

Abstract
The PKA catalytic (C) subunit is inhibited by two classes of functionally non-redundant regulatory subunits, RI and RII. Unlike RI-subunits, RII-subunits are both substrates and inhibitors. Because RIIβ knockout mice have important disease phenotypes, the RIIβ holoenzyme is a target for developing isoform-specific agonists and/or antagonists. We also know little about the linker region that connects the inhibitor site to the N-terminal dimerization domain although this linker determines the unique globular architecture of the RIIβ holoenzyme. To understand how RIIβ functions as both an inhibitor and substrate and to elucidate the structural role of the linker, we engineered different RIIβ constructs. In the absence of nucleotide RIIβ(108–268) that contains a single cyclic nucleotide binding domain bound C-subunit poorly whereas with AMP-PNP, a non-hydrolyzable ATP analog, the affinity was 11nM. The RIIβ(108–268) holoenzyme structure (1.62Å) with AMP-PNP/Mn2+, showed that we trapped the RIIβ-subunit in an enzyme:substrate complex with the C-subunit in a closed conformation. The enhanced affinity afforded by AMP-PNP/Mn2+ may be a useful strategy for increasing affinity and trapping other protein substrates with their cognate protein kinase. Because mutagenesis predicted that the region N-terminal to the inhibitor site might dock differently to RI and RII, we also engineered RIIβ(102–265) that contained six additional linker residues. The additional linker residues in RIIβ(102–265) increased the affinity to 1.6 nM suggesting that docking to this surface may also enhance catalytic efficiency. In the corresponding holoenzyme structure this linker docks as an extended strand onto the surface of the large lobe. This hydrophobic pocket, formed by the αF-αG loop and conserved in many protein kinases, also provides a docking site for the amphipathic helix of PKI. This novel orientation of the linker peptide provides the first clues as to how this region contributes to the unique organization of the RIIβ holoenzyme.
Keywords: PKA, cAMP, SPR, Affinity, Kinase, Linker, Crystal Structure, RIIβ, Holoenzyme
Introduction
To understand how protein phosphorylation mediates signal transduction requires an understanding of how a kinase is activated, how it recognizes its substrates, how it is inhibited, and how it is localized. cAMP-dependant protein kinase (PKA) is assembled as a fully active kinase that is then regulated and localized in large part by its regulatory (R) subunits. The dimeric R-subunits are highly dynamic and multifunctional proteins. They serve as scaffolds to localize the kinase, they are receptors for cAMP, and they are kinase inhibitors. At the N-terminus is a dimerization/docking (D/D) domain that binds to A Kinase Anchoring Proteins (AKAPs) and targets PKA to specific cellular locations1–3. At the C-terminus are two tandem cAMP binding (CNB) domains (CNB-A and CNB-B)4–6. In the flexible linker that joins the D/D domains to the CNB domains is an inhibitor site that docks to the active site of the catalytic (C) subunit in the inactive tetrameric holoenzyme7,8. Cooperative binding of cAMP to the two CNB domains leads to the allosteric release of inhibition. There are four non-redundant R-subunit isoforms, RIα, RIβ, RIIα, and RIIβ9,10, and isoform diversity is an important way to achieve specificity in PKA signaling.
Targeted gene knockouts reveal the unique importance of RIIβ in adipocytes and neuronal tissues. While knockouts of RIα are embryonically lethal11, RIIβ knockout mice have small fat cells and resistance to obesity. These lean mice are also resistant to diet-induced diabetes12, and RIIβ knockout in obesity model agouti mice restores the normal lean phenotype13. The knockout mice also have increased alcohol consumption and a resistance to alcohol-induced motor defects14. The balance of type I and type II activity and expression also plays an important role in cell growth and differentiation15,16. Disruption of this balance is implicated in several disease mechanisms, including the autoimmune disease, systemic lupus erythematosus (SLE/Lupus), Carney complex, and breast cancer17–21. To understand the role that RIIβ plays in disease and to realize the potential for designing isoform-specific agonists and antagonists, it is important to elucidate the structural and functional features that make RIIβ unique. While the structure of the cAMP bound RIIβ subunit was solved, the structure of an RIIβ:C complex has been missing.
One of the key features that distinguish RI and RII subunits is the inhibitor sequence. All R-subunits inhibit the catalytic subunit with sub-nM affinity and are activated by cAMP. However, RII-subunits have a P-site serine in their inhibitor site that can be autophosphorylated22,23, while RI-subunits have a glycine or alanine at the P-site and are pseudosubstrates. Two holoenzyme structures have been solved so far. RIα(91–379):C was solved in the presence of nucleotide and has the C-subunit in a closed conformation24 while RIIα(90–400):C was solved in the absence of nucleotide and has the C-subunit in an open conformation25. Comparing these two structures highlights the importance of the inhibitor site and in particular the P-site residue. This difference in the P-site residue was also found to be important for dissociation of the regulatory and catalytic subunits when RI and RII holoenzymes were expressed in cells26,27.
To fully understand PKA signaling, it is essential to know how the full length tetrameric holoenzymes are assembled. Surprisingly the architecture of each tetramer differs, and most striking are the differences between the RIIα and RIIβ holoenzymes. Based on small angle Xray scattering (SAXS), free RIIα and RIIβ dimers are both fully extended and almost rod-like with the CNB domains tethered by a flexible linker to the D/D domains28. This is in contrast to the RIα subunits which are Y-shaped29. The RIIα holoenzyme remains extended and rod-like allowing the attached C-subunits to be tethered by a flexible linker. In contrast, the RIIβ holoenzyme is compact and quite globular. This difference in holoenzyme architectures is determined by the linker that lies between the inhibitor site and the D/D domain28,29. To understand the isoform-specific features of the RIIβ holoenzyme, and, in particular, how the linker contributes to the unique architecture of the RIIβ holoenzyme, we engineered several different RIIβ constructs and then tried to crystallize holoenzyme complexes both in the presence and absence of AMP-PNP, a non-hydrolyzable analog of ATP, in hopes of trapping RIIβ in a substrate-like complex.
The constructs containing both CNB domains bound with high affinity to the C-subunit both in the absence and presence of AMP-PNP, but never gave high quality crystals. Although the single domain construct bound poorly to the C-subunit on its own, the addition of AMP-PNP/Mn2+ enhanced the affinity by several orders of magnitude, and adding six additional linker residues increased the affinity further to 1.6 nM. This effect of AMP-PNP for enhancing the affinity is quite striking and may be relevant for other protein substrates of protein kinases. The structure of the RIIβ(108–268) holoenzyme, solved at 1.6 Å resolution, closely resembles an enzyme:substrate complex, while the longer construct shows how the extra linker residues dock in a novel way onto the surface of the kinase large lobe. They occupy the same site as PKI but bind as an extended strand in contrast to PKI, which binds as an amphipathic helix. These results confirm earlier chemical foot-printing and mutagenesis, which predicted that the linker region immediately preceding the inhibitor site in RI and RII would bind differently to the C-subunit in the two holoenzymes30. Although the structure places only a few additional residues at the N-terminus compared to our previous structures of RIα and RIIα, the importance of this linker region for organization of the novel RIIβ tetramer is quite profound. It is the first clue as to the molecular role of any PKA linker.
Results
Designing RIIβ Constructs that Bind to the Catalytic Subunit
To understand how isoform differences contribute uniquely to the regulation of PKA activity requires structures of not only individual subunits but also of holoenzyme complexes. Holoenzyme structures are also a prerequisite for designing isoform-specific agonists and antagonists. Given the unique disease phenotypes associated with RIIβ knockout mice, we thus sought to crystallize holoenzyme complexes of RIIβ. We used mass spectrometry to guide us in engineering monomeric forms of RIIβ that were stable, expressed at high levels, and formed a stable complex with the C-subunit. The resulting constructs were then characterized using SPR and crystallography.
We initially designed a construct that contained the inhibitor site and both cAMP binding domains (residues 106 to 416). However, MS analysis showed that this construct degraded easily at both its N and C-termini and identified RIIβ(108–402) to be a stable degradation product. Although this construct readily formed stable holoenzyme both in the presence and absence of AMP-PNP, extensive crystallization trials yielded crystals that diffracted only to 7 Å.
