

Abstract
This Letter describes the continued optimization of an MLPCN probe molecule (ML012) through an iterative parallel synthesis approach. After exploring extensive modifications throughout the parent structure, we arrived at a more highly M1-selective antagonist, compound 13l (VU0415248). Muscarinic subtype selectivity across all five human and rat receptors for 13l, along with rat selectivity for the lead compound (ML012), is presented.
Keywords: Muscarinic acetylcholine receptor 1, M1, Antagonist, ML012, VU0415248
Acetylcholine (ACh) is a critical neurotransmitter with diverse functions both within the central nervous system (CNS) and in peripheral signaling pathways.1–4 ACh carries out its functions by interacting with two very distinct groups of receptors; a set of ligand-gated ion channels—the nicotinic acetylcholine receptors (nAChRs)- and a set of family A, G protein-coupled receptors (GPCRs)—the muscarinic acetylcholine receptors (mAChRs). The muscarinic cohort of acetylcholine receptors are further divided into five subtypes (M1–5).5 These five subtypes can be further classified into two subsets based on their G protein-coupling partners, with the M1, 3, 5 receptors preferentially coupling to Gq/11 which stimulates PLC and calcium mobilization, and with the M2,4 receptors preferentially coupling to Gi/o to inhibit adenylate cyclase (AC), thereby decreasing cAMP production.4 These mAChRs are widely expressed throughout the body with varying degrees of expression levels for specific subtypes based on the particular site or organ.6 As a result, mAChRs play significant roles in a wide range of physiological functions such as memory and attention, motor control, nociception, regulation of sleep-wake cycles, cardiovascular function, secretory functions, and mediators of inflammation, renal and GI function, among many others.6,7
While it is believed that the M1,4,5 receptors are the most important for CNS targets, what remains unknown are the specific functions that are regulated by each individual receptor subtype.7 This is a direct result of the highly conserved orthosteric binding site for the endogenous ligand (ACh), which is shared across all five mAChRs; this high conservation has severely confounded the development of muscarinic ligands with a high degree of selectivity for a single subtype.8 A lack of subtype selectivity, however, has not precluded the use of mAChR-acting pharmaceuticals for a wide range of indications but many of the undesirable side effects for these nonselective medications can be attributed to activity at the other mAChRs (often M2 and M3), limiting their clinical utility. Additionally, the limited selectivity displayed by earlier mAChR ligands has complicated the interpretation of both animal model studies as well as human clinical trials with respect to the specific mAChR subtype being responsible for the given desired (or undesired) result. For example, xanomeline, a reported M1- and M4-selective agonist, showed clinical efficacy in Phase II trials for Alzheimer’s disease and schizophrenia,9,10 but also has nearly equivalent agonist activity at M3.8 Even in the absence of off-target mAChR activity, the debate still remains as to whether a single mAChR subtype (M1 or M4) is responsible for the very encouraging positive outcomes, in these trials.
Single and double mAChR knock out (KO) mice for each of the muscarinic receptor subtypes have yielded a great deal of insight into the specific roles played by each receptor, but these results must always be viewed with the caveat that compensatory changes may have occurred during the development of the mutant KO mice.8 More highly selective mAChR ligands would allow for a better understanding of the individual roles for each of the five mAChRs. There are a number of ways to potentially engender mAChR subtype selectivity among synthetic ligands. First, the more classical approach, by simultaneously binding to the orthosteric site while at the same time extending beyond this highly conserved region of the receptor and into adjacent areas which may be less structurally conserved among the other mAChRs (vide infra).11 Second, by binding to a completely distinct region of the mAChR (an allosteric site), which is distinct from the orthosteric binding site and which might also show less amino acid similarity between the five mAChRs. This allosteric approach has been highly successful for a number of the individual mAChRs: M1,12–14 M4,15 and M5.16
In a previous publication from our laboratories, we described the highly selective M1 antagonist ML012 (VU0255035, Fig. 1).11 Although acting at the orthosteric site on all five mAChRs, ML012 achieved M1 selectivity in the range of 45- to 159-fold over M2–5. Additionally, ML012reduced pilocarpine-induced seizures in rodent models at doses that did not induce deficits in contextual freezing, demonstrating that selective M1 antagonists may have greater therapeutic potential than the non-selective muscarinic antagonists presently available. Given the potential for M1 antagonists in such indications as Parkinson’s disease, movement disorders, and Fragile X syndrome,17,18 we have continued the optimization of ML012 and now detail those efforts in this Letter.
