5SNa+ [M+Na]+: 504.1063, found 504.1059.
Pentacycle 36
To a bright red THF (0.2 mL) solution of Ru(acac)3 (1.0 mg, 0.0025 mmol) was added LiCl (2.0 mg, 0.047 mmol) and the reaction vessel was purged with N2. EtMgBr (0.01 mL, 1.0 M in THF, 0.01 mmol) was added and the reaction mixture turned dark brown. The reaction was stirred at ambient temperature for 15 min at which point a 0.5 mL THF solution of triflate 34 (5.0 mg, 0.01 mmol) was added and the reaction mixture was again purged with N2 and placed in an oil-bath preheated to 60 °C. After 20h the reaction was cooled to ambient temperature and filtered over a 1″ silica plug (pipette) and concentrated. The crude material was purified via preparatory-TLC (plate size 10 cm × 10 cm; eluent 4:1 hexanes:EtOAc) to afford pentacycle 36 as a white solid (50–70%). IR (Neat film, NaCl): 2959, 2922, 2853, 1752, 1716, 1465, 1446, 1419, 1376, 1365, 1251, 1208, 1143 cm−1. 1H NMR (CDCl3) δ 10.06 (1H, s), 7.04 (1H, d, J = 8.0 Hz), 6.92 (1H, d, J = 8.0 Hz), 6.89 (1H, s), 6.08 (1H, d, J = 1.4 Hz), 5.70 (1H, d, J = 9.0 Hz), 3.71 (3H, s), 3.52 (1H, d, J = 19.8 Hz), 3.23 (1H, dd, J = 19.8, 9.0 Hz), 3.18 (1H, br s), 1.91 (3H, s), 1.68 (3H, s), 1.33 (3H, s). 13C NMR (CDCl3) δ 211.0, 198.1, 141.8, 136.1, 130.5, 130.2, 129.8, 129.7, 127.7, 125.9, 123.1, 122.8, 121.9, 108.1, 74.1, 64.6, 41.4, 34.4, 33.6, 33.0, 32.9, 20.8. HRMS m/z calc’d for C22H21NO2Na [M+Na]+: 354.1465, found 354.1463.
Supplementary Material
Scheme 5.

Successful Synthesis of the Functionalized Welwitindolinone Core
Acknowledgments
Generous financial grant from the National Cancer Institute of the NIH (R01 CA101438) is gratefully acknowledged. J.A.M gratefully acknowledges postdoctoral fellowship support (#PF-04-016-01-CDD) from the American Cancer Society. We thank Dr. I. M. Steele and Dr. A. Jurkiewicz for X-ray crystallographic and NMR spectroscopic assistance, respectively.
Footnotes
This paper is submitted in celebration of the 90th birthday of Professor Gilbert Stork, whose unequaled creativity and boundless generosity has inspired generations of chemists, including the authors of the present publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) MacKay JA, Bishop RL, Rawal VH. Org Lett. 2005;7:3421–3424. doi: 10.1021/ol051043t. [DOI] [PubMed] [Google Scholar]; (b) Bhat V, Allan KM, Rawal VH. J Am Chem Soc. 2011;133:5798–5801. doi: 10.1021/ja201834u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bhat V, MacKay JA, Rawal VH. Org Lett. 2011;13:3214–3217. doi: 10.1021/ol201122f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bhat V, Rawal VH. Chem Commun. 2011;47:9705–9707. doi: 10.1039/c1cc13498a. [DOI] [PubMed] [Google Scholar]
- 2.(a) Stratmann K, Moore RE, Bonjouklian R, Deeter JB, Patterson GMLS, Shaffer Smith CD, Smitka TA. J Am Chem Soc. 1994;116:9935–9942. [Google Scholar]; (b) Jimenez JI, Huber U, Moore RE, Patterson GML. J Nat Prod. 1999;62:569–572. doi: 10.1021/np980485t. [DOI] [PubMed] [Google Scholar]