Previous studies of RIα indicated that a construct that extends from the inhibitor site through CNB-A contributes most of the affinity between RIα and C31 and also releases the C-subunit in a cAMP-dependent manner. To define the minimal high-affinity binding region for RIIβ, we also engineered a similar shorter RIIβ construct that we hoped would retain the ability to inhibit the C-subunit. The first attempt to form an RIIβ construct lacking the B-domain, RIIβ(108–281), was also susceptible to degradation, and MS analysis indicated that RIIβ(108–268) was a stable product. This mutant was also purified and was stable to further degradation.
Formation of Holoenzyme Complexes Shows Dependence on AMP-PNP
Initial attempts to form complexes with the C-subunit showed striking differences between the two RIIβ constructs. RIIβ(108–402) containing both CNB domains formed a tight complex, based on gel filtration, in the absence of nucleotide, whereas the smaller construct that only contained the first CNB domain did not. Because the RIIβ subunit is not only an inhibitor that binds to the C-subunit with high affinity but also a substrate that can be phosphorylated and subsequently released, we attempted to stabilize this complex using a non-hydrolyzable analog of ATP, AMP-PNP, to prevent phosphoryl transfer during the formation of the RIIβ:C complex, and this complex eluted as a single sharp peak from a gel filtration column.
Surface plasmon resonance (SPR) was used to quantify the effect of AMP-PNP on holoenzyme formation (Table 1). Although RIIβ(108–402) with both CNB domains bound to the C-subunit with a nanomolar affinity in the absence of AMP-PNP (Kd= 1.6 nM), this interaction, surprisingly, was further enhanced by AMP-PNP (Kd= 0.2 nM). Binding of the smaller construct containing only the first CNB domain, RIIβ(108–268), was undetectable by SPR without Mn2+/AMP-PNP; however, with Mn2+/AMP-PNP, the Kd was dramatically improved to 11.3 nM. In summary, nucleotide and divalent cations are essential for forming stable complexes between RIIβ and the C-subunit when only the first CNB of RIIβ is present whereas they are not required, but simply enhance the affinity when both CNB domains are present. However, this dependence on ATP is only seen when a non-hydrolyzable ATP analog is used.
Table 1. Affinities of the RIIβ constructs for the Catalytic Subunit.
Affinities of the different RIIβ constructs for the catalytic subunit were measured by surface plasmon resonance (SPR). RIIβ(108–268) and RIIβ(102–265) were both measured in the presence of Mn2+:AMP-PNP.
|
| ||||
|---|---|---|---|---|
| Complex | RIIβ (108–402) APO |
RIIβ (108–402) AMP-PNP |
RIIβ (108–268) AMP-PNP |
RIIβ (102–265) AMP-PNP |
| kassoc (M−1s−1) | 1.5 (±0.2) × 105 | 4.2 (±0.6) × 105 | 3.1 (±0.2) × 106 | 8.6 (±1.0) × 106 |
| Kdiss (s−1) | 2.4 (±0.1) × 10−4 | 8.4 (±2.6) × 10−5 | 35 (±0.1) × 10−3 | 4.9 (±0.6) × 10−3 |
| KD | 1.6 nM | 0.2 nM | 11.3 nM | 0.6 nM |
Crystallization of an RIIβ Holoenzyme Complex
Because we ultimately want to understand how RIIβ and C interact as a substrate-kinase complex and as an inhibitor-kinase complex, we attempted to crystallize both complexes in the presence and absence of AMP-PNP. Only the shorter construct in the presence of Mn2+:AMP-PNP yielded diffraction quality crystals. The crystal structure of RIIβ(108–268):C in the presence of AMP-PNP and 2 Mn2+ ions was solved at 1.62 Å resolution. Data statistics are shown in Table 2.
Table 2.
Data Collection and Refinement Statistics
| RIIβ(108–268):C | RIIβ(102–265):C | |
|---|---|---|
| Data collection | ||
| Space group | C2 | C2 |
| Cell dimensions (Å) | ||
| a | 173.3 | 179.3 |
| b | 65.7 | 67.4 |
| c | 47.2 | 47.3 |
| β (°) | 99.9 | 99.5 |
| No. of molecule per asymmetrical unit | 1 | 1 |
| Resolution (Å) | 1.62 | 2.70 |
| Rmerge | 0.068 (0.48) | 0.069 (0.25) |
| Completeness (%) | 99.4 (99.1) | 95.2 (88.9) |
| I/sigma | 30.3 (6.5) | 32.5 (9.7) |
| No. reflections | 88992 | 25383 |
| Refinement | ||
| Resolution (Å) | 50.0–1.62 | 50.0–2.70 |
| Rwork/Rfree (%) | 20.6/23.0 | 24.1/31.9 |
| No. of protein residues | 500 | 506 |
| No. of ligand/ion | 3 | 3 |
| No. of water | 398 | 43 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.005 | 0.008 |
| Bond angles (°) | 1.2 | 1.4 |
| Ramachandran angles (%) | ||
| most favored | 92.0 | 78.1 |
| disallowed | none | none |
Values in parentheses are for highest-resolution shell: (1.62–1.68 Å) for RIIβ(108–268):C; (2.70–2.78 Å) for RIIβ(102–265):C
Overall Structure
The overall structure of the RIIβ(108–268) holoenzyme complex is shown in Figure 1. Unlike the RIIα holoenzyme, the catalytic subunit assumes a closed conformation. There are two major changes that take place in the RIIβ subunit as a consequence of forming holoenzyme. The essential change for kinase inhibition is that the disordered inhibitor site becomes ordered by locking into the active site cleft of the C-subunit. We confirm here that this basic mechanism of inhibition (Figure 2) is conserved for all of the R-subunits24,25, and this feature is also seen in the PKI inhibitors that have been crystallized bound to the C-subunit32,33. In each case, the inhibitor site forms a short β strand that fits into the active site cleft and forms an antiparallel β sheet with the P+1 loop of the C-subunit. Two conserved arginines at the P-2 and P-3 position of the inhibitor sequence bind to conserved acidic residues near the active site cleft while the C-terminus of the inhibitor sequence (P+1 residue) docks to a hydrophobic pocket that is formed by the C-subunit P+1 loop. Additionally, in the holoenzymes Y247 in the αG helix of the C-subunit contributes to the P+1 binding site by nucleating an extended hydrophobic interface with the CNB-A domain. Y226 in the phosphate binding cassette (PBC) of the RIIβ subunit also contributes directly to the P+1 site. Interactions of the inhibitor site, shown in Figures 2 and 3, clearly define the fundamental mechanism for inhibition and activation of PKA where cAMP (a small ligand) and the C-subunit (a large protein) compete for the PBC of the R-subunit.
Figure 1. RIIβ Holoenzyme Overall Structure.
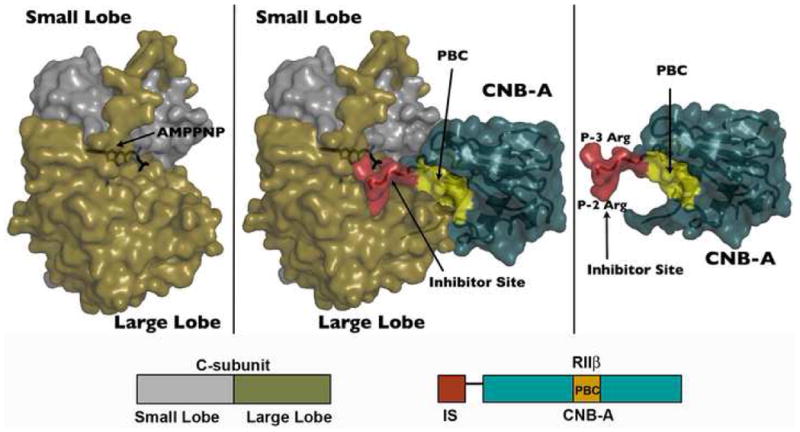
The crystal structure of RIIβ(108–268) holoenzyme is shown as space filling surface. The holoenzyme is shown in the middle. catalytic subunit (small lobe in grey, large lobe in tan) alone on the left, and regulatory subunit (turquoise) alone on the right. The inhibitor site (red) and phosphate binding cassette, PBC (yellow) of the regulatory subunit are highlighted.
Figure 2. Mechanism for Inhibition of the Catalytic Subunit.