Figure 1.
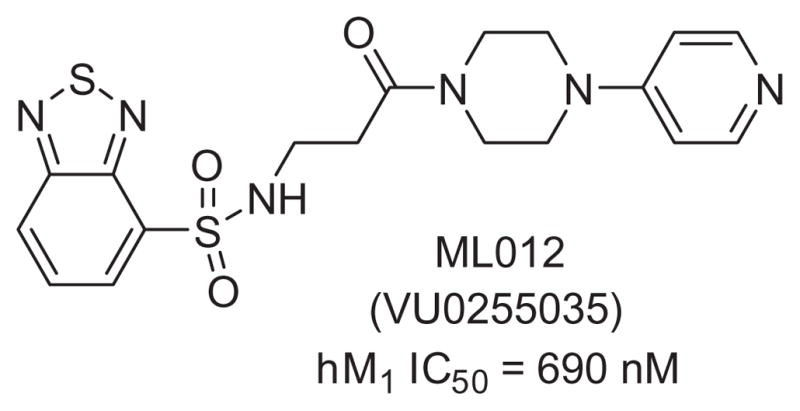
The MLPCN probe molecule ML012.
With ML012 as our starting point, we utilized an iterative parallel synthesis approach to explore structure–activity-relationships (SAR) at both ends of the molecule by employing the synthesis routes depicted in Scheme 1, while simultaneously making more speculative modifications within single compounds. To explore SAR around the Eastern portion, we started with β-alanine methyl ester 1. Sulfonation to provide 2, was then followed by saponification and amide coupling to provide amides (3) appearing in Table 1 containing numerous variations in aryl groups (Ar′). Alternatively, starting from the N-Boc protected β-alanine 4, peptide coupling with a limited number of optimized Eastern portions provided compound 5, which was then treated with HCl subsequent to sulfonation with a variety of sulfonyl chlorides to provide the sulfonamides (3) appearing in Table 2, which now display a wide range of Ar groups on the Western side.
Scheme 1.
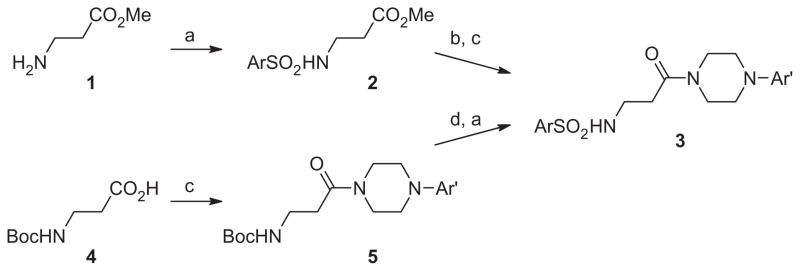
Reagents: (a) ArSO2Cl, NEt3, DCM; (b) NaOH/H2O/THF; (c) amine, EDCI, HOBt, NEt3; (d) 4 M HCl in dioxane.
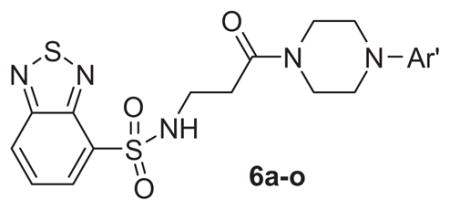
Table 1.