- 3.For approaches toward the welwitindolinones (1–3), see: Konopelski JP, Deng H, Schiemann K, Keane JM, Olmstead MM. Synlett. 1998:1105–1107.Wood JL, Holubec AA, Stoltz BM, Weiss MM, Dixon JA, Doan BD, Shamji MF, Chen JM, Heffron TP. J Am Chem Soc. 1999;121:6326–6327.Kaoudi T, Ouiclet-Sire B, Seguin S, Zard SZ. Angew Chem, Int Ed. 2000;39:731–733.Deng H, Konopelski JP. Org Lett. 2001;3:3001–3004. doi: 10.1021/ol016379r.Jung ME, Slowinski F. Tetrahedron Lett. 2001;42:6835–6838.López-Alvarado P, García-Granda S, Àlvarez-Rúa C, Avendaño C. Eur J Org Chem. 2002:1702–1707.Ready JM, Reisman SE, Hirata M, Weiss MM, Tamaki K, Ovaska TV, Wood JL. Angew Chem, Int Ed. 2004;43:1270–1272. doi: 10.1002/anie.200353282.Baudoux J, Blake AJ, Simpkins NS. Org Lett. 2005;7:4087–4089. doi: 10.1021/ol051239t.Greshock TJ, Funk RL. Org Lett. 2006;8:2643–2645. doi: 10.1021/ol0608799.Lauchli R, Shea KJ. Org Lett. 2006;8:5287–5289. doi: 10.1021/ol0620747.Xia J, Brown LE, Konopelski JP. J Org Chem. 2007;72:6885–6890. doi: 10.1021/jo071156l.Richter JM, Ishihara Y, Masuda T, Whitefield BW, Llamas T, Pohjakallio A, Baran PS. J Am Chem Soc. 2008;130:17938–17954. doi: 10.1021/ja806981k.Boissel V, Simpkins NS, Bhalay G, Blake AJ, Lewis W. Chem Commun. 2009:1398–1400. doi: 10.1039/b820674k.Boissel V, Simpkins NS, Bhalay G. Tetrahedron Lett. 2009;50:3283–3286.Tian X, Huters AD, Douglas CJ, Garg NK. Org Lett. 2009;11:2349–2351. doi: 10.1021/ol9007684.Trost BM, McDougall PJ. Org Lett. 2009;11:3782–3785. doi: 10.1021/ol901499b.Brailsford JA, Lauchli R, Shea KJ. Org Lett. 2009;11:5330–5333. doi: 10.1021/ol902173g.Freeman DB, et al. Tetrahedron. 2010;66:6647–6655. doi: 10.1016/j.tet.2010.04.131.Heidebrecht RW, Jr, Gulledge B, Martin SF. Org Lett. 2010;12:2492–2495. doi: 10.1021/ol1006373.Ruiz M, López-Alvarado P, Menéndez JC. Org Biomol Chem. 2010;8:4521–4523. doi: 10.1039/c0ob00382d.
- 4.Total syntheses of welwitindolinone A: Baran PS, Richter JM. J Am Chem Soc. 2005;127:15394–15396. doi: 10.1021/ja056171r.Reisman SE, Ready JM, Hasuoka A, Smith CJ, Wood JL. J Am Chem Soc. 2006;128:1448–1449. doi: 10.1021/ja057640s.For the synthesis of welwitindolinone D (3), see ref 1b. (d) While the present manuscript was under review for publication, a report appeared in J. Am. Chem. Soc., ASAP on the total synthesis of welwitindolinone C (2): Huters AD, Quasdorf KW, Styduhar ED, Garg NK. J Am Chem Soc. 2011 doi: 10.1021/ja206538k.
- 5.For a review on the Vilsmeier–Haack reaction, see: Meth-Con O, Stanforth SP. Comp Org Syn. 1991;2:777–794.For an example of deconjugative methylation of a cyclic enal, see: Burnell RH, Côté C, Théberge N. J Nat Prod. 1993;56:1459–1467.
- 6.TFEF = 2,2,2-trifluoroethylformate. Zayia GH. Org Lett. 1999;1:989–991.
- 7.Sakagami M, Muratake H, Natsume M. Chem Pharm Bull. 1994;42:1393–1398. [Google Scholar]
- 8.See Supporting Information for synthesis.
- 9.Funk and coworkers have also reported difficulties with a similar acylation step. See: ref 3i.
- 10.For a full discussion of this observation, see ref 1d.