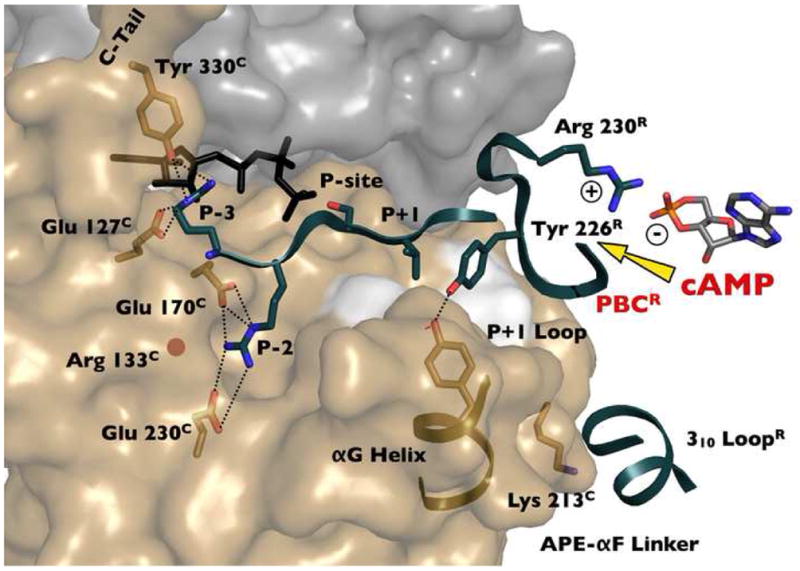
The inhibitor peptide and the tip of the phosphate binding cassette (PBC) of RIIβ dock to the active site cleft of the catalytic subunit in the holoenzyme conformation of RIIβ. cAMP activation thus involves a competition between cAMP and the C-subunit for binding the PBC of domain A. The docking of the 310 loop in RIIβ to the surface surrounding Lys213C in the catalytic subunit is also shown.
Figure 3. RIIβ Holoenzyme Assumes a Closed Confirmation.
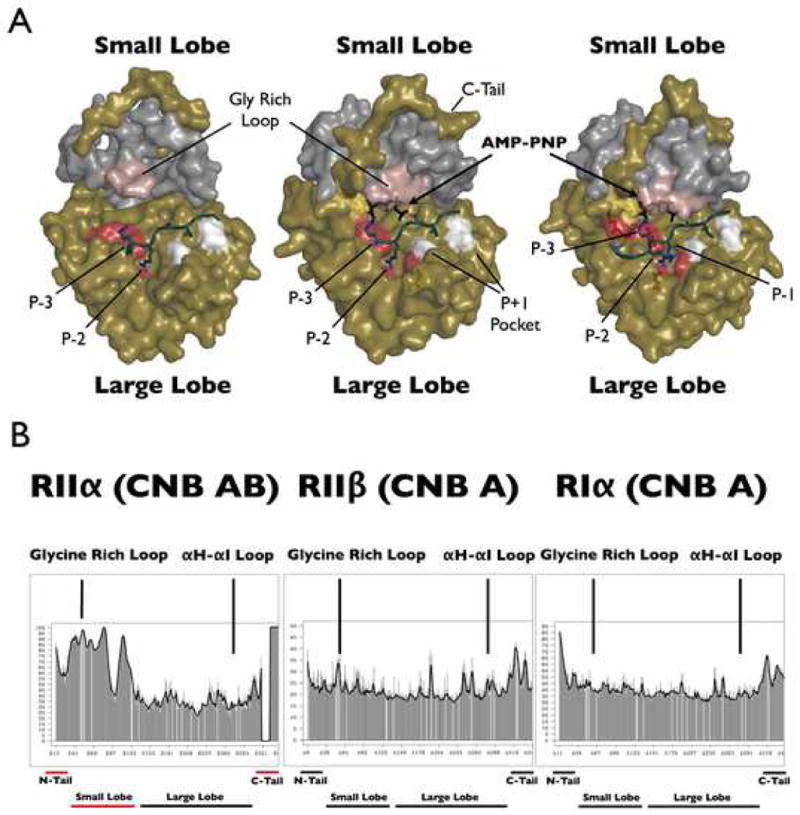
Panel A shows three holoenzyme structures with the trapped transition state that is present in the RIIα holoenzyme structure (middle). The RIIα holoenzyme (left) is in an open form whereas the RIIβ and RIα complexes (right) are in a closed conformation. The color designations are as follows: E230C, E170C, and E203C are shown in red and provide the acidic docking surface for the P-3 and P-2 arginines; the P+1 pocket (L198C and P202C) is in white and provide the hydrophobic site for the P+1 residue; Y330C in the C-tail is yellow; the glycine rich loop is in pink; AMP-PNP is in black. The temperature factors analysis of the catalytic subunit in three holoenzyme structures is shown in panel B. Shown on the left is RIIα (90–400):C in the absence of ATP (2QVS), with the disordered/mobile regions underlined in red; in the middle is RIIβ(108–268):C:AMP-PNP; on the right is RIα (91–244):C:AMP-PNP (1U7E).
The other major conformational change in the CNB-A domain is that the kinked B/C helix extends into a single long helix (Figure 4). This B/C helix in domain A transmits the allosteric signal that couples cAMP binding to the release of kinase inhibition, and by comparing the structures of the two conformational states of the B/C helix in RIIβ with RIα, we see some important differences. This is the first time we can compare the same RII subunit in both its cAMP bound and holoenzyme states. The extended B/C helix is conserved in both RIIβ and RIα holoenzymes; however, its position in the cAMP bound conformations differs5,6. The distance that the B/C helix moves upon cAMP binding is greater in RIα than in RIIβ. In addition, to recruit the B/C helix to the PBC, the C helix must twist as well as bend. The C helix is twisted approximately 60° in RIα but only 30° in RIIβ, and it is this twist that determines the position of domain B in the cAMP bound conformation. Another difference is that the C-terminus of the C helix is more frayed in the RIIβ holoenzyme.
Figure 4. Conformational Changes of the αB/C Helix of RIIβ.
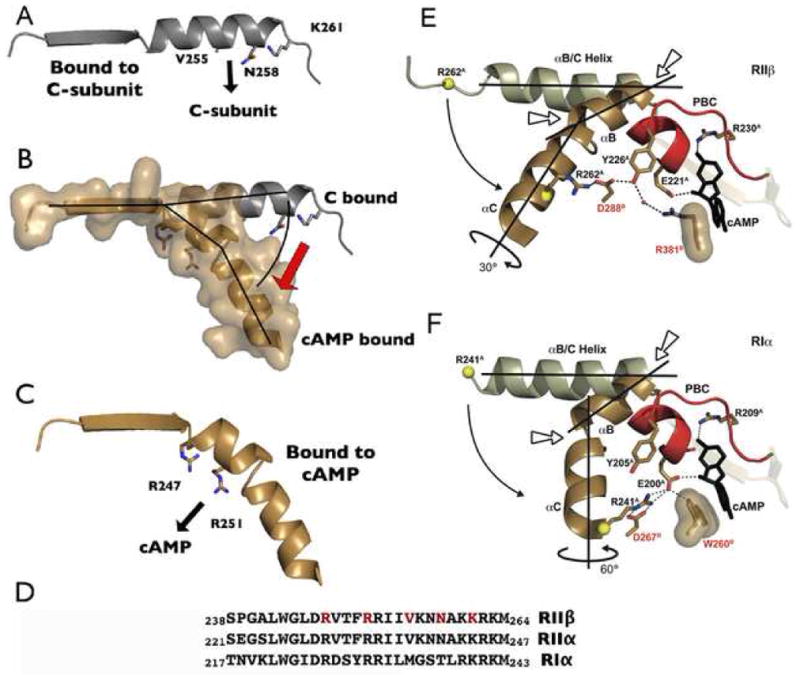
Panel A shows the αB/C helix of RIIβ in the C-subunit bound state with residues that interact with the C-subunit highlighted. Panel B compares the same helix in the cAMP-bound (PDB code 1CX4) (tan) and C-bound (grey) states. Panel C shows the conformation of the αB/C helix when it is bound to cAMP with residues that interact with the PBC highlighted. Panel D shows the amino acid sequence of this region in RIα, RIIα, and RIIβ. Interacting residues indicated above are highlighted in red. Panels E and F compare the conformational change of the αB/C helix in RIIβ (E) and RIα (F). cAMP bound RIIβ (1CX4) and RIα (1RGS) are shown in brown, with the PBC in red. Two kink points are indicated by short arrows. The key residues are also highlighted; two from domain B are labeled in red. RIIβ(108–268) holoenzyme and RIα (91–379) holoenzyme (2QCS) are shown in white. The curved arrows indicate the movement between the holoenzyme states to the cAMP bound states. The C helix is twisted approximately 60° in RIα but only 30° in RIIβ.