Structures and activities of M1 antagonist analogs 6a–o
| ||
|---|---|---|
| Compd | Ar′ | hM1 IC50 (μM)a |
| ML012 |
|
0.69 |
| 6a |
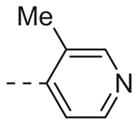
|
2 |
| 6b |
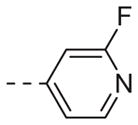
|
>10 |
| 6c |
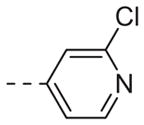
|
>10 |
| 6d |
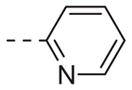
|
>10 |
| 6e |
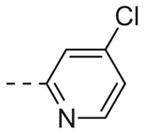
|
4.5 |
| 6f |
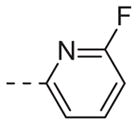
|
>10 |
| 6g |
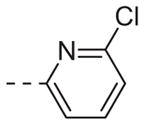
|
>10 |
| 6h |
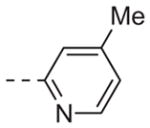
|
>10 |
| 6i |
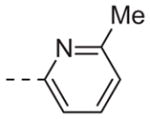
|
>10 |
| 6j |
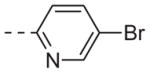
|
0.32 |
| 6k |
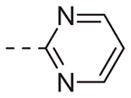
|
>10 |
| 6l |
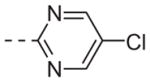
|
>10 |
| 6m |
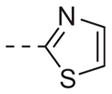
|
>10 |
| 6n |
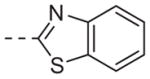
|
>10 |
| 6o |
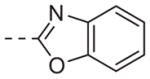
|
>10 |
Average of at least three independent determinations. Standard deviations ranging from 9–19% of the value reported.
Table 2.
Structures and activities of M1 antagonist analogs 13a–r
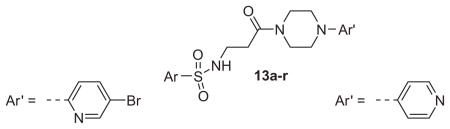
| |||||
|---|---|---|---|---|---|
| Compd | Ar′ | hM1 IC50a (μM) | Compd | Ar′ | hM1 IC50a (μM) |
| 13a |
|
1.1 | 13b |
|
2.6 |
| 13c |
|
>10 | 13d |
|
>10 |
| 13e |
|
>10 | 13f |
|
>10 |
| 13g |
|
4.8 | 13h |
|
>10 |
| 13i |
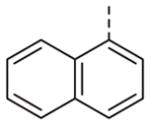
|
>10 | 13j |
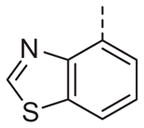
|
>10 |
| 13k |
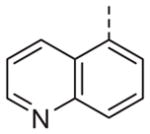
|
2.1 | 13l |
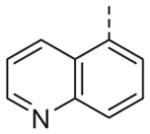
|
0.4 |
| 13m |
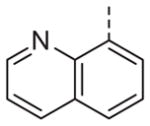
|
>10 | 13n |
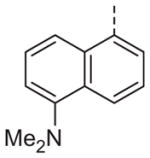
|
>10 |
| 13o |
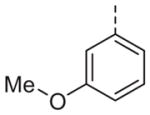
|
>10 | 13p |
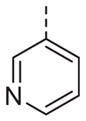
|
>10 |
| 13q |
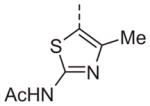
|
>10 | 13r |
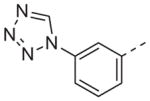
|
>10 |
Average of at least three independent determinations. Standard deviations ranging from 5–23% of the value reported.
Table 1 shows the SAR developed around a variety of aryl groups attached to the piperazine. With ML012 as our benchmark (hM1 IC50 = 0.69 μM),19 it can be seen that additional substitutions on the 4-pyridyl ring only resulted in decreased antagonist activity (6a–c), as measured by their ability to inhibit the calcium mobilization induced by ACh. Very few analogs around a 3-pyridyl moiety (structures and data not shown) were prepared, but these compounds only served to confirm exploratory SAR demonstrating that a 3-pyridyl was the least tolerated.11 Alternatively, substitutions around a 2-pyridyl scaffold were more fruitful. As a starting point, the unsubstituted 2-pyridyl analog 6d possessed inhibitory activity roughly 14-fold less than ML012, but a modest improvement resulted from chlorination para to the nitrogen, as in compound 6e. Halogenation at the ortho-position caused a decrease in activity (6f and 6g), as did methylation at either the para- or the ortho-position (6h and 6i). The largest improvement in activity resulted from the substitution of a bromine at the meta-position shown in compound 6j. This M1 antagonist displayed activity (IC50 = 0.32 μM) 2-fold greater than ML012 and provided an alternative Eastern aryl group for additional rounds of SAR. Replacing the terminal pyridine with a variety of heterocycles met with similarly unforgiving steep SAR (6k–o).
