

Abstract
Imaging membranes in live cells with nanometer-scale resolution promises to reveal ultrastructural dynamics of organelles that are essential for cellular functions. In this work, we identified photoswitchable membrane probes and obtained super-resolution fluorescence images of cellular membranes. We demonstrated the photoswitching capabilities of eight commonly used membrane probes, each specific to the plasma membrane, mitochondria, the endoplasmic recticulum (ER) or lysosomes. These small-molecule probes readily label live cells with high probe densities. Using these probes, we achieved dynamic imaging of specific membrane structures in living cells with 30–60 nm spatial resolution at temporal resolutions down to 1–2 s. Moreover, by using spectrally distinguishable probes, we obtained two-color super-resolution images of mitochondria and the ER. We observed previously obscured details of morphological dynamics of mitochondrial fusion/fission and ER remodeling, as well as heterogeneous membrane diffusivity on neuronal processes.
Keywords: nanoscopy, diffraction limit, photoswitchable dye, stochastic optical reconstruction microscopy, photoactivation localization microscopy
Membrane structures, such as the plasma membrane, endosomes, lysosomes, mitochondria, the Golgi, and the endoplasmic recticulum (ER), perform a variety of functions in eukaryotic cells. To accommodate these functions, cellular membranes fold into various shapes, often with highly curved morphologies and nanometer-scale dimensions (1, 2). Examples include the filopodia protruding from the plasma membrane, the cristae of mitochondria, the cisternae of the Golgi, and the meshwork of the ER (1, 2). These intricate membrane structures often undergo rapid remodeling. Filopodia extend from and retract into the cell via cytoskeletal assembly and disassembly (1). ER tubules are pulled out of membrane sheets by molecular motors or polymerizing cytoskeletal filaments (2). Mitochondria constantly fuse and divide while moving along microtubules (3). Such dynamic processes play essential roles for maintaining the morphology and functions of these subcellular structures and organelles.
Ultrastructural characterization of membrane organelles has mainly relied on electron microscopy (EM), which provides both membrane contrast and nanometer-scale resolution. However, EM cannot be used to image live specimens and hence does not provide dynamic information directly. This difficulty may be overcome by the recently developed super-resolution fluorescence microscopy methods (4–7). Among these methods, stochastic optical reconstruction microscopy (STORM) (8) and (fluorescence) photoactivation localization microscopy [(F)PALM] (9, 10) take advantage of the use of photoswitchable probes and high-precision localization of single molecules to surpass the diffraction limit. During the imaging process, only a sparse subset of the probes are switched on at any time such that their positions can be determined with high precision by finding the centroids of their images. After the coordinates of a sufficient number of probe molecules are accumulated, a super-resolution image can be constructed from these molecular coordinates. It has been further demonstrated that many conventional dyes can be used for super-resolution imaging based on photoswitching/bleaching and localization of single molecules (11–20). Using single-molecule-based super-resolution methods, 3D resolutions down to approximately 10 nm have been demonstrated for fixed samples (21–26), and live-cell imaging has also been achieved with spatial resolutions of 20–60 nm at time resolutions ranging from 0.5 s to 1 min (27–32).
To date, super-resolution imaging has been demonstrated mostly on protein-based fluorescent labels such as proteins tagged with dyes or genetically encoded fluorescent proteins. However, membrane proteins are often not distributed uniformly on the membrane, but instead form localized domains nonideal for general membrane imaging. Moreover, since probe density is a key determinant of the image resolution (27, 31), protein labels need to be expressed at high levels to achieve high resolutions, which can cause overexpression artifacts. An alternative to protein labels is small-molecule probes that directly bind to membrane structures, which have been widely used for specific labeling of membrane organelles (33). The cell permeability, small size, and high affinity of these probes for specific membrane organelles facilitate high-density labeling of live cells. Moreover, these small-molecule probes are commercially available and easy to use. However, it is unknown whether these probes are photoswitchable and suitable for super-resolution imaging.
In this work, we demonstrate super-resolution fluorescence imaging of membranes in live cells by using photoswitchable membrane probes. We identified the photoswitching capabilities of eight small-molecule probes, commonly used for labeling the plasma membrane, mitochondria, the ER, or lysosomes. Using these probes, we achieved 30–60 nm spatial resolution with temporal resolution down to 1–2 sec, and tens of independent snapshots when imaging live cells. The different spectral properties of these probes also allowed two-color super-resolution imaging of mitochondria and the ER. Utilizing these imaging capabilities, we captured ultrastructural dynamics of the plasma membrane, mitochondria, and the ER.