The RIIβ (AMP-PNP) Holoenzyme Closely Resembles an Enzyme:Substrate Complex
This RIIβ holoenzyme defines a new and physiologically relevant state for an RII holoenzyme. By using AMP-PNP, we were able to trap the complex in a conformational state where the RII subunit is recognized as a substrate, as shown in Figure 3. This RIIβ(108–268) holoenzyme structure represents the C-subunit in a form after the binding of ATP and the R-subunit, but before subsequent transfer of the phosphate to the inhibitor site of RIIβ. It allows us for the first time to appreciate the critical role of the nucleotide. In the RIIα holoenzyme, crystallized in the absence of ATP, the C-subunit is in an open conformation and is inhibited by interactions with only the large lobe25. The RIIβ holoenzyme represents a “trapped” state where the Ser-OH is present but the γ-phosphate of the AMP-PNP cannot be cleaved. A comparison of the temperature factors in the three holoenzyme complexes highlights the novelty of this RIIβ complex (Figure 3). The low temperature factors in the small lobe are consistent with its closed conformation. In contrast, the small lobe of the RIIα complex that lacks nucleotide is in an open conformation, has high temperature factors in the small lobe and C-tail, and appears to be quite dynamic25.
In this conformation we are viewing the RIIβ subunit as a substrate while in the nucleotide free RIIα holoenzyme structure the R-subunit is seen only as an inhibitor. In the RIIβ complex we see how the nucleotide is essential for bridging the small and large lobes together into a closed conformation. The adenine ring completes the catalytic hydrophobic spine, described recently34, so that the phosphate can be poised for transfer. This conformation would be essential for autophosphorylation of RIIβ. Contacts between the small lobe and the RIIβ subunit closely resemble contacts observed in the RIα holoenzyme structure. For example, the C-terminal tail of the catalytic subunit is fully ordered, and the P-3 arginine in the inhibitor site engages both the small lobe and the nucleotide. This is similar to RIα and opposed to RIIα. By using AMP-PNP, the intermediate state for transferring the γ-phosphate of ATP to the P-site serine has been captured in the RIIβ structure, and these interactions likely account for the increased affinity conferred by AMP-PNP. Although in most ways this RIIβ structure resembles a transition state, one important aspect is different. The glycine-rich loop is not anchored to the γ-phosphate of ATP; it is over 5 Å away. As seen in Figure 5, this loop is not fully closed, and this is also reflected in the temperature factors (Fig. 3), as well as in the previous HD/MS analysis35.
Figure 5. Glycine Rich Loop in RIIβ Holoenzyme Represents a Transition-Like State.
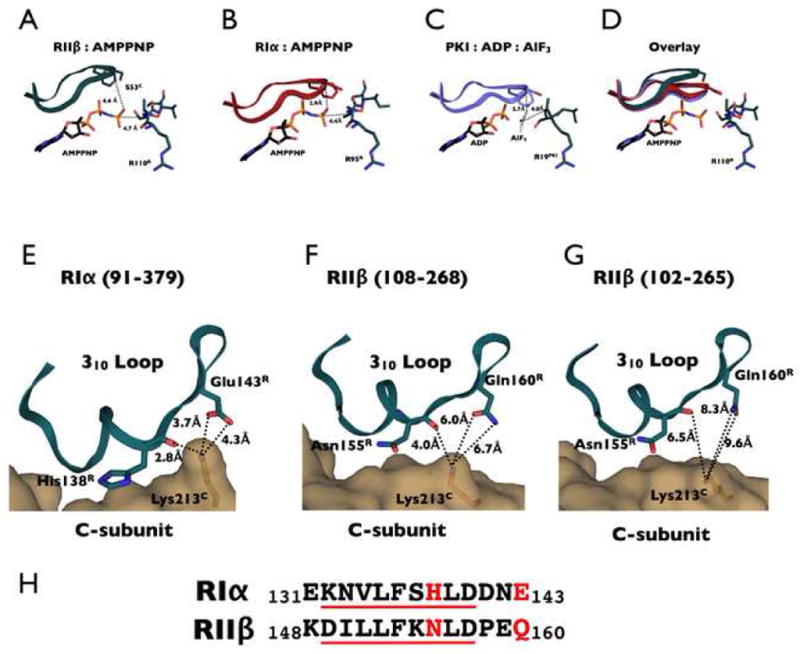
The upper panel shows the glycine rich loop in the RIIβ complex (A) which is opened by approximately 4 Å compared to RIα holoenzyme (B) and PKI:ADP:AlF3 PKA complex (C). Glycine rich loop structures are overlaid in (D). The lower panel shows the interaction between Lysine 213C in the C-subunit and the 310 loop of CNB domain A in RIα (91–379) (E), RIIβ(108–268) (F) and RIIβ(102–265) (G). The C-subunit is shown as a tan surface representation, and the R-subunit is shown as a dark teal cartoon. Lysine 213C and interacting residues in the R-subunits are shown as sticks. The distances to K213C shown in panel F for RIIβ are very similar for the RIIα holoenzyme that also lacks the N-terminal residues that precede the inhibitor site. The respective sequences of the R-subunits are aligned in panel H.
Additional Residues in the Linker Contribute Further Stability to the AMP-PNP Holoenzyme Complex
Previous mutagenesis30 and small angle X-ray scattering analysis28,29 indicated that the region that precedes the inhibitor sequence may also contribute to isoform specificity. Sequence comparisons in this region, corresponding to the P-4 through the P-10 residue, show that RI and RII subunits are quite different from each other, but conserved in each isoform. To determine whether this segment of RIIβ interacts in a specific way with the C-subunit that is distinct from RIα, we engineered a longer form of RIIβ, RIIβ(102–265). Surprisingly, in comparison to RIIβ(108–268), addition of this 6-residue segment increased the affinity for the C-subunit by almost 20-fold. In the presence of AMP-PNP, RIIβ(108–268) had an affinity of 11.3 nM, while adding six residues (PVINRF) increased the affinity to 0.6 nM (Table 1). We thus confirmed the importance of the linker region that precedes the inhibitor site.
To understand the molecular basis for this enhanced affinity, the holoenzyme structure of RIIβ(102–265) in complex with the C-subunit, AMP-PNP, and two Mn2+ ions was solved to 2.70 Å resolution. This RIIβ complex structure has the exact same space group, crystal packing, and overall structure as the RIIβ(108–268) complex (for statistics see Table 2), but the additional six residues at the N-terminus of RIIβ are ordered (Figure 6). They dock to a hydrophobic surface on the C-subunit, the same surface required by PKI to achieve its high affinity binding. RIIβ, however, utilizes an extended strand to bind this hydrophobic surface, in contrast to PKI which utilizes an amphipathic helix to dock to the same site. This demonstrates a remarkable diversity of this binding pocket on the surface of the large lobe of the C-subunit to accept peptides that assume different conformations.
Figure 6. Variation in Positioning of the Region That Lies N-terminal to the Inhibitor Site.
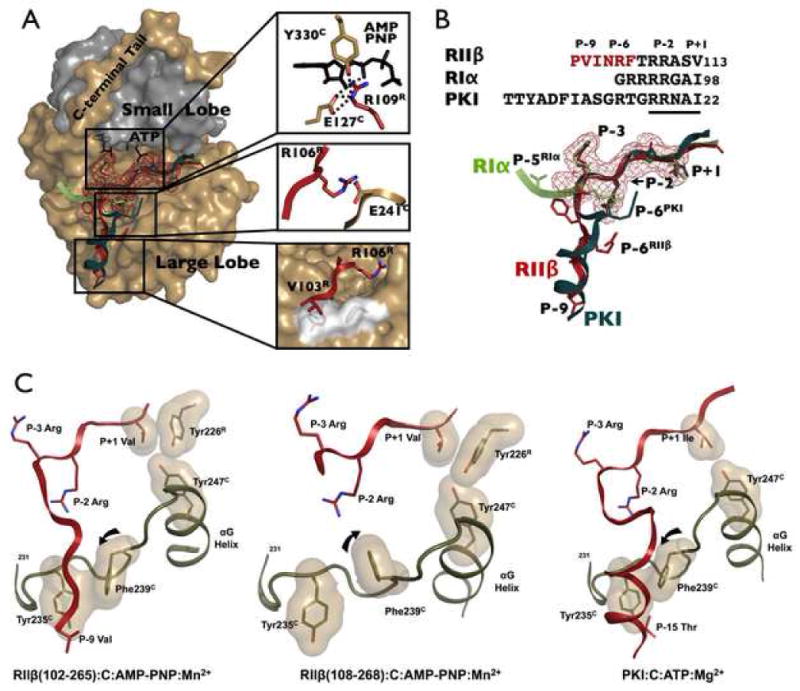
(A) Highlighted N-terminal binding regions of PKI (dark teal), RIα (light green) and RIIβ (red) demonstrate differential binding between inhibitors. RIIβ and PKI dock to the C-lobe, while RIα docks to the C-terminal tail and N-lobe. Surface mesh representation is shown for the highly conserved P-3 to P+1 region. Details of RIIβ binding peptide are highlighted. (B) The sequence and detailed structural comparison of the inhibitor peptides of PKI, RIα and RIIβ. (C) The αF-αG loop (tan) creates a hydrophobic pocket that recognizes different substrates (red). Tyr235C and Phe239C are highly conserved residues in this pocket. The region of RIIβ that lies N-terminally to the inhibitor site docks as a strand to this pocket (left) while the amphipathic helix of PKI (right) docks to the same surface. In both cases the peptides dock against Phe239C. In RIIβ(108–268) this site is unoccupied (middle) and the side chain of Phe239C is rotated away from Tyr235C. In the two RIIβ holoenzymes, one can see how Tyr247C in the G-helix interfaces with the P+1 Val and with the Tyr226R in the PBC of RIIβ.
