

Abstract
PV1 is an endothelial‐specific protein with structural roles in the formation of diaphragms in endothelial cells of normal vessels. PV1 is also highly expressed on endothelial cells of many solid tumours. On the basis of in vitro data, PV1 is thought to actively participate in angiogenesis. To test whether or not PV1 has a function in tumour angiogenesis and in tumour growth in vivo, we have treated pancreatic tumour‐bearing mice by single‐dose intratumoural delivery of lentiviruses encoding for two different shRNAs targeting murine PV1. We find that PV1 down‐regulation by shRNAs inhibits the growth of established tumours derived from two different human pancreatic adenocarcinoma cell lines (AsPC‐1 and BxPC‐3). The effect observed is because of down‐regulation of PV1 in the tumour endothelial cells of host origin, PV1 being specifically expressed in tumour vascular endothelial cells and not in cancer or other stromal cells. There are no differences in vascular density of tumours treated or not with PV1 shRNA, and gain and loss of function of PV1 in endothelial cells does not modify either their proliferation or migration, suggesting that tumour angiogenesis is not impaired. Together, our data argue that down‐regulation of PV1 in tumour endothelial cells results in the inhibition of tumour growth via a mechanism different from inhibiting angiogenesis.
Keywords: angiogenesis, fenestrae, vesiculo‐vacuolar organelles, caveolae, pancreatic cancer, transendothelial channels, tumour microenvironment
Introduction
The vertebrate gene Plasmalemmal Vesicle Associated Protein (PLVAP) encodes for PV1, a 60kd, single span, type II integral membrane N‐glycosylated protein 1, 2, 3. PLVAP/PV1 expression is restricted to a subset of normal microvascular endothelial cells (ECs) and, in adult organisms, PV1 is not expressed in the ECs of all large vessels with the exception of the endocardium lining the heart chambers 1, 3, 4, 5. In the ECs of normal capillaries of selected organs, PV1 is essential for the formation of the stomatal diaphragms of caveolae and the diaphragms of fenestrae, transendothelial channels and vesiculo‐vacuolar organelles 2, 4, 6, 7, 8. Formation of the diaphragms seems to be the only cellular function of PV1 9.
Endothelial diaphragms are present in ECs of tumour vessels undergoing angiogenesis (reviewed in 10) and PV1 was reported to be highly expressed in the ECs of human solid tumours, pancreatic cancer included 11, 12, 13, 14 15. PV1 expression on tumour ECs was known for more than two decades, through data obtained with the PAL‐E and MECA32 monoclonal antibodies (mAb), shown recently to bind to human and mouse PV1 respectively 16, 17(reviewed in 10). In vitro data suggest that PV1 plays an active role in pathological angiogenesis by facilitating human EC tube formation in Matrigel 11, 12 and EC migration induced by angiogenic growth factors such as VEGF and HGF11, 12, 13, 15, 18. However, the usefulness of PV1 as a novel anti‐cancer therapeutic target is unclear 12, 19.
Besides its structural role and postulated roles in cell migration and angiogenesis, PV1 may also have a role in inflammation 10, 17. PV1 blockade in mice using anti‐PV1 antibodies results in obvious impairment of recruitment of neutrophils and macrophages at inflammation sites 17. PV1 down‐regulation by siRNA in TNFα‐activated ECs in vitro inhibits diapedesis of leucocytes without affecting their adhesion to and rolling on activated ECs under flow 17.
Pancreatic ductal adenocarcinoma (PDAC) ranks fourth among cancers as a cause of death in the United States and Europe with a median survival of 6 months 20. Five‐year survival is <5% and is limited to stage I and II patients who can benefit from pancreas resection in combination with chemotherapy and radiotherapy 21. Late stage (III and IV) unresectable PDAC patients have access only to palliative chemotherapy, yielding a median survival rate of 6–11 months 22, 23. So far, all PDAC therapies are short‐lived and associated with significant toxicities. Thus, pancreatic cancer patients are prime candidates for the benefit of synergistic adjuvant therapies to increase efficacy and/or manage toxicity.