While not conducted in an exhaustive fashion, various replacements for the piperazine heterocycle were explored. However, none of the more commonly employed surrogates shown in Fig. 2 provided antagonists with appreciable activity.
Figure 2.
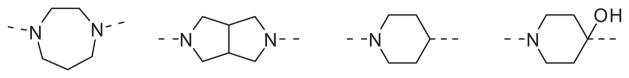
Piperazine replacements that did not maintain potency (hM1 IC50 >10 μM).
The β-alanine central linker was also explored with respect to the various modifications shown in Fig. 3. The reduced amide analog of ML012, compound 7 (Fig. 3), while still an antagonist, showed a greater than 14-fold loss in potency (hM1 IC50 >10 μM, but only 17% ACh activity remaining at the highest concentration tested (30 μM)). Methylation at the α-position of compound 13b (See Table 2) provided a pair of inactive enantiomers (8). However, fluorination at this location did not remove all activity, but instead provided a racemic mixture with somewhat reduced inhibitory efficacy (9, 25% activity remaining at 30 μM) relative to the non-fluorinated analog, 13b (hM1 IC50 = 4.5 μM). It should not be overlooked that compound 9 was a mixture of two enantiomers while compound 13b was a single achiral molecule. Given the roughly 2-fold decrease in potency for 9, there remains the possibility that one enantiomer of 9 had about the same potency as its non-fluorinated progenitor 13b, while the other enantiomer of 9 could be inactive. Regardless, substitution by either of these groups was not obviously beneficial with respect to potency. The individual β-substituted enantiomers, 10 and 11, were prepared from commercially available starting materials, and while both enantiomers were less potent than their unsubstituted analog 13b, there was some evidence for enantiospecific activity between the two compounds. The (R)-enantiomer (10) was completely inactive at the highest concentration tested (30 μM), while the (S)-enantiomer (11) demonstrated about 30% inhibition of the acetylcholine response at the same concentration. The free N–H of the sulfonamide was important for M1 antagonist activity since N-methylation of ML012 to provide compound 12 resulted in a greater than 14-fold loss in potency (hM1 IC50 >10 μM).
Figure 3.
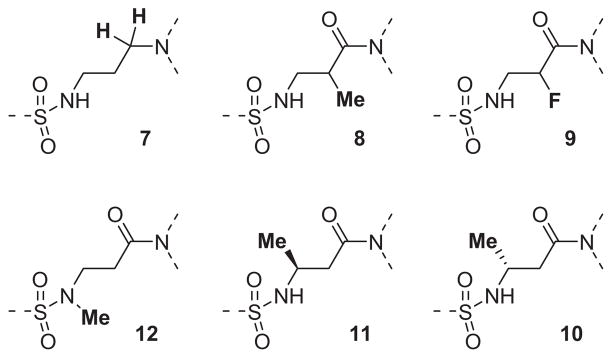
A collection of central linker modifications.
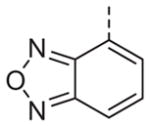
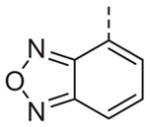
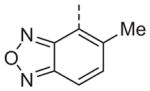
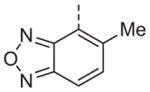
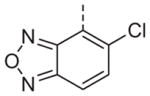
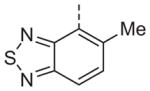
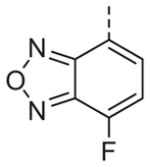
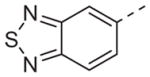
Now focusing on the Western sulfonamide region, and employing our two best Eastern aryl groups, SAR was similarly restrictive (Table 2—Ar′= 4-pyridyl (analogs on the right) and 5-bromo-2-pyridyl (analogs on the left)). The slight change associated with replacing the benzothiadiazole with a benzoxadiazole in compounds 13a and 13b resulted in a 3-fold and 4-fold decrease in activity relative to their sulfur analogs 6j and ML012, respectively. Substitution on either of these bicyclic heterocycles at the 5- or 7-positions resulted in weakly active or inactive compounds (13c–g). Moving the sulfonamide from the 4-position to the 5-position on the benzothiadiazole was also not tolerated, as can be seen with compound 13h. Although it was not surprising that removing all heteroatoms from the sulfonamide aryl ring would result in a complete loss of activity (13i), it was somewhat surprising that the benzothiazole sulfonamide 13j was also inactive. This was particularly unexpected considering the discovery that a single nitrogen atom reintroduced into the naphthalene sulfonamide, to arrive at the quinoline sulfonamides 13k and 13l, could provide compounds with activity on par with, or better than, their benzothiadiazole and benzoxadiazole analogs, and highlights the importance of this nitrogen’s positioning. This nitrogen effect was very specific for mimicking the interactions presumably occurring with the N-1 nitrogen of the benzothiadiazole. Hence, the introduction of a nitrogen atom on the other side of the naphthalene, to mimic the N-3 nitrogen of the benzothiadiazole, resulted in a very weakly active compound (quinoline13m). Further attempts to present heteroatoms in this approximate location generally met with no success (13n–r).