The extended P-9 to P-7 region of RIIβ (V103-I104-N105) makes mostly hydrophobic contacts with the large lobe of the C-subunit. The region of the C-subunit that contacts this segment of RIIβ is the αF-αG loop (Y235-P-P-F-F239), which forms a hydrophobic pocket that is also used by PKI to achieve high affinity binding and is actually highly conserved in many kinases (Figure 6 and S1). The PKI peptide forms an amphipathic α helix in the P-8 to P-16 region and the hydrophobic surface of this helix, nucleated by F10PKI and Y7PKI, binds to the large lobe of the C-subunit. RIIβ binds to the same hydrophobic surface on the C-subunit; however, instead of forming a helix, RIIβ forms a strand with the P-9 Val and P-8 Ile dominating the hydrophobic interaction. Like PKI, RII-subunits have a P-6 Arg and in both cases this arginine binds to an acidic residue on the C-subunit. The RI-subunit, however, utilizes different sets of interactions to bind to the C-subunit. RIα has two additional arginines that precede the inhibitor site. However, the P-5 Arg goes to D328C in the C-terminal tail, while the P-4 Arg goes to E203C. Thus the N-terminus of RIα further strengthens interactions with the C-terminal tail and reinforces the closed conformation, while the same region in RIIβ further anchors the RIIβ subunit to the large lobe.
The N-terminus of the C-subunit is Ordered Differently in the AMP-PNP Holoenzyme Complex
The first 8 residues of the N-terminus of the C-subunit in both RIIβ holoenzyme structures are disordered whereas in both RIα and RIIα holoenzymes the first 14 residues of the C-subunit are disordered. The only structure solved so far where this region was ordered was the myristylated mammalian C-subunit36 where the myristic acid moiety is folded into a hydrophobic pocket. When detergents are included in the crystallization buffer, the detergent occupies this pocket37. As shown in Figure S2, in the RIIβ holoenzyme complex, R262R from a symmetrically related molecule fits precisely into the myristic acid pocket. Crystallization of the RIIβ holoenzyme was only achieved using C-subunit that lacked a phosphate on S10C. The presence of the phosphate would likely disorder the N-terminus of the C-subunit and may prevent the crystal packing that we observe. The dynamic properties of the myristyl moiety and the C-tail are also very different when the C-subunit is bound to RI vs. RII. Based on fluorescence anisotropy, binding of RII causes the myristylated N-tail to become more dynamic and flexible, and this is also the form that interacts with membrane vesicles38. In contrast, the N-terminus of the C-subunit remains buried in the RIα holoenzyme. This structure of the RIIβ complex provides for the first time some clues as to how the N-terminus of the C-subunit might be ordered differently depending on its binding partner. In this structure the N-terminus reaches over to interact directly with the beginning of the C-tail as it starts to wrap around the kinase core. It will be important to solve additional structures with myristylated C-subunit to better define this important region where the N- and C-tails converge.
Isoform-specific Features of the R:C Interface
Another critical interacting motif for the R and C subunits is the 310 loop in RIIβ (residues 132–140). This segment, in addition to the PBC, is a conserved feature of the cyclic nucleotide binding domain39. The hydrophobic residues in this region are highly conserved and form part of the contact with the C-subunit, specifically to the hydrophobic surface on the C-subunit that is created by the αG helix and the region that links the APE motif to the αF helix. Just beyond the 310 loop in RIα is E143R that forms an essential ion pair with K213C in the RIα holoenzyme. This ion pair was identified as a critical docking site for RIα prior to the structure solution40. Because the equivalent of E143R in RIIβ is a glutamine, Q160R, the ion pair is missing and the nature of this interface is different and weaker, as shown in Figure 5. In addition, His138R, which contributes significantly to the hydrophobic interface in RIα, is replaced with an Asn in RIIβ. These two changes substantially alter the interface and are likely to be significant isoform differences. Another surprising observation is that this interface changes somewhat in the longer RIIβ holoenzyme structure (Fig. 6) suggesting that docking to the αF-αG loop may be inversely correlated with docking to this surface.
Discussion
RIIβ is an important physiological inhibitor of the PKA catalytic subunit as demonstrated by the complex phenotypes associated with its disruption17–21. To understand the unique features of RIIβ and for eventually designing isoform-specific agonists and antagonists, it is essential to have structures that represent RIIβ in its different conformational states. While we had a cAMP bound structure of RIIβ5, until now a holoenzyme complex has been missing. To trap RIIβ in a holoenzyme complex, we engineered several deletion mutants. RIIβ(108–402) which contains both CNB domains bound very tightly to the catalytic subunit in the absence of ATP (Kd = 1 nM), like full length RIIβ; however, a shorter construct containing only the first CNB domain, RIIβ(108–268), bound poorly. We thus attempted to use AMP-PNP and Mn2+ to stabilize the RIIβ(108–268):C complex, and the nucleotide surprisingly reduced the Kd to 11 nM. The structure of this complex showed that RIIβ was indeed bound as a PKA substrate with its inhibitor site docked firmly to the active site cleft. The catalytic subunit was in a closed conformation with the tails recruited to the kinase core in a manner similar to the RIα and PKI complexes. This is in contrast to the complex of RIIα and nucleotide free catalytic subunit, where the tails of the C-subunit were disordered and the core was in an open conformation (Figure 3),
The linker regions in RIα, RIIα and RIIβ create very different architectures for the tetramer holoenzymes, however, until now little was known about how these linker regions interact with the catalytic subunit. Earlier mutagenesis and chemical foot-printing suggested that the linker region immediately preceding the inhibitor site in RI and RII subunits might interact differently with the catalytic subunit30. Specifically, mutagenesis of Asp328C in the C-tail interfered with binding of RIα while mutation of Arg133C in the large lobe interfered selectively with RIIα binding. Based on these results, we predicted that the region N-terminal to the inhibitor site in RIIβ might dock in a specific way to the large lobe. To test this prediction, we engineered RIIβ(102–265) that contained six additional residues at the N-terminus. Surprisingly, the Kd for RIIβ(102–265) was reduced to 0.6 nM in the presence of AMP-PNP confirming that this region was indeed important for RIIβ:C interactions. The data also suggest that this segment from the N-Linker may actually enhance catalytic efficiency by increasing the affinity of the complex for ATP. A recent kinetic analysis of fungal regulatory subunits also demonstrated that the linker region contributes to enhanced catalytic efficiency41.
The structure of the RIIβ(102–265) holoenzyme complex showed that the additional 6 linker residues interacted with the αF-αG loop on the large lobe as we had predicted; however, these residues formed a strand which was unexpected. We believe that this surface constitutes an important and versatile docking site that can be used quite creatively by the C-subunit to recognize different substrates and inhibitors. Docking is also independent of secondary structure. As seen in Figure 6, the strand that links the αF and αG helices, Tyr235-Pro-Pro-Phe-Phe239 in PKA, creates a hydrophobic pocket. Tyr235C and Phe239C line this hydrophobic pocket, and this motif is actually conserved in many protein kinases42. This site is thus likely to be an important docking site for many protein kinase43,44. When this site is not occupied by peptide, as in the RIIβ(108–268) holoenzyme, Phe239C is rotated upwards towards the catalytic loop while binding of peptide to this surface locks the side chain into an alternative conformation (Figures 6 and S1). The position of the Phe239C side chain thus serves as an indicator of occupancy of this site.