Results
Imaging the Plasma Membrane with Lipophilic Cyanine Dyes.
Carbocyanine dyes with long alkyl chains such as DiI, DiD, and DiR have been widely used as labels for the plasma membrane (see Fig. S1A for chemical structures). Due to their lipophilic nature, incubating live cells with dye solutions for a few minutes is sufficient to label the plasma membrane with high probe density (33). Interestingly, we found that DiI, DiD, and DiR exhibited photoswitching behaviors without an exogenous switching agent. Under 561-nm (for DiI), 657-nm (for DiD), or 752-nm (for DiR) illumination, these probes fluoresced and rapidly switched off to a dark state; the dark-state molecules could then be reactivated to the fluorescent state by 405-nm illumination (Fig. 1A). Similar laser excitation schemes have been used for switching other dyes (13, 17). This reversible photoswitching behavior were suitable for STORM imaging of the plasma membrane (Fig. 2A and Fig. S2 A and B).
Fig. 1.

Photoswitching behavior of small-molecule probes for four membrane structures in live cells. BS-C-1 cells were labeled with (A) DiI for the plasma membrane, (B) MitoTracker Red for mitochondria, (C) ER-Tracker Red for the ER, (D) LysoTracker Red for lysosomes. Top row: Images taken with weak 561-nm illumination to excite fluorescence from these probes without switching them off appreciably. Middle row: Images taken after strong 561-nm illumination (approximately 10 kW/cm2), which turned the probes off efficiently. Bottom row: Images taken with weak 561-nm light after 405-nm light was used to turn the probes on again. Image contrasts in the middle and bottom rows were either the same as the images in the top row or increased by 6 times, as indicated. The incomplete recovery of ER- and LysoTrackers immediately after switching-off did not substantially degrade the STORM images due to the high labeling density. The recovery of ER- and LysoTrackers slowly increased and reached about 100% after 15 min of 405-nm illumination. Scale bars, 1 μm.
Fig. 2.

STORM images of four membrane organelles/structures in live cells. Conventional (i) and STORM (ii) images of (A) the plasma membrane labeled with DiI in a hippocampal neuron, (B) mitochondria labeled with MitoTracker Red in a BS-C-1 cell, (C) the ER labeled with ER-Tracker Red in a BS-C-1 cell, and (D) lysosomes labeled with LysoTracker Red in a BS-C-1 cell. The conventional fluorescence images were taken immediately before STORM imaging with a low excitation intensity to avoid switching off the dyes appreciably. The STORM images were acquired in 15 sec (A), 10 sec (B and C) and 1 sec (D). Arrowheads in A indicate spine necks along with their measured widths. Scale bars, 1 μm.
We performed STORM imaging of the plasma membrane on live hippocampal neurons using DiI (Fig. 2A). DiI-labeled neurons were imaged with 561-nm illumination at a 500-Hz camera frame rate. A roughly constant number of activated DiI molecules per camera frame were maintained with ramping 405-nm illumination. The fast switching rates of DiI allowed us to collect a super-resolution image in 15 sec or less. Compared to the raw and deconvolved conventional images, substantial resolution improvement was observed in the STORM image (Fig. 2A and Fig. S3A). The enhanced resolution allowed us to measure the widths of the dendritic spine necks in live neurons (Fig. 2A).
During STORM imaging, individual DiI molecules were switched off in approximately two camera frames, emitting 720 photons per frame on average. By measuring the spread of the localization distributions of individual fixable DiI molecules in fixed cells, we determined the localization precision to be 17 nm, measured in SD, corresponding to an image resolution of 40 nm measured in full width at half maximum (Table S1). Another key factor affecting the final image resolution is the label or localization density within the structure. Because DiI molecules diffuse on the plasma membrane in live cells, a single probe molecule is able to sample different locations on the membrane and contribute multiple independent localizations for mapping out the underlying structure. Based on the Nyquist sampling criterion, which equates the resolution limit to twice the average distance between neighboring localizations, a Nyquist resolution limit can be defined as 2/(localization density)1/2 for 2D images (27). For the live neuron images, we found the Nyquist resolution limit to be approximately 40 nm for the 15-sec STORM snapshots (eight independent snapshots) and approximately 70 nm for 5-sec snapshots (24 independent snapshots).