To evaluate whether or not PV1 plays a role in angiogenesis in vivo and the potential of PV1 as a therapeutic target in PDAC treatment, we first tested its role in tumour growth in two different xenograft models of PDAC. We show that PV1 down‐regulation by a single intratumoural delivery of PV1shRNA using lentiviruses results in reduced tumour growth in these two models. Because of the sequence mismatch between human and mouse PV1, we show that this effect is clearly the result of PV1 down‐regulation in tumour stroma, which is of mouse origin. Moreover, in both tumours, PV1 is expressed only in ECs of tumour vessels and not expressed in tumour or stromal cells at protein or mRNA level. Taken together, these data argue that PV1 expression in tumour ECs is required for tumour growth in vivo. In addition, we show that deletion of PV1 in tumour ECs may not result in decreased tumour angiogenesis implicating additional mechanisms in PV1‐promoted tumour growth.
Materials and methods
Materials
All general reagents were from Thermo‐Fisher (Pittsburgh, PA, USA), unless otherwise stated.
Antibodies
Rat anti‐mouse CD31 (PECAM‐1) clone MEC13.3, unlabeled or conjugated to APC, was from BioLegend (San Diego, CA, USA); Mouse anti‐β‐actin mAb (AC40) from Sigma‐Aldrich (St. Louis, MO, USA); Biotinylated rabbit anti‐mouse IgG from Vector Laboratories (Burlingame, CA, USA); HRP‐conjugated goat anti‐rat IgG‐HRP, and goat anti‐mouse IgG‐HRP from Biodesign (Kennebunk, ME, USA); Goat anti‐chicken IgG‐Alexa 568 and Alexa 647, (Fab)2 fragment from Invitrogen (Eugene, OR, USA). Rat anti‐mouse PV‐1 mAb clone MECA‐32 mAb 9 and chicken anti‐human PV1C pAb 6 were as previously described.
Primary antibody labelling with fluorophores
Affinity purified primary antibodies, rat anti‐mouse PV‐1 mAb clone MECA‐32 were labelled with Alexa 568 or 647 fluorophores (Molecular Probes; Invitrogen), as per manufacturer's instructions.
Cells and cell culture
Mouse lung endothelial cells (MLEC) isolated from lungs of wild‐type mice or Cav1‐knockout mice (MLEC‐Cav1KO), and immortalized with polyoma middle T antigen, and flow sorted for CD31 and PV1 expression, are described before 9. MLEC were cultured on plastic in MLEC growth medium consisting of a modified endothelial growth medium 2 (EGM2) (Lonza, Walkersville, MD, USA) formulation, from which the serum coming with the kit was omitted and replaced with heat‐inactivated foetal bovine serum (FBS) (Hyclone, Logan, UT, USA) to 15% final concentration. The formulation was also supplemented with 100 μg/ml of each penicillin, streptomycin and glutamine (Invitrogen, Carlsbad, CA, USA), as described 9. Bovine aortic (BAEC), human umbilical vein (HUVEC) and human lung microvascular (HLMVEC) endothelial cells were obtained from Lonza and were cultured in EGM2 or EGM‐2MV medium, respectively. The MECA‐32 hybridoma (developed by the laboratory of E. Butcher 24 at Stanford University) was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA, USA).
AsPC‐1 and BxPC‐3 human pancreatic cancer cell lines, obtained from ATCC (Manassas, VA, USA), were grown in RPMI (Mediatech Inc., Herndon, VA, USA), with 10% FBS (Omega Scientific, Tarzana, CA, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (complete medium).
PV1 siRNA sequences
PV1 siRNA‐1, ‐2, ‐3, ‐4 (84094: PLVAP) were obtained from Dharmacon (Lafayette, CO, USA). The PV1 siRNA‐5 duplex (ID85339; Ambion, Austin, TX, USA) 8 and Luciferase negative control siRNA 25 were previously reported.
PV1‐knockdown effectiveness was evaluated in MLEC transfected with Oligofectamine (Invitrogen) in EBM2 culture medium lacking FBS and antibiotics, as per manufacturer's instructions. siRNAs were used at a final concentration of 5 nM and PV1 knockdown assessed 24, 72 and 96 h after transfection, using Western blotting with anti‐PV‐1 mAb MECA32. PV1‐shRNA sequences are described in Table 1 and were designed as in 26.
Table 1.