Although only six additional residues are positioned in the longer construct, these residues likely play a critical role in determining the overall architecture of the RIIβ tetrameric holoenzyme and may also determine how the linker interfaces with the dimerization domain to form a compact tetramer. Furthermore, placement of this region of the linker will be unique for RII subunits since the comparable region of the RI subunits, which contain multiple arginines, cannot dock to this same surface.
The inhibitor site is one of the critical regions that distinguishes the functionally non-redundant RI and RII isoforms of the PKA regulatory subunits. Both subunits have an inhibitor site that is disordered in the cAMP-bound conformation, and in this state the cyclic nucleotide binding domains appear to be uncoupled from the D/D domains at the N-terminus. Upon formation of the holoenzyme, this inhibitor site docks to the active site cleft of the C-subunit and recruits both CNB domains to the R:C interface. Docking of the inhibitor site to the C-subunit renders the C-subunit inactive and unable to bind other substrates. RI subunits, which have a pseudosubstrate inhibitor site, have an absolute requirement for ATP and Mg2+ to form a high affinity complex with the catalytic subunit; there is no acceptor for the γ-phosphate of ATP so it is trapped at the interface. In contrast, the RII subunits are not only inhibitors of C, they are also substrates. They use ATP to phosphorylate the inhibitor site, and this facilitates dissociation of the RII-subunit45. Here we used a non-hydrolyzable ATP analog, AMP-PNP, to trap the RIIβ holoenzyme in a closed conformation that resembles an intermediate transition state. With this strategy we also discovered novel RII-specific interactions of the extended RII linker region with the large lobe of the C-subunit. Targeting this novel site may provide a strategy for developing isoform-specific agonists or antagonists that are independent of the cyclic nucleotide binding pockets. So far Rp-cAMP is the only known antagonist of PKA46.
RII subunits in mammalian cells are typically targeted via AKAPs to specific membrane sites, and phosphatases are often recruited to the same scaffold. The kinases and the phosphatases thus serve as coordinated switches to dynamically regulate the phosphorylation state of a particular site in a channel or a receptor. However, the phosphorylation state of the RII subunits is also likely to be regulated47. Phosphorylation of RII subunits appears to be to slow down the rate of reassociation23, and recent results using GFP tagged RI and RII subunits demonstrated that reassociation of RI and RII subunits in cells is different and influenced dramatically by whether the P site residue is a substrate or a pseudosubstrate26,27. However, both forms of RIIβ, phosphorylated and dephosphorylated, form a tight inhibited complex with the catalytic subunit (Kd = 0.1 nM vs 1 nM, respectively). Of course, neither of these RII:C complexes will bind ATP or ADP to form a stable complex as we see here with the “trapped” complex. Thus, under physiological conditions the N and C lobes in an RII holoenzyme will likely be uncoupled and devoid of nucleotide, leaving the tails, as well as the N-lobe, free to interact in principle with other proteins. A future challenge, in addition to determining when RII subunits become phosphorylated in cells and how that phosphorylation is regulated, is to establish whether the conformation and/or function of the full-length tetrameric RIIβ holoenzyme is influenced by the phosphorylation state of the RIIβ subunit.
Materials and Methods
Engineering and Expression of R-subunits
Expression and purification of the R-subunits was carried out as previously described48. In short, following E. coli expression, cells were lysed and clarified. After 60% ammonium sulfate precipitation, the pellet was resuspended and then bound to cAMP-Sepharose resin overnight. After washing with 0.7 M NaCl, R-subunit was eluted with 30 mM cGMP at pH 5.5. Buffer consisted of 50 mM MES, 200 mM NaCl, 5 mM dithiolthreitol, 2 mM EDTA and 2 mM EGTA.
The RIIβ(108–268) and RIIβ(102–265) genes were PCR cloned using a longer E. coli based expression vector from Rattus norvegicus cDNA as template, and inserted into a commercial expression vector, pRSET (Invitrogen). The construct was expressed as an untagged protein, as purification was possible through use of a cAMP-Sepharose resin described previously49,50. Precast Nupage SDS gels (Invitrogen) were used for all SDS gel analysis. Samples were prepared using a commercial 4X sample buffer (Invitrogen), and 5% 1 M dithiolthreitol was added prior to heating for 10 min at 70 °C.
RIIβ:C Purification and Crystallization
RIIβ(108–268) was chosen as the minimal high affinity binding protein for crystallization based on previous results. RIIβ(108–268) and C-subunit were purified as described previously using cAMP resin and cGMP elution51. RIIβ was purified twice through Superdex 75 gel filtration for optimal purity and to remove cGMP. AMP-PNP and MnCl2 were added to the C-subunit52, followed by a 1.2 fold molar excess of R-subunit. The complex was spin dialyzed 3 times into the holoenzyme AMP-PNP buffer containing 10 mM MOPS pH 7.0, 50 mM NaCl, 1mM tris(carboxyethyl)phosphine, 1mM MnCl2 and 0.2 mM AMP-PNP. The complex was then gel filtered through Superdex 75 16/60 in the same buffer to remove excess R-subunit. The protein was concentrated to approximately 10 mg/ml for crystallization.
For crystallization of PKA type IIβ holoenzyme vapor diffusion under oil (VDUO) crystallization was used. This technique incorporates a layer of oil over drops of crystallization reagent and protein sample mixed together using 12 channel pipettes in 96 well plates. Commercial PACT and JCSG+ crystallization screens (Molecular Dimensions) were chosen for initial screening53. Crystals were transferred into a drop containing the initial crystallization condition + 10% glycerol. This drop served to remove excess oil from the crystal and allowed the soaking of the glycerol cryo-protectant. Crystals were then flash frozen in liquid nitrogen.
RIIβ(102–265) holoenzyme was formed as described above for RIIβ(108–268):C complex. Final crystallization conditions were 6mg/mL RIIβ(102–265):C, 8% PEG 3350, 40 mM Bis Tris pH 7.5, 0.05 mM Na Acetate.
Data Collection and Processing
Initial crystal screening was carried out at 100 K at the UCSD chemistry home source before shipping to the Advanced Light Source (ALS) for data collection. The home source has a rotating anode CuKα source emitting a wavelength of 1.52 Å. Images were collected on a Mar 345 image plate detector. The best diffraction for RIIβ(108–268) holoenzyme was obtained from crystals grown at 4° C from crystallization solution containing 200 mM NaBr, 20% PEG 3350. Three data sets were collected at the ALS beamline 8.2.2, and then processed using HKL2000 to a space group of C2. Initial phases were obtained using the PKI bound C-subunit (PDB code 1ATP) as a search model by the CCP4 package program PHASER. The refinement was done using the REFMAC5 program in CCP4. Data and refinement statistics are shown in Table 2.
Surface Plasmon Resonance (SPR)
Affinities between the R and C-subunits at 25°C were determined using surface plasmon resonance (SPR) to quantify the on and off rates of the R-subunit in solution binding to an immobilized C-subunit. A Biacore 3000 instrument (Biacore Inc.) was used for all interaction analysis, and Biacore BiAanalysis software was used for data analysis. Data were fit using both standard 1:1 Langmuir binding model with and without limiting mass-transfer correction terms. By checking the absolute values of kon and koff in both fitting models, results were confirmed as accurate and mass transfer effects minimal.
C-subunit was attached to a carboxylated gold sensor CM-5 chip through amine coupling at a concentration of 0.01 mg/mL. Immobilization levels of ~300–500 response units (R.U.) were achieved. Regeneration of the C chip was achieved by application of 50 μL of 100 μM cAMP in the running buffer. Buffers used for analysis were 20 mM HEPES pH 7.0, 150 mM KCl, 1mM tris(2-carbxyethyl)phosphine and 0.05% P20 surfactant for the Apo buffer. AMP-PNP (0.2 mM) and 1 mM MnCl2 were added for the AMP-PNP buffer.
Supplementary Material
The two residues form a hydrophobic pocket that is used by PKI (top left) to achieve high affinity binding as an α helix, or by RIIβ(102–265) as a β strand (boxed in blue dash line). The orientation of the side chain of Phe239 appears to be a marker for occupancy of this surface pocket. The 2Fo-Fc density maps of the Y235C-P-P-F-F239C regions in both RIIβ(108–268) (left) and RIIβ(102–265) (right, with the presence of the N-terminal region of V103R-I-N-R-F107R) holoenzymes, contoured at 1σ, are shown on the bottom. The Phe239 side chain flip is highlighted by an arrow.