Time-resolved STORM images of DiI captured extension and retraction of filopodia or dendritic spines (Fig. 3A). In addition to the morphological dynamics of the plasma membrane, molecular motion within the membrane can also be monitored by tracking individual probe molecules. The use of photoswitchable probes allows a high density of molecular trajectories to be accumulated over time (34). In Fig. 3B, we present a subset of the DiI trajectories that lasted for 15 frames or more, from which we calculated the local diffusion coefficients (See Fig. S4 for the full distribution of the trace length). Diffusion of DiI was slower in thin dendritic structures such as filopodia or spines, in comparison to the mobility measured in the shaft (Fig. 3C), consistent with previous results (35). Moreover, the high density of molecular traces allowed us to determine the local distribution of diffusivity within filopodia or spines (Fig. 3D). The mobility of DiI decreased as the molecules approached the tips of filopodia or spines (Fig. 3E), possibly due to the higher local membrane curvature (36) and the different composition of membrane proteins at these locations.
Fig. 3.

Plasma membrane dynamics in dendrites of a live hippocampal neuron at 37 °C. (A) Morphological changes of dendritic structures captured by a series of 10-sec STORM snapshots. Green arrowhead: A growing spine or filopodium. Blue arrowhead: An extending filopodium. Purple arrowhead: A retracting filopodium. (B–E) High-density single-particle-tracking of DiI. (B) Molecular trajectories lasting at least 15 camera frames (2 ms per frame) colored by their diffusion coefficients, D, according to the color map on the Right. (C) Diffusion coefficients in different dendritic structures. Error bars indicate standard errors: N = 613 traces for shaft; N = 90 traces for spine/filopodia. (D) A zoom-in of the boxed region in B. (E) Local distribution of diffusion coefficients within the filopodium in the dashed box in D. Error bars indicate standard errors (N = 7–18). Scale bars, 1 μm.
Imaging Mitochondria with Cationic Rosamine and Carbocyanine Fluorophores.
Mitochondria consist of two layers of membranes: the outer membrane forms a smooth tubular outline of the organelle; the inner membrane forms deeply invaginated tubules and lamellae called cristae. Many mitochondrial probes are cell-permeant cations that accumulate on the inner membrane of mitochondria via electrostatic interactions (33). We identified three photoswitchable mitochondrial probes that can be classified into two groups: (i) MitoTracker Orange and MitoTracker Red are cationic rosamine dyes; (ii) MitoTracker Deep Red is a cationic carbocyanine (Fig. S1B). Similar to DiI, cells could be labeled with these probes simply by incubating with dye solutions for a few minutes. They could be excited to fluoresce and turned off by 561-nm (for MitoTracker Orange/Red) or 657-nm (for MitoTracker Deep Red) illumination, and reactivated by 405-nm illumination (Fig. 1B). All three dyes could be used for STORM imaging of mitochondria (Fig. 2B and Fig. S2 C and D).
We obtained STORM images of mitochondria in live BS-C-1 cells using MitoTracker Red with a procedure similar to that used in DiI imaging at a 500-Hz frame rate (Fig. 2B). Per imaging frame, each dye molecule emitted on average 790 photons and was localized with 13-nm localization precision, corresponding to 30-nm resolution (Table S1). Images acquired in 10 sec exhibited localization densities that correspond to a Nyquist resolution limit of approximately 30 nm with < 15% variation among different cells and experiments. Such resolutions could be achieved for up to 30 independent snapshots per movie. Compared to the raw and deconvolved conventional images, the STORM images showed substantially improved resolution (Fig. 2B and Fig. S3B).
Because mitochondria tend to move fast in cells, we further improved the time resolution by increasing the camera frame rate to 900 Hz and by activating a higher density of probes per frame such that their images partially overlap. We analyzed the overlapping images of individual molecules by a multiemitter fitting algorithm (37). Under these imaging conditions, we obtained a Nyquist-based resolution of approximately 40 nm in 2 sec. The improved temporal resolution allowed us to capture mitochondrial fission and fusion intermediates (Fig. 4).
Fig. 4.

Mitochondrial dynamics in a live BS-C-1 cell. (A) A 6-sec STORM snapshot shows thin tubes connecting neighboring mitochondria (indicated by yellow arrowheads). (B) Width distribution of inter-mitochondria tubules taken from multiple cells. Black bars: All tubules. Green bars: Tubules prior to fission. Red bars: Tubules after fusion. (C) Fission (green arrowheads) and fusion (red arrowheads) events captured by a time-series of 2-sec STORM snapshots (i) and corresponding conventional images (ii). While the images were acquired continuously (Movie S1), only every other 2-sec snapshot is displayed here to save space. Scale bars, 500 nm.