PV1‐shRNA sequences used for insertion in lentiviruses
| Gene targeted | Strand | shRNA sequence |
|---|---|---|
| Luciferase | sensea | 5′–TCTTACGCTGAGTACTTCGATTCAAGAGATCGAAGTACTCAGCGTAAGTTTTTTC–3′ |
| antisense | 5′–TCGAGAAAAAACTTACGCTGAGTACTTCGATCTCTTGAATCGAAGTACTCAGCGTAAGA–3′ | |
| PV1‐1 | sense | 5′–TGCGAGCTGGAGGCGCGTAATTCAAGAGATTACGCGCCTCCAGCTCGCTTTTTTC–3′ |
| mouse 1080‐1099 | antisense | 5′–TCGAGAAAAAAGCGAGCTGGAGGCGCGTAATCTCTTGAATTACGCGCCTCCAGCTCGCA–3′ |
| PV1‐2 | sense | 5′–TGAGGTGGATGTGCGCATTATTCAAGAGATAATGCGCACATCCACCTCTTTTTTC–3′ |
| mouse 1124‐1142 | antisense | 5′‐TCGAGAAAAAAGAGGTGGATGTGCGCATTATCTCTTGAATAATGCGCACATCCACCTCA‐3′ |
| PV1‐4 | sense | 5′–TAGGAGAAGTTCCAGGCGGATTCAAGAGATCCGCCTGGAACTTCTCCTTTTTTTC–3′ |
| mouse 690‐708 | antisense | 5′ TCGAGAAAAAAAGGAGAAGTTCCAGGCGGATCTCTTGAATCCGCCTGGAACTTCTCCTA‐3′ |
| PV1‐5 | sense | 5′–TGGAATTCAAGAAAAGGATCTTCAAGAGAGATCCTTTTCTTGAATTCCTTTTTTC–3′ |
| mouse 1258‐1276 | antisense | 5′‐TCGAGAAAAAAGGAATTCAAGAAAAGGATCTCTCTTGAAGATCCTTTTCTTGAATTCCA‐3′ |
siRNA sequences used as template are in bold.
Lentivirus production and EC infection
Oligonucleotides corresponding to the short hairpin RNA (shRNA) sequence of interest were annealed and cloned into the lentivirus vector, pLentiLox 3.7 (pll3.7) 26 (Addgene, Cambridge, MA, USA). Virus stocks were prepared by co‐transfecting pll3.7 with three packaging plasmids pMDLg/pRRE, CMV‐VSVG and RSV‐Rev, (Addgene) into 293T cells 25, 26.
Mouse lung endothelial cells were incubated (4 hrs) with lentiviruses encoding for PV1shRNA or controls (empty virus, shLuc) at 1 and 10 MOI, with or without 8 μg/ml polybrene in infection medium (EBM‐2), followed by incubation (24–48 hrs) with MLEC growth medium; infectivity was measured by flow cytometry using GFP expression as readout. The ability of pll3.7‐based lentiviruses to infect AsPC‐1 and BxPC‐3 tumour cells was already demonstrated 25.
PV1‐shRNA validation – immunofluorescence on cells and confocal microscopy
Mouse lung endothelial cells were infected with lentiviruses encoding for shLuc, shPV1‐1 and shPV1‐5 at an MOI = 0.1 and allowed to express shRNA for 48 hrs. Cells were labelled live (30 min., 10°C, EBM2 + 1% BSA) with fluorescent anti‐PV1 (1.5 μg/ml), rinsed (3×, RT) in PBS containing calcium and magnesium (PBS‐CM) and chased (37°C, MLEC growth medium), rinsed (3×, RT) in PBS‐CM, fixed (10 min., RT) in 4% paraformaldehyde in PBS‐CM, rinsed again in PBS‐CM, mounted in PermaFluor and examined by confocal fluorescence microscopy using a Zeiss 510 Meta confocal system (Carl Zeiss, Thornwood, NY, USA). Image stacks were acquired with the pinhole set at 1 Airy unit and transformed through the maximum‐intensity projection function to obtain global images of the cells using ImageJ software (NIH, Bethesda, MD, USA).
Flow cytometry
Labelled cells were analysed using a FacsCalibur or a CANTO flow cytometer controlled by either CellQuest or DIVA software respectively (BD Biosciences, San Diego, CA, USA). Data analysis was carried out using FlowJo 9.4 software (Tree Star, Ashland, OR, USA). Each experiment had four replicates per time‐point and was repeated three times. Median fluorescence from at least 5000 live cells was calculated in each sample. Statistical significance was calculated using Student's t‐test.