Ultimately, 6j and 13l represented the only compounds with improved antagonist activity relative to ML012. The quinoline sulfonamide 13l was deemed more attractive due to more pronounced improvements in its calculated and physical properties when compared to either ML012 or 6j. Although not a major difference, 13l possesses the lowest molecular weight among the three compounds (MW ML012 = 432, 6j = 511 and 13l = 425). Both 6j and 13l displayed more moderate cLogP values (1.88 and 1.04, respectively) relative to ML012 (cLogP = 0.37). The quinoline sulfonamide compound 13l no longer contained the electron deficient benzothiadiazole found in both ML012 and 6j, which subsequently reduced its polar surface area to 96 Å2 and favorably compared to the other two compounds which both had a polar surface area of 108 Å2. With these properties in mind, compound 13l was profiled against both the rat and human M1–5 receptors in a functional assay for calcium mobilization. Figure 4A shows these results for the rat (r) M1–5 receptors. Despite being presumably an orthosteric ligand, based on its close structural relationship to ML012, 13l shows very high selectivity for the M1 receptor over the M2–5 subtypes with a similar level of selectivity observed across the human subtypes (hM2–5 IC50s >30 μM, data not shown). For comparison, ML012 was profiled against the complete set of rat M1–5 receptors and these results appear in Figure 4B (data not available at the time of our initial publication on ML012). Gratifyingly 13l represented an improvement over ML012 with respect to muscarinic subtype selectivity at the rat receptor, and to a lesser extent a small improvement with respect to potency (rM1 IC50 = 0.18 μM for 13l and rM1 IC50 = 0.24 μM for ML012). Additionally, the quinoline nitrogen at its specific location indicated the presence of an important interaction between compound 13l and the M1 receptor which does not appear to be present in the M2–5 receptor subtypes. We next evaluated 13l for its potential as an improved in vivo tool, but were disappointed in the compound’s poor pharmacokinetic properties. Consistent with in vitro DMPK predictions of high clearance, 13l displayed an IV clearance value of 77 mL/min/kg, roughly hepatic blood flow, and a shorter half life (t1/2 = 40 min) relative to ML012 in male Sprague– Dawley rats.11 In tandem, 13l was found to display very low estimated bioavailability and undetectable CNS exposure in the rat (% F <10 and undetectable CNS concentrations when dosed orally at 10 mg/kg).
Figure 4.
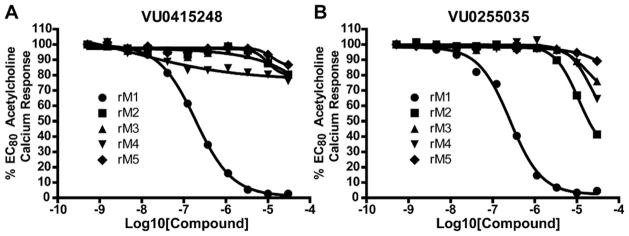
Compound 13l (VU0415248) and ML012 (VU0255035) selectively antagonize the M1 mAChR relative to the M2–5 mAChRs. CRCs were performed in the presence of an EC80 concentration of ACh for each receptor in a calcium mobilization assay. Data were normalized to the maximum response to 30 μM ACh and are presented as a percentage of the EC80 ACh response. A, selectivity for 13l across the rat mAChRs (rM1 EC50 = 0.18 μM). B, selectivity for ML012 across the rat mAChRs (rM1 EC50 = 0.24 μM).