Time-lapse STORM images revealed thin, extended tubular intermediates connecting neighboring mitochondria both prior to fission and after fusion (Fig. 4A and Movie S1). The average width of these tubular structures was 104 ± 15 nm (SD, N = 35) (Fig. 4B). Notably, such tubules tend to have uniform widths over an extended length of several hundred nanometers. Multiple fission and fusion events (Fig. 4C) were captured in the movie. The tubular intermediates for the fusion and fission events had similar widths [Fig. 4B, 102 ± 16 nm (SD, N = 24) prior to fission; 108 ± 12 nm (SD, N = 11) after fusion]. These thin tubular structures could persist for more than 10 sec. While fission and fusion events could also be observed in the conventional fluorescence movies, the tubular intermediates were not clearly resolved in the diffraction-limited images (Fig. 4C, ii).
Given the extended length of the tubular intermediate with a relatively constant width, such membrane tube is likely constricted by a protein tubular structure. A potential candidate protein is Drp1, a dynamin-family protein required for mitochondrial division in mammalian cells (3, 38). Indeed, in the in vitro assemblies of liposome and Dnm1, the Drp1-homolog in yeast, Dnm1 was found to assemble outside membrane tubes and constrict them to a lumenal diameter of approximately 90 nm (39), similar to the widths of the fusion and fission intermediates measured here.
Imaging the ER and Lysosomes with BODIPY Dye-Conjugates.
BODIPY dyes are neutral and nonpolar. Therefore, BODIPY conjugates tend to be more cell permeant than charged or polar dyes, and are often used to label intracellular structures (33). We tested two BODIPY-conjugated probes: (i) ER-Tracker Red, a BODIPY TR conjugate of glibenclamide which binds to potassium channels enriched in the ER; (ii) LysoTracker Red, a BODIPY 564/570 linked to a weak base that is highly selective for the acidic membrane of lysosomes (Fig. S1 C and D) (33). Again, labeling procedures for these probes are similar to that of DiI. We found both BODIPY TR and BODIPY 564/570 to photoswitch in live cells; they could be imaged and switched off by 561-nm illumination and reactivated by 405-nm light (Fig. 1 C and D).
STORM imaging of the ER and lysosomes were conducted using a procedure similar to that used for DiI or MitoTracker Red (Fig. 2 C and D). At a 500-Hz frame rate, ER-Tracker Red and LysoTracker Red emitted an average of 820 photons per frame, providing a localization precision of 13–15 nm, corresponding to a resolution of 30–35 nm (Table S1). We obtained STORM images within 10 sec for the ER and 1 sec for lysosomes with localization densities that correspond to a Nyquist resolution limit of 30–40 nm for 30–80 snapshots. STORM images of the ER showed an intricate meshwork of tubules and sheets (Fig. 2C). While these structures were largely resolvable in conventional images, the width of the ER tubules were often below the diffraction limit, but could be determined from STORM images (Fig. 2C). Likewise, lysosomes were often resolvable from each other in conventional images, but their sizes and shapes were better determined in STORM images (Fig. 2D).
Dynamics of ER-remodeling were observed using ER-Tracker Red (Fig. 5). In a time-series of 10-sec snapshots (Fig. 5A and Movie S2), we observed extending ER tubules (Fig. 5A). When multiple snapshots were collapsed into a single image with each localization colored by the time of appearance (Fig. 5B), extending tubules could be easily identified by color. The newly extended tubules appeared thinner than older tubules (Fig. 5C); the average widths were 84 ± 15 nm (SD, N = 22) for the newly extended tubes and 127 ± 32 nm (SD, N = 34) for tubules that existed for at least 2 min. When we increased the dye activation rate as in the case of mitochondrial imaging, we were able to increase the imaging speed to 2 sec per image without compromising the spatial resolution (Fig. 5 D and E and Movie S3).
Fig. 5.

ER dynamics in live BS-C-1 cells. (A) A time-series of 10-sec STORM snapshots. Blue arrowheads: Tips of extending tubules. (B) A composite image containing all snapshots in A with each localization colored by its time of appearance according to the color map on the Right. (C) Distribution of the widths of ER tubules. Green bars: Newly extended tubules. Red bars: Old tubules that had already existed for at least 2 min. (D) A time-series of 2-sec STORM snapshots. Blue arrowheads: Extending tubules. Purple arrowheads: Retracting tubules. Yellow arrowheads: Extending sheets. (E) A composite image containing all snapshots in D with each localization colored by the time of its appearance. Scale bars, 500 nm.