Crystal packing interactions in the RIIβ holoenzyme structure reveal R262R from an adjacent molecule docks into the myrstic acid binding pocket of the C-subunit. The N and C-terminal tails of the C-subunit are shown as ribbons, and the remainder of the C-subunit is surface rendered. R262R, F18C, Y306C and the myristic acid aligned from PDB 1CDK are shown as sticks. Blue mesh represents the surface of R262R.
Acknowledgments
We specifically thank Alexander Kornev for critical review, structure analysis and assistance with preparation of the manuscript. We also thank Nick Nyguen at the UCSD Chemistry X-ray source and Corie Ralston at the beamline 8.2.2 at the Advanced Light Source for assistance with data collection. Thanks to Ganapathy Sarma for assistance in model building, and Sventja von Daake in manuscript and figure preparation. We thank Ganesh Anand for HD/MS analysis of complexes and Mike Deal for assistance with protein purification. This Investigation was supported by a Ruth L. Kirschstein National Research Service Award (NIH/NCI T32 CA009523) to S.H.J.B. and NIH grant (GM34921) to S.S.T.
Abbreviations
- PKA
cAMP-dependant protein kinase
- SPR
surface plasmon resonance
- CNB
cAMP binding domain
- C
catalytic subunit
- R
regulatory subunit
Footnotes
Accession Numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers: 3IDB and 3IDC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Banky P, Newlon MG, Roy M, Garrod S, Taylor SS, Jennings PA. Isoform-specific differences between the type Ialpha and IIalpha cyclic AMP-dependent protein kinase anchoring domains revealed by solution NMR. J Biol Chem. 2000;275:35146–35152. doi: 10.1074/jbc.M003961200. [DOI] [PubMed] [Google Scholar]
- 2.Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newlon MG, Roy M, Morikis D, Hausken ZE, Coghlan V, Scott JD, Jennings PA. The Molecular Basis for Protein Kinase A Anchoring Revealed by Solution NMR. Nat Struct Bio. 1999;6:222–227. doi: 10.1038/6663. [DOI] [PubMed] [Google Scholar]
- 4.Berman HM, Ten Eyck LF, Goodsell DS, Haste NM, Kornev A, Taylor SS. The cAMP binding domain: an ancient signaling module. Proc Natl Acad Sci U S A. 2005;102:45–50. doi: 10.1073/pnas.0408579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diller TC, Madhusudan, Xuong NH, Taylor SS. Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type II beta regulatory subunit. Structure. 2001;9:73–82. doi: 10.1016/s0969-2126(00)00556-6. [DOI] [PubMed] [Google Scholar]
- 6.Su Y, Dostmann WRG, Herberg FW, Durick K, Xuong NH, Ten Eyck LF, Taylor SS, Varughese KI. Regulatory (RIa) Subunit of Protein Kinase A: Structure of Deletion Mutant with cAMP Binding Domains. Science. 1995;269:807–819. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- 7.Gangal M, Li F, Jones JM, Deich J, Lovett K, Taylor SS, Johnson DA. Consequences of cAMP and Catalytic Subunit Binding on the Flexibility of the A-Kinase of the Regulatory Subunit. Biochemistry. 2000;39:15626–15632. doi: 10.1021/bi002196l. [DOI] [PubMed] [Google Scholar]
- 8.Zawadzki KM, Pan CP, Barkley MD, Johnson D, Taylor SS. Endogenous tryptophan residues of cAPK regulatory subunit type IIbeta reveal local variations in environments and dynamics. Proteins. 2003;51:552–561. doi: 10.1002/prot.10326. [DOI] [PubMed] [Google Scholar]
- 9.Skalhegg BS, Tasken K, Hansson V, Huitfeldt HS, Jahnsen T, Lea T. Location of cAMP-dependent protein kianse type I with the TCR-CD3 complex. Science. 1994;263:84–87. doi: 10.1126/science.8272870. [DOI] [PubMed] [Google Scholar]
- 10.Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent Protein Kinase: Framework for a Diverse Family of Regulatory Enzymes. Annu Rev Biochemistry. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 11.Cummings DE, Brandon EP, Planas JV, Motamed K, Idzerda RL, McKnight GS. Genetically lean mice result from targeted disruption of the RII beta subunit of protein kinase A. Nature. 1996;382:622–626. doi: 10.1038/382622a0. [DOI] [PubMed] [Google Scholar]
- 12.Schreyer SA, Cummings DE, McKnight GS, LeBoeuf RC. Mutation of the RIIbeta subunit of protein kinase A prevents diet-induced insulin resistance and dyslipidemia in mice. Diabetes. 2001;50:2555–2562. doi: 10.2337/diabetes.50.11.2555. [DOI] [PubMed] [Google Scholar]
- 13.Czyzyk TA, Sikorski MA, Yang L, McKnight GS. Disruption of the RIIbeta subunit of PKA reverses the obesity syndrome of Agouti lethal yellow mice. Proc Natl Acad Sci U S A. 2008;105:276–281. doi: 10.1073/pnas.0710607105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thiele TE, WIllis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS. High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci. 2000;20:1–6. doi: 10.1523/JNEUROSCI.20-10-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffin KJ, Kirschner LS, Matyakhina L, Stergiopoulos S, Robinson-White A, Lenherr S, Weinberg FD, Claflin E, Meoli E, Cho-Chung YS, Stratakis CA. Down-regulation of regulatory subunit type 1A of protein kinase A leads to endocrine and other tumors. Cancer Res. 2004;64:8811–8815. doi: 10.1158/0008-5472.CAN-04-3620. [DOI] [PubMed] [Google Scholar]
- 16.Lohmann SM, Walter U. Regulation of the Cellular and Subcellular Concentrations and Distribution of Cyclic Nucleotide Protein Kinases. Adv Cyc Nuc Prot Phos Res. 1984;18:63–117. [PubMed] [Google Scholar]
- 17.Bartlett JM, Hulme MJ, Miller WR. Analysis of cAMP RI alpha mRNA expression in breast cancer: evaluation of quantitative polymerase chain reaction for routine use. Br J Cancer. 1996;73:1538–1544. doi: 10.1038/bjc.1996.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casey M, Vaughan CJ, He J, Hatcher CJ, Winter JM, Weremowicz S, Montgomery K, Kucherlapati R, Morton CC, Basson CT. Mutations in the protein kinase A R1alpha regulatory subunit cause familial cardiac myxomas and Carney complex. J Clin Invest. 2000;106:R31–38. doi: 10.1172/JCI10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kammer GM. Deficient protein kinase a in systemic lupus erythematosus: a disorder of T lymphocyte signal transduction. Ann N Y Acad Sci. 2002;968:96–105. doi: 10.1111/j.1749-6632.2002.tb04329.x. [DOI] [PubMed] [Google Scholar]
- 20.Kirschner LS, Carney JA, Svetlana DP, Taymans SE, Giatzkis C, Cho YS, Cho-Chung YS, Stratakis CA. Mutations of the Gene Encoding the Protein Kinase A Type I-a Regulatory Subunit in Patients with Carney Complex. Nature Genetics. 2000;26:89. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 21.Taimi M, Breitman TR, Takahashi N. Cyclic AMP-dependent protein kinase isoenzymes in human myeloid leukemia (HL60) and breast tumor (MCF-7) cells. Arch Biochem Biophys. 2001;392:137–144. doi: 10.1006/abbi.2001.2443. [DOI] [PubMed] [Google Scholar]
- 22.Erlichman J, Rosenfield R, Rosen OM. Phosphorylation of a cAMP-Dependent Protein Kinase From Bovine Cardiac Muscle. J Biol Chem. 1974;249:5000–5003. [PubMed] [Google Scholar]
- 23.Rangel-Aldao R, Rosen OM. Dissociation and reassociation of the phosphorylated and nonphosphorylated forms of adenosine 3′:5′-monophosphate-dependent protein kinase from bovine cardiac muscle. J Biol Chem. 1976;251:3375–3380. [PubMed] [Google Scholar]