- 11.(a) Reich HJ. J Org Chem. 1974;39:428–429. [Google Scholar]; (b) Sharpless KB, Lauer RF. J Org Chem. 1974;39:429–430. [Google Scholar]; (c) Nicolaou KC, Petasis NA, Claremon DA. Tetrahedron. 1985;41:4835–4841. [Google Scholar]
- 12.(a) Jung ME, Wasserman JI. Tetrahedron Lett. 2003;44:7273–7275. [Google Scholar]; (b) Jung ME, Murphy JM. Tetrahedron Lett. 2007;48:8388–8391. [Google Scholar]
- 13.Lambert JB, Wang GT, Finzel RB, Teramura DH. J Am Chem Soc. 1987;109:7838–7845. [Google Scholar]
- 14.Moughamir K, Mezgueldi B, Atmani A, Mestdagh H, Rolando C. Tetrahedron Lett. 1998;3:59–62. [Google Scholar]
- 15.(a) Su W, Jin C. Org Lett. 2007;9:993–996. doi: 10.1021/ol062991c. [DOI] [PubMed] [Google Scholar]; (b) Spaggiari A, Vaccari D, Davoli P, Torre G, Prati F. J Org Chem. 2007;72:2216–2219. doi: 10.1021/jo061346g. [DOI] [PubMed] [Google Scholar]; (c) Isaacs NS, Kirkpatrick D. J Chem Soc Chem Comm. 1972:443–444. [Google Scholar]; (d) Mateeva ED, Feshin DB, Zefirov NS. Russ J Org Chem. 2001;37:63–66. [Google Scholar]; (e) Kamei K, Maeda N, Tatsuoka T. Tetrahedron Lett. 2005;46:229–232. [Google Scholar]
- 16.Wulff WD, Peterson GA, Bauta WE, Chan K–S, Faron KL, Gilbertson SR, Kaeslar RW, Yang DC, Murray CK. J Org Chem. 1986;51:277–279. [Google Scholar]
- 17.Shirakawa E, Imazaki Y, Hayashi T. Chem Commun. 2009:5088–5090. doi: 10.1039/b907761h. [DOI] [PubMed] [Google Scholar]
- 18.Shen X, Hyde AM, Buchwald SL. J Am Chem Soc. 2010;132:14076–14078. doi: 10.1021/ja107481a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.It is interesting to note that in Garg’s recent synthesis of welwitindolinone C (2) (see reference 4d), no such rearrangement is observed on a substrate which differs from 34only in that it has an unsubstituted C11-position (H in place of CHO). Evidently, the aldehyde functionality in 34weakens the C11-C12 bond so as to facilitate the Cope pathway. The differences in the reactivity of these two closely related substrates provides yet another example of how small structural changes can dramatically alter reactivity.
- 20.For a review on the Cope rearrangement, see: Rhoads SJ, Raulins NR. Org React. 1975;22:1.
- 21.(a) Marvell EN, Lin C. J Am Chem Soc. 1978;100:877–883. [Google Scholar]; (b) Lambert JB, Fabricius DM, Hoard JA. J Org Chem. 1979;44:1480–1485. [Google Scholar]; (c) Seki K, Tooya M, Sato T, Ueno M, Uyehara T. Tetrahedron Lett. 1998;39:8673–8676. [Google Scholar]; (d) Marvell EN, Almond SW. Tetrahedron Lett. 1979;20:2779–2780. [Google Scholar]
- 22.The reverse aromatic Cope rearrangement has, in fact, been used successfully: Kawasaki T, Nonaka Y, Watanabe K, Ogawa A, Higuchi K, Terashima R, Masuda K, Sakamoto M. J Org Chem. 2001;66:1200–1204. doi: 10.1021/jo0014921.
- 23.For convenience, intermediate 37 is shown to have already undergone oxidative addition by the transition metal. It is also possible that the rearrangement takes place prior to oxidative addition.
- 24.Similar rearrangements, albeit in simple acyclic systems, have been observed, see: Nishiyama T, Esumi T, Iwabuchi Y, Irie H, Hatakayema S. Tetrahedron Lett. 1998;39:43–46.Ogasawara M, Ikeda H, Hayashi T. Angew Chem, Int Ed. 2000;39:1042–1044.Djahanbini D, Cazes B, Gore J. Tetrahedron Lett. 1984;25:203–206.Djahanbini D, Cazes B, Gore J. Tetrahedron. 1987;43:3441–3452.For a review, see: Ogasawara M, Hayashi T. In: Modern Allene Chemistry. Krause N, Hashmi ASK, editors. Vol. 1. Wiley-VCH; Weinheim: 2004. pp. 93–140.
- 25.This axiom is a variation of Woodward’s pithy statement: “…there are no general reactions.” See: Woodward RB. In: Milligan WO, editor. Organic Synthesis; Proceedings of the Robert A. Welch Foundation Conference on Chemical Research, XII; Huston, TX. 1969. pp. 3–6.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.