Two-Color Imaging of Mitochondria and the ER.
The different spectral properties of the probes described above allow for multicolor imaging. For instance, the emission maxima of MitoTracker Red and ER-Tracker Red differ by 16 nm. They can be distinguished using a ratiometric method by splitting their emission into a short- and a long-wavelength channel; the probe identity can then be determined from the intensity ratio of these two channels (30, 32, 40).
Using this approach, we obtained two-color STORM images of mitochondria and the ER in live cells (Fig. 6 and Movie S4). Although we achieved a comparable Nyquist resolution (25–35 nm) to that of the single-color images, the image quality was somewhat degraded from single-color images due to the color cross-talk (16% from ER to mitochondria, 28% from mitochondria to ER with the color assignment defined in Fig. S5).
Fig. 6.
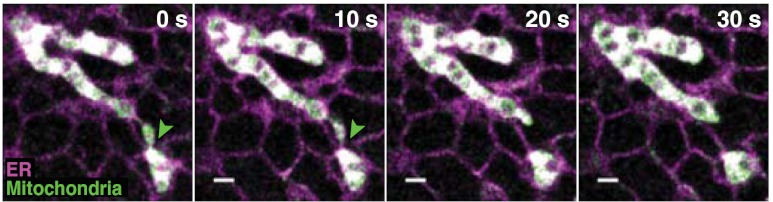
Two-color STORM images of mitochondria (green) and the ER (magenta) in a live BS-C-1 cell. The snapshots are 10 sec long. The ER tubules at the mitochondrial fission site are indicated by green arrowheads. Scale bars, 500 nm.
Correlative dynamics of the two structures were observed from these images. ER tubules were often present at the sites of mitochondrial contraction and fission (Fig. 6), suggesting that ER tubules are involved in constriction of mitochondria. This observation is consistent with the previous hypothesis that ER-induced mitochondrial constriction facilitates the recruitment of Drp1 at the mitochondrial fission site (41).
Discussion
Cell membranes often exhibit nanometer-scale morphologies and undergo dynamic remodeling. Super-resolution fluorescence imaging can reveal previously unknown ultrastructural dynamics of these structures. Here, we identified several photoswitchable, small-molecule membrane probes and demonstrated their utility in super-resolution STORM imaging of the plasma membrane, mitochondria, the ER, and lysosomes in living cells. The images revealed nanometer-scale morphological dynamics of neuronal processes, mitochondria, and the ER, as well as nonuniform membrane diffusivity in neurons.
STORM imaging of these probes was performed in buffered medium supplemented with an oxygen scavenger system, but no exogenous chemical additives such as thiol reagents was added. Without the oxygen scavenger, the probes photoswitched similarly but bleached substantially faster, and therefore the overall observation time was reduced by 10–20 fold. Although these membrane probes all photoswitched under the same buffer conditions, their switching mechanisms could potentially be different. The carbocyanine dyes (DiI, DiD, DiR, and MitoTracker Deep Red) were possibly switched to a dark state by forming a thiol adduct with endogenous thiols (e.g. Glutathione exists in animal cells at approximately 5 mM), as shown for Cy5 (42). The rosamine dyes (MitoTracker Orange/Red) may be switched to long-lived radical species, similar to the structurally related rhodamine dyes (43). The switching mechanism of the BODIPY dyes (ER/LysoTracker Red) is difficult to speculate on, as no dyes in this class have been previously shown to photoswitch. Further work is needed to characterize the dark states of these fluorophores.