Generation of stable MLEC cell lines expressing PV1shRNA
Mouse lung endothelial cells were infected with a low (0.1–0.5) MOI of lentiviruses encoding for PV1shRNA or controls (empty virus, shLuc) in presence of 8 μg/ml polybrene in infection medium, allowed to express the shRNAs for 48 hrs when the cells were sorted based on GFP expression. Transduction efficiency in these conditions was ~1–5% and purposely designed to limit the number of multiple lentiviral integrations in the genome. The cell populations obtained were labelled MLEC‐shPV1‐1, ‐2, ‐4, ‐5, MLEC‐shLuc MLEC‐EV (empty vector).
RNA isolation
Total RNA was isolated using RNAeasy kit (Qiagen, Valencia, CA, USA) and quality controlled using Bioanalyzer (Agilent/Stratagene, Santa Clara, CA, USA) and NanoDrop (Thermo‐Fisher).
Real‐time quantitative PCR
Quantitative real‐time PCR was performed as before 9, using the Gene Expression Assays mouse PV1 (Mm00453379_m1), mouse Actin B (Mm00607939_s1), human PV1 (Hs00229941_m1), human Actin B (Hs03023880_g1) (Applied Biosystems, Foster City, CA, USA). Reactions were performed in triplicate, utilizing cDNA corresponding to 10 ng RNA. Data were analysed on the basis of the relative expression method with the formula 2−ΔΔ CT, where ΔΔC T = ΔC T (sample) − ΔC T (calibrator = average C T values of all samples), and ΔC T is the C T of the housekeeping gene [beta‐Actin] subtracted from the C T of the target gene.
Evaluation of cell‐surface PV1 levels by flow cytometry
Adherent MLEC stably expressing different shRNAs were labelled (30 min., 4°C) live with 1.5 μg/ml MECA‐32‐Alexa 647 mAb in MLEC growth medium, rinsed (3×, RT) in PBS and non‐enzymatically detached using EDTA (Cell Dissociation Solution; Sigma‐Aldrich). Cells were then mixed with an equal volume of 1% BSA in PBS, and kept on ice in the dark until examined using flow cytometry.
Western blotting
Equal amounts (20 μg/lane) of MLEC proteins were immunoblotted with MECA‐32 and mouse anti‐ β‐actin monoclonal antibody (clone AC40), as described. 9. Signal quantization by densitometry on TIFF files was carried out using GelEval v1.35 software (FrogDance, Dundee, UK).
Pancreatic tumour xenograft model
Female athymic mice (Nu/Nu, Charles River) were injected subcutaneously into the dorsal flank area with 1 × 106 of either ASPC‐1 or B×PC‐3 cells. For each cell type, the mice were divided randomly into four equal groups of EIGHT mice to be left untreated or injected with shPV1‐1‐LV, shPV1‐5‐LV or shLuc‐LV. Once tumours reached a volume of 50 mm3 (8–10 days after injection of the cells), they were injected with 4.107 viral particles in 50 μl of OptiMem (Invitrogen). Tumour diameters were measured every 3 days. Tumour volumes were calculated as π/4 × width × height × length of the tumour. Experiments were terminated when the tumour diameter reached 15 mm following the procedures approved by the Dartmouth College IACUC.
Statistics on tumour growth
Data were analysed using anova and Tukey HSD test for parametric data, or the Kruskall and Wallis test for non‐parametric data using the Dunn‐Benferroni test for multiple comparisons (VassarStats website). P < 0.05 was taken as the level of significance.
Colocalization of PV1 and CD31 in tumour samples by confocal microscopy
AsPC‐1 and BxPC‐3 tumours were induced as described and allowed to grow for 21 days. One hr before harvesting, mice were injected via the tail vein with 10 mg/kg Dark Red Fluorescent (660⁄680) FluoSpheres® in PBS (Molecular Probes/Invitrogen, Cat# F8789) to label the intravascular space. The mice were killed with CO2, tumours harvested unfixed, embedded in optimal cutting temperature compound (OCT). Co‐immunofluorescence experiments on methanol fixed 10 μm thick sections were performed with 1.5 μg/ml of Alexa 647‐coupled MECA‐32 mAb and Alexa488‐coupled rat anti‐CD31 MEC13.3 mAb and examined by confocal fluorescence microscopy using a Zeiss 510 Meta confocal system. The controls consisted of non‐immune rat IgG2a (clone 2A3) labelled with Alexa 647 or 488. Choice of fluorophores ensured lack of spectral overlap between the labels to virtually eliminate co‐localization artefacts.