- 24.Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 25.Wu J, Brown SH, von Daake S, Taylor SS. PKA type IIalpha holoenzyme reveals a combinatorial strategy for isoform diversity. Science. 2007;318:274–279. doi: 10.1126/science.1146447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin BR, Deerinck TJ, Ellisman MH, Taylor SS, Tsien RY. Isoform-specific PKA dynamics revealed by dye-triggered aggregation and DAKAP1alpha-mediated localization in living cells. Chem Biol. 2007;14:1031–1042. doi: 10.1016/j.chembiol.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 27.Diskar M, Zenn HM, Kaupisch A, Prinz A, Herberg FW. Molecular basis for isoform-specific autoregulation of protein kinase A. Cell Signal. 2007;19:2024–2034. doi: 10.1016/j.cellsig.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Vigil D, Blumenthal DK, Taylor SS, Trewhella J. Solution scattering reveals large differences in the global structures of type II protein kinase A isoforms. J Mol Biol. 2006;357:880–889. doi: 10.1016/j.jmb.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Vigil D, Blumenthal DK, Heller WT, Brown S, Canaves JM, Taylor SS, Trewhella J. Conformational differences among solution structures of the type Ialpha, IIalpha and IIbeta protein kinase A regulatory subunit homodimers: role of the linker regions. J Mol Biol. 2004;337:1183–1194. doi: 10.1016/j.jmb.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 30.Cheng X, Phelps C, Taylor SS. Differential binding of cAMP-dependent protein kinase regulatory subunit isoforms Ialpha and IIbeta to the catalytic subunit. J Biol Chem. 2001;276:4102–4108. doi: 10.1074/jbc.M006447200. [DOI] [PubMed] [Google Scholar]
- 31.Anand G, Taylor SS, Johnson DA. Cyclic-AMP and Pseudosubstrate Effects on Type-I A-Kinase Regulatory and Catalytic Subunit Binding Kinetics. Biochemistry. 2007;46:9283–9291. doi: 10.1021/bi700421h. [DOI] [PubMed] [Google Scholar]
- 32.Knighton DR, Zheng J, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal Structure of the Catalytic Subunit of cAMP-dependent Protein Kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 33.Knighton DR, Zheng J, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Structure of a Peptide Inhibitor Bound to the Catalytic Subunit of Cyclic Adenosine Monophosphate-Dependent Protein Kinase. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 34.Kornev AP, Haste NM, Taylor SS, Ten Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci U S A. 2006;103:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anand GS, Hotchko M, Brown SH, Ten Eyck LF, Komives EA, Taylor SS. R-subunit isoform specificity in protein kinase A: distinct features of protein interfaces in PKA types I and II by amide H/2H exchange mass spectrometry. J Mol Biol. 2007;374:487–499. doi: 10.1016/j.jmb.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng J, Knighton DR, Xuong NH, Taylor SS, Sowadski JM, Ten Eyck LF. Crystal structures of the myristylated catalytic subunit of cAMP-dependent protein kinase reveal open and closed conformations. Protein Sci. 1993;2:1559–1573. doi: 10.1002/pro.5560021003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knighton DR, Bell SM, Zheng J, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. 2.0 Å Refined Crystal Structure of the Catalytic Subunit of cAMP-dependent Protein Kinase Complexed with a Peptide Inhibitor and Detergent. Acta Cryst. 1993;D49:357–361. doi: 10.1107/S0907444993000502. [DOI] [PubMed] [Google Scholar]
- 38.Gangal M, Clifford T, Deich J, Cheng X, Taylor SS, Johnson DA. Mobilization of the A-kinase N-myristate through an isoform-specific intermolecular switch. Proc Natl Acad Sci U S A. 1999;96:12394–12399. doi: 10.1073/pnas.96.22.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kornev AP, Taylor SS, Ten Eyck LF. A generalized allosteric mechanism for cis-regulated cyclic nucleotide binding domains. PLoS Comput Biol. 2008;4:e1000056. doi: 10.1371/journal.pcbi.1000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibson RM, Ji-Buechler Y, Taylor SS. Interaction of the regulatory and catalytic subunits of cAMP-dependent protein kinase. Electrostatic sites on the type Ialpha regulatory subunit. J Biol Chem. 1997;272:16343–16350. doi: 10.1074/jbc.272.26.16343. [DOI] [PubMed] [Google Scholar]
- 41.Rinaldi J, Ocampo J, Rossi J, Moreno S. A novel activating effect of the regulatory subunit of protein kinase A on catalytic subunit activity. Arch Biochem Biophys. 2008;480:95–103. doi: 10.1016/j.abb.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 42.Kannan N, Neuwald AF. Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component? J Mol Biol. 2005;351:956–972. doi: 10.1016/j.jmb.2005.06.057. [DOI] [PubMed] [Google Scholar]
- 43.Thompson EE, Kornev AP, Kannan N, Kim C, Ten Eyck LF, Taylor SS. Comparitive Surface Geomerty of the Protein Kinase Family. Protein Science. doi: 10.1002/pro.209. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levinson NM, Seeliger MA, Cole PA, Kuriyan J. Structural basis for the recognition of c-Src by its inactivator Csk. Cell. 2008;134:124–134. doi: 10.1016/j.cell.2008.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herberg FW, Taylor SS. Physiological inhibitors of the catalytic subunit of cAMP-dependent protein kinase: effect of MgATP on protein-protein interactions. Biochemistry. 1993;32:14015–14022. doi: 10.1021/bi00213a035. [DOI] [PubMed] [Google Scholar]
- 46.Rothermel JD, Botelho LHP. A mechanistic and kinetic analysis of the interactions of the diastereoisomers of adenosine 3,5 -(cyclic)phosphorothioate with purified cyclic AMP-dependent protein kinase. Biochem J. 1988;251:757–762. doi: 10.1042/bj2510757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scholten A, van Veen TA, Vos MA, Heck AJ. Diversity of cAMP-dependent protein kinase isoforms and their anchoring proteins in mouse ventricular tissue. J Proteome Res. 2007;6:1705–1717. doi: 10.1021/pr060601a. [DOI] [PubMed] [Google Scholar]
- 48.Diller TC, Xuong NH, Taylor SS. Type IIB Regulatory Subunit of cAMP-Dependent Protein Kinase: Purification Strategies to Optimize Crystallization. Protein Expression and Purification. 2000;20:357–364. doi: 10.1006/prep.2000.1312. [DOI] [PubMed] [Google Scholar]
- 49.Saraswat LD, Filutowics M, Taylor SS. Expression and Mutagenesis of the RI-subunit of cAMP-Dependent Protein Kinase in E. coli. In: Corbin JA, Johnson RA, editors. Methods in Enzymology. New York: Academic Press, Inc; 1988. pp. 325–336. [DOI] [PubMed] [Google Scholar]
- 50.Wu J, Jones JM, Xuong NH, Ten Eyck LF, Taylor SS. Crystal structures of RIalpha subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry. 2004;43:6620–6629. doi: 10.1021/bi0302503. [DOI] [PubMed] [Google Scholar]
- 51.Wu J, Brown S, Xuong NH, Taylor SS. RIalpha subunit of PKA: a cAMP-free structure reveals a hydrophobic capping mechanism for docking cAMP into site B. Structure. 2004;12:1057–1065. doi: 10.1016/j.str.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 52.Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- 53.Newman J, Egan D, Walter TS, Meged R, Berry I, Ben Jelloul M, Sussman JL, Stuart DI, Perrakis A. Towards rationalization of crystallization screening for small- to medium-sized academic laboratories: the PACT/JCSG+ strategy. Acta Crystallogr D Biol Crystallogr. 2005;61:1426–1431. doi: 10.1107/S0907444905024984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The two residues form a hydrophobic pocket that is used by PKI (top left) to achieve high affinity binding as an α helix, or by RIIβ(102–265) as a β strand (boxed in blue dash line). The orientation of the side chain of Phe239 appears to be a marker for occupancy of this surface pocket. The 2Fo-Fc density maps of the Y235C-P-P-F-F239C regions in both RIIβ(108–268) (left) and RIIβ(102–265) (right, with the presence of the N-terminal region of V103R-I-N-R-F107R) holoenzymes, contoured at 1σ, are shown on the bottom. The Phe239 side chain flip is highlighted by an arrow.
Crystal packing interactions in the RIIβ holoenzyme structure reveal R262R from an adjacent molecule docks into the myrstic acid binding pocket of the C-subunit. The N and C-terminal tails of the C-subunit are shown as ribbons, and the remainder of the C-subunit is surface rendered. R262R, F18C, Y306C and the myristic acid aligned from PDB 1CDK are shown as sticks. Blue mesh represents the surface of R262R.