When performing super-resolution imaging of live cells using these probes, one has to be cautious about potential artifacts arising from phototoxicity and motion blurring. We performed STORM imaging using an excitation intensity of ≤ 10 kW/cm2 at 561, 657, or 752 nm, and weak 405-nm activation intensities (typically 0–3 W/cm2), in standard culture medium supplemented with the oxygen scavenger system. Previously, under similar illumination and buffer conditions, we found that the cell morphology and dynamics remain unperturbed for up to 20 min (31). However, mitochondria are known to be relatively sensitive to the oxidative and phototoxic stress (44). So we further examined whether the imaging conditions exert adverse effects on mitochondrial morphology. Under our strongest illumination conditions, mitochondrial morphology appeared normal for about 1 min. After 1 min, the shape of mitochondria began to change appreciably, evolving from an elongated, tubular shape into a more spherical shape, which is often observed for mitochondria under stress (Movie S5) (44). Hence, under these illumination conditions, we can only observe mitochondria for about 1 min before phototoxicity substantially perturbs the mitochondrial morphology. Fortunately, entire time courses of mitochondrial fusion and fission events can often be observed within 1 min, allowing studies of fusion and fission dynamics. For studying dynamics over a longer time, we can strobe the illumination at the cost of the time resolution. For example, when turning on illumination for one-fourth of the time with an excitation sequence of two frames of illumination followed by six dark frames, we were able to elongate the observation time to approximately 10 min without substantially perturbing the mitochondrial morphology (Movie S6). The plasma membrane and ER were less sensitive to illumination. We did not observe appreciable perturbation to the morphologies of these structures for the entire imaging time of up to 7 min even with the strongest continuous illumination that we used.
Another potential artifact to consider is motion blurring. In single-molecule-based super-resolution imaging, two types of motion can affect the image quality. The first type is the diffusion of the probe molecules on the membrane structure. On the plus side, diffusion allows each probe molecule to sample different locations of the membrane structure in different camera frames and to contribute multiple independent localizations, thereby increasing the localization density. On the flip side, a membrane probe can diffuse a substantial distance even within a single camera frame, and the measured localization represents the average position of the probe. Consequently, the detected probe position tends to converge to the center of the confining membrane structure, which could artificially shrink the apparent size of the structure (Fig. S6A). The extent of shrinkage depends on the diffusion coefficient of the probe, the size of the confining structure, and the camera frame rate (Fig. S6 B and C). For example, the diffusion coefficient of MitoTracker Red, measured to be 0.3 μm2/s, resulted in minimal artifacts at the frame rates used here (500 and 900 Hz), but reducing the frame rate to 100 Hz would lead to an appreciable shrinkage (Fig. S6B). The diffusion coefficient of the plasma membrane probe DiI, 1.1 μm2/s, resulted in more noticeable effects even at 500 Hz; a 100-nm wide tube was shrunken by approximately 20% (Fig. S6C). Probe diffusion could also blur sharp features of the membrane structure because the average probe position within a frame could lie outside the structure when the structure exhibits sharp bending. To illustrate this effect, we simulated how probe diffusion blurs a mesh structure with various grid sizes (Fig. S6 D and E). At 500 Hz, the diffusivity of MitoTracker Red made a meshwork with 10-nm grids unresolvable, while the faster diffusion of DiI increased the unresolvable grid size to approximately 20 nm. These numbers are relatively small compared to the image resolution reported in this work, indicating that probe diffusion did not substantially blur structures in our images.
The second type of motion to consider is the movement of the membrane structure itself. As for any live-cell imaging, the image acquisition time should be sufficiently short that the displacement of the structure during this time is small compared to the resolution. Neuronal processes and ER tubules typically move slowly enough for our image resolutions. For example, the widths of ER tubules measured from 15-sec STORM images of live cells (108 ± 31 nm, SD, N = 111) were essentially identical to those determined from fixed-cell images (109 ± 22 nm, SD, N = 246). Mitochondria are more dynamic, moving with speeds of approximately 20 nm/s during fusion and approximately 50 nm/s at other times (45). For a spatial resolution of 30–60 nm and a time resolution of 2 sec, movement speeds up to 15–30 nm/s are tolerable and allowed us to capture mitochondria fission/fusion dynamics without substantial motion blur. Indeed, STORM images of mitochondria revealed fission/fusion intermediate structures previously unobserved in cells. The widths of the tubular fission/fusion intermediates measured in live cells (104 ± 15 nm, SD, N = 35) is similar to the width of the tubular structure of mitochondrial dynamin assembled in vitro (39), suggesting a molecular underpinning of the fission/fusion intermediates. We also observed intermitochondria tubes in fixed cells with a nearly identical width (102 ± 28 nm, SD, N = 44), suggesting that the tubular structures were not substantially blurred by motion. On the other hand, the cristae structure was partially blurred. Only the cristae with relatively large spacing appeared to be resolved in the live-cell STORM images (Fig. 4), as in previous live-cell images by structured illumination microscopy (46). The more tightly packed cristae with smaller spacing, observed in EM and fixed-cell STORM images (Fig. S7), were not well resolved in the live-cell images.