Tumour vascular area density
Tumours processed as described above were stained with a rat anti‐CD31 mAb (clone MEC13.3) (BD Biosciences) using a Vectastain ABC Peroxidase kit (Vector Laboratories). Sections were examined using an Olympus BX‐61 upright microscope equipped with an Olympus DP70 camera.
For quantitative morphometric analysis of CD31 staining in tumour samples, 10 random fields/slide at 20× magnification were quantified for DAB signal using Image‐Pro Plus program (Version 7.0; Media Cybernetics, Silver Spring, MD, USA). Three tumours per group were analysed. Vascular area density 27, 28 was calculated as the ratio of CD31 positive pixels to the total area of the image, as described before 25.
Cell proliferation assays
To assess doubling time, cells were seeded in six well plates in triplicate at a density of 104 cells/well and the doubling time was calculated over 5 days. Every 24 hrs, cell numbers were determined by counting the cells using a haemocytometer. The experiments were repeated twice.
To assess cell proliferation based on DNA content, we employed an established assay 29, 30. Briefly, MLEC were seeded at 1000 cell/well in 96‐well plates (n = 8 wells per each time‐point per genotype), cultured in full growth medium and collected every day for 7 days. Each day the wells were briefly rinsed in PBS, and the cells frozen until the last time‐point was collected. Cells were lysed by osmotic shock with low (1×) strength SSC and then incubated with 1 mg/ml Hoechst 33258 (Enzo Life Sciences, Farmingdale, NY, USA). The signal was quantitated by spectrophotometry using a Synergy HT plate reader (BioTek, Winooski, VT, USA).
Adenovirus constructs and transduction
Adenoviral AdEasy‐1 system (Agilent/Stratagene) was used to generate adenoviruses expressing human PV1‐3xHA (AdPV1HA). Briefly, the sequence coding for human PV1‐3xHA was PCR amplified, inserted into the multiple cloning site of the pShuttle vector and the resulting construct was recombined with pAdEasy‐1 vector by co‐transfection in the BJ5183 E. coli strain (Agilent/Stratagene). Positive clones were linearized with Pac I restriction enzyme and transfected into 293 cells for Adenovirus production using the CsCl2 density gradient method or the VivaPure AdenoPACK kits (VivaScience, Sartorius, Germany).
BAECs and HUVECs were transduced (4–6 hrs, 37°C) with adenoviruses at an MOI of 10 PFUs/cell for AdGFP or 50–200 viral particles/cell for AdNull and AdPV1HA in EBM2 supplemented with 2% FBS, cells were rinsed twice in EGM2 and incubated (24 hrs, 37°C) in growth medium. The cells were routinely used 24 hrs after infection. Empty adenovirus (AdNull) or adenovirus encoding GFP (AdGFP) at the same MOI were used as controls.
Expression of PV1‐HA was determined in HUVEC or BAEC transduced in 100 mm dishes with an adenoviral MOI of 1. Twenty‐four hrs after transduction, the cells were processed for Western blotting, as described above for MLEC, using 25 μg of total protein per well and using chicken anti‐human PV1C pAb.
Endothelial cell migration assays
For scratch‐wound assay, cells were grown to confluence in triplicate in six well plates, the monolayer was scratched in five different places using a pipette tip and cell debris removed by rinsing the cells with medium. Images of the cells were acquired at 2, 4, 8, 12 and 20 hrs using a Leica DMIL inverted microscope equipped with a 10× phase contrast objective and a QImaging 12‐bit CCD camera, and analysed with ImageJ. Fifteen images per well were acquired at each time‐point. Cell migration was quantified by determining the area of the wound at noted time‐points at predetermined sites.
Electric cell‐substrate impedance sensing (ECIS) automated cell‐migration assay was performed on an ECIS system (Applied BioPhysics) equipped with a “wound healing module” and using 8‐chambered gold‐plated electrode arrays (8W10E). Cells (5 × 104/well) were plated on sterile 8W10E electrodes pre‐coated with 0.1% porcine gelatin (collagen I), grown to full confluence and transduced with adenoviruses. Immediately after adenoviral transduction, the electrode arrays were mounted on the ECIS system within an incubator (37°C, 5% CO2) and connected to its recorder device. The cell monolayer was wounded by electric pulses, the medium changed and the transendothelial electrical resistance, an index of endothelial cell barrier function, was measured in real time and recorded for 50 hrs in 5‐min. intervals.