In summary, we have expanded the SAR surrounding ML012, resulting in the development of 13l (VU0415248) as a more potent in vitro tool with improved selectivity for the M1 receptor. On-going work which builds on the SAR described herein may provide compounds with both improved selectivity and PK parameters. This work will be reported in due course. ML012 is an MLPCN probe and freely available upon request.20
Acknowledgments
The authors thank Seaside Therapeutics, NIMH(RO1MH082867), NIH (U54MH084659) and NINDS (P50NS071669) for support of our Program in the development of subtype selective mAChR antagonists.
References and notes
- 1.Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987;237:527. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- 2.Bonner TI, Young AC, Brann MR, Buckley NJ. Neuron. 1988;1:403. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]
- 3.Wess J. Annu Rev Pharmacol Toxicol. 2004;44:423. doi: 10.1146/annurev.pharmtox.44.101802.121622. [DOI] [PubMed] [Google Scholar]
- 4.Langmead CJ, Watson J, Reavill C. Pharmacol Ther. 2008;117:232. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Wess J. Crit Rev Neurobiol. 1996;10:69. doi: 10.1615/critrevneurobiol.v10.i1.40. [DOI] [PubMed] [Google Scholar]
- 6.Wess J, Eglen RM, Gautam D. Nat Rev Drug Discov. 2007;6:721. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 7.Bridges TM, LeBois EP, Hopkins CR, Wood MR, Jones JK, Conn PJ, Lindsley CW. Drug News Perspect. 2010;23:229. doi: 10.1358/dnp.2010.23.4.1416977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinrich JN, Butera JA, Carrick T, Kramer A, Kowal D, Lock T, Marquis KL, Pausch MH, Popiolek M, Sun SC, Tseng E, Uveges AJ, Mayer SC. Eur J Pharmacol. 2009;605:53. doi: 10.1016/j.ejphar.2008.12.044. [DOI] [PubMed] [Google Scholar]
- 9.Bymaster FP, Whitesitt CA, Channon HE, DeLapp N, Ward JS, Calligaro DO, Shipley LA, Buelke-Sam JL, Bodick NC, Farde L, Sheardown MJ, Olesen PH, Hansen KT, Suzdak PD, Swedberg MDB, Sauerberg P, Mitch CH. Drug Dev Res. 1997;40:158. [Google Scholar]
- 10.Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Am J Psychiatry. 2008;165:1033. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 11.Sheffler DJ, Williams R, Bridges TM, Lewis LM, Xiang Z, Kane AS, Byun NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Lindsley CW, Conn PJ. Mol Pharmacol. 2009;76:356. doi: 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bridges TM, Kennedy JP, Noetzel MJ, Breininger ML, Gentry PR, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:1972. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reid PR, Bridges TM, Sheffler DJ, Cho HP, Lewis LM, Days E, Daniels JS, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Wood MR. Bioorg Med Chem Lett. 2011;21:2697. doi: 10.1016/j.bmcl.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuduk SD, Chang RK, Di Marco CN, Ray WJ, Ma L, Wittmann M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. ACS Med Chem Lett. 2010;1:263. doi: 10.1021/ml100095k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey JK, Conn PJ, Lindsley CW. J Pharmacol Exp Ther. 2008;327:941. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges TM, Kennedy JP, Hopkins CR, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:5617. doi: 10.1016/j.bmcl.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veeraragavan S, Bui N, Perkins JR, Yuva-Paylor LA, Carpenter RL, Paylor R. Psychopharmacology. 2011 doi: 10.1007/s00213-011-2276-6. Article ASAP. [DOI] [PubMed] [Google Scholar]
- 18.Healy A, Rush R, Ocain T. ACS Chem Neuro. 2011 doi: 10.1021/cn200019z. Article ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Although the IC50 for ML012 has been previously reported as 132nM (Ref. 11), the inherent variability associated with functional assays supports that the most recent IC50 = 690nM for ML012 is equally valid and can serve as a baseline comparator between the two papers.
- 20.For information on the MLPCN and information on how to request probe compounds, such as ML012, see: http://mli.nih.gov/mli/mlpcn/.