Finally, since biological structures are typically three-dimensional, extending the high image resolution into the third dimension could help resolve these structures better. Indeed, the cristae of mitochondria in fixed cells were better resolved in the 3D STORM images than in the corresponding 2D projection images (Fig. S7). The z coordinates of the molecules can be obtained by a variety of means for 3D super-resolution imaging (21–26), and indeed 3D STORM has been demonstrated in live cells (31). However, the use of diffusing membrane probes presents an additional challenge, especially when the shapes of the single-molecule images are used to determine their z-positions. Further increasing the camera frame rate or strobing the excitation laser should help overcome this problem and allow 3D super-resolution imaging of membrane organelles in living cells.
Materials and Methods
Sample Preparation.
Immediately before imaging, BS-C-1 cells and hippocampal neurons were labeled by incubating live cells with culture medium containing one of the membrane probes for 0.5–3 min. For two-color imaging, cells were labeled sequentially with MitoTracker Red and then ER-Tracker Red.
Image Acquisition and Analysis.
Labeled cells were imaged with continuous illumination at 561, 657, or 752 nm for imaging and 405 nm for probe activation in an oblique-incidence geometry on an Olympus IX-71 inverted microscope. The single-molecule images were recorded at 500–900 Hz frame rates on an EMCCD camera (iXon 860; Andor). For ratiometric two-color imaging, the fluorescence was split by a 624-nm longpass dichroic mirror into two channels and imaged on two halves of the same camera. The movies were analyzed by custom-written STORM analysis software as we previously described (31), or using the DAOSTORM (37) or a similar multiemitter fitting software (47). See SI Materials and Methods for details.
Supplementary Material
ACKNOWLEDGMENTS.
This work is supported in part by the US National Institutes of Health and the Collaborative Innovation Award of Howard Hughes Medical Institute (X.Z.), and National Natural Science Foundation of China and National Basic Research Program of China (G.-Q.B). X.Z. is a Howard Hughes Medical Institute investigator. S.-H.S is in part supported by the Mary Fieser fellowship. J.C.V. is supported in part by a Burroughs-Wellcome Career Award at the Scientific Interface.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. S.W.H. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201882109/-/DCSupplemental.
References
- 1.McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 2.Shibata Y, Hu J, Kozlov MM, Rapoport TA. Mechanisms shaping the membranes of cellular organelles. Annu Rev Cell Dev Biol. 2009;25:329–354. doi: 10.1146/annurev.cellbio.042308.113324. [DOI] [PubMed] [Google Scholar]
- 3.Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 4.Hell SW. Microscopy and its focal switch. Nat Methods. 2009;6:24–32. doi: 10.1038/nmeth.1291. [DOI] [PubMed] [Google Scholar]
- 5.Heintzmann R, Gustafsson MGL. Subdiffraction resolution in continuous samples. Nat Photonics. 2009;3:362–364. [Google Scholar]
- 6.Patterson G, Davidson M, Manley S, Lippincott-Schwartz J. Superresolution imaging using single-molecule localization. Annu Rev Phys Chem. 2010;61:345–367. doi: 10.1146/annurev.physchem.012809.103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: Super-resolution imaging of cells. Cell. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 10.Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bock H, et al. Two-color far-field fluorescence nanoscopy based on photoswitchable emitters. Appl Phys B. 2007;88:161–165. [Google Scholar]
- 12.Heilemann M, et al. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- 13.Folling J, et al. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- 14.Steinhauer C, Forthmann C, Vogelsang J, Tinnefeld P. Superresolution microscopy on the basis of engineered dark states. J Am Chem Soc. 2008;130:16840–16841. doi: 10.1021/ja806590m. [DOI] [PubMed] [Google Scholar]
- 15.Baddeley D, Jayasinghe ID, Cremer C, Cannell MB, Soeller C. Light-induced dark states of organic fluochromes enable 30 nm resolution imaging in standard media. Biophys J. 2009;96:L22–L24. doi: 10.1016/j.bpj.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhuang XW. Nano-imaging with STORM. Nat Photonics. 2009;3:365–367. doi: 10.1038/nphoton.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang XW. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burnette DT, Sengupta P, Dai YH, Lippincott-Schwartz J, Kachar B. Bleaching/blinking assisted localization microscopy for superresolution imaging using standard fluorescent molecules. Proc Natl Acad Sci USA. 2011;108:21081–21086. doi: 10.1073/pnas.1117430109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simonson PD, Rothenberg E, Selvin PR. Single-molecule-based super-resolution images in the presence of multiple fluorophores. Nano Lett. 2011;11:5090–5096. doi: 10.1021/nl203560r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cox S, et al. Bayesian localization microscopy reveals nanoscale podosome dynamics. Nat Methods. 2012;9:195–200. doi: 10.1038/nmeth.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juette MF, et al. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat Methods. 2008;5:527–529. doi: 10.1038/nmeth.1211. [DOI] [PubMed] [Google Scholar]