Results
PV1 down‐regulation by lentiviral‐encoded shRNA
Small interfering siRNA sequences directed against mouse PV1, previously reported 6, 8 or designed de novo, were transfected in MLEC and PV1 knockdown determined by immunoblotting. As all the PV1 siRNAs were able to down‐regulate PV1 (data not shown), these sequences were used to design the shRNA sequence to be introduced into the pll3.7 vector for lentivirus (LV) generation. The lentiviral vector also encodes for the green fluorescent protein (GFP) for ease of identification of transduced cells. The siRNA and shRNA sequences used are shown in Table 1.
We first determined that all LVs generated were able to infect mouse ECs at low MOI, as shown by flow cytometric quantification of GFP‐expressing cells (Fig. 1A) or using fluorescence confocal microscopy (Fig. 1B).
Figure 1.
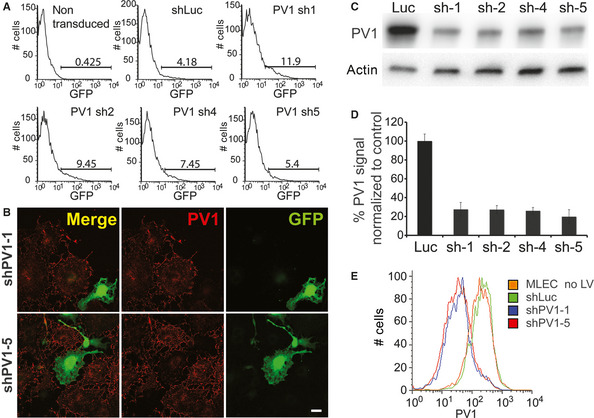
PV1 knockdown by lentiviral shRNA. (A) Flow cytometry analysis demonstrating the ability of each of the generated LVs to infect mouse ECs, as demonstrated by GFP expression. The cells were incubated with a LV dose of ~0.1 MOI. The GFP‐positive cells were used for sorting to generate stable cell lines expressing the various shRNAs. (B) Laser confocal microscopy demonstrating the down‐regulation of PV1 signal in cells transduced with low MOI of shPV1‐1‐LV (top panels) or shPV1‐5‐LV (bottom panels). PV1 signal was detected with the anti‐PV1 MECA‐32 mAb directly coupled to a fluorophore (red). Transduced cells were identified by their expression of GFP (green). The images represent maximum‐intensity projections of confocal stacks encompassing all the cell thickness, acquired with a 60× objective and no zoom. Non‐transduced cells serve as an internal control for normal PV1 levels. Bars 20 μm. (C) Total cellular‐PV1 protein levels in MLEC stably transduced with LVs encoding for shLuc, shPV1‐1, ‐2, ‐4 and ‐5. Equal amounts (20 μg/lane) of proteins were resolved by SDS‐PAGE and immunoblotted with anti‐PV1 and anti‐actin antibodies, as described in Methods. (D) Quantization of PV1 level on the surface of mouse ECs transduced with LVs encoding for shLuc, shPV1‐1, ‐2, ‐4 and ‐5. The data represent median fluorescence (n = 4, P < 0.01, error bars ‐stdev). (E) Examples of flow cytometry data showing PV1 levels on the surface of MLEC parental (orange trace), MLEC‐shLuc (green trace), MLEC‐shPV1‐1 (blue trace), MLEC‐shLuc (red trace).
Next, the effectiveness of PV1 down‐regulation in MLEC stably transduced with shRNA‐LVs targeting PV1 or luciferase (shLuc‐LV) was tested by multiple complementary approaches. We chose to study long‐term, stable expression of shRNAs, as it is more relevant to the in vivo experimental approach. According to real‐time PCR (not shown) and Western blotting results (Fig. 1C), total cellular levels of PV1 were decreased by more than 70% for all the PV1‐shRNA‐expressing cell lines. As PV1 function occurs at the cell surface 9, we determined the effect of different shRNAs on plasma membrane levels of PV1 protein. We show using confocal microscopy (Fig. 1B) and flow cytometry (Fig. 1E) that all mouse PV1‐shRNA‐LVs efficiently silenced PV