- 23.Pavani SRP, et al. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc Natl Acad Sci USA. 2009;106:2995–2999. doi: 10.1073/pnas.0900245106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shtengel G, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci USA. 2009;106:3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aquino D, et al. Two-color nanoscopy of three-dimensional volumes by 4Pi detection of stochastically switched fluorophores. Nat Methods. 2011;8:353–359. doi: 10.1038/nmeth.1583. [DOI] [PubMed] [Google Scholar]
- 26.Xu K, Babcock HP, Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat Methods. 2012;9:185–188. doi: 10.1038/nmeth.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biteen JS, et al. Super-resolution imaging in live Caulobacter crescentus cells using photoswitchable EYFP. Nat Methods. 2008;5:947–949. doi: 10.1038/NMETH.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wombacher R, et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat Methods. 2010;7:717–719. doi: 10.1038/nmeth.1489. [DOI] [PubMed] [Google Scholar]
- 30.Testa I, et al. Multicolor fluorescence nanoscopy in fixed and living cells by exciting conventional fluorophores with a single wavelength. Biophys J. 2010;99:2686–2694. doi: 10.1016/j.bpj.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones SA, Shim SH, He J, Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nat Methods. 2011;8:499–508. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gunewardene MS, et al. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophys J. 2011;101:1522–1528. doi: 10.1016/j.bpj.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson I, Spence MTZ. Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies. 11th Ed. Carlsbad, CA: Life Technologies; 2010. [Google Scholar]
- 34.Manley S, et al. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- 35.Ashby MC, Maier SR, Nishimune A, Henley JM. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Domanov YA, et al. Mobility in geometrically confined membranes. Proc Natl Acad Sci USA. 2011;108:12605–12610. doi: 10.1073/pnas.1102646108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holden SJ, Uphoff S, Kapanidis AN. DAOSTORM: An algorithm for high-density super-resolution microscopy. Nat Methods. 2011;8:279–280. doi: 10.1038/nmeth0411-279. [DOI] [PubMed] [Google Scholar]
- 38.Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. 2012;13:75–88. doi: 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mears JA, et al. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol. 2011;18:20–26. doi: 10.1038/nsmb.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bossi M, et al. Multicolor far-field fluorescence nanoscopy through isolated detection of distinct molecular species. Nano Lett. 2008;8:2463–2468. doi: 10.1021/nl801471d. [DOI] [PubMed] [Google Scholar]
- 41.Friedman JR, et al. ER Tubules Mark Sites of Mitochondrial Division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dempsey GT, et al. Photoswitching mechanism of cyanine dyes. J Am Chem Soc. 2009;131:18192–18193. doi: 10.1021/ja904588g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van de Linde S, et al. Photoinduced formation of reversible dye radicals and their impact on super-resolution imaging. Photochem Photobiol Sci. 2011;10:499–506. doi: 10.1039/c0pp00317d. [DOI] [PubMed] [Google Scholar]
- 44.Bereiter-Hahn J, Jendrach M. Mitochondrial dynamics. Int Rev Cell Mol Biol. 2010;284:1–65. doi: 10.1016/S1937-6448(10)84001-8. [DOI] [PubMed] [Google Scholar]
- 45.Liu X, Weaver D, Shirihai O, Hajnoczky G. Mitochondrial ‘kiss-and-run’: Interplay between mitochondrial motility and fusion-fission dynamics. Embo J. 2009;28:3074–3089. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shao L, Kner P, Rego EH, Gustafsson MGL. Super-resolution 3D microscopy of live whole cells using structured illumination. Nat Methods. 2011;8:1044–1046. doi: 10.1038/nmeth.1734. [DOI] [PubMed] [Google Scholar]
- 47.Babcock H, Sigal YM, Zhuang X. A high-density 3D localization algorithm for stochastic optical reconstruction microscopy. Optical Nanoscopy. 2012;1:6. doi: 10.1186/2192-2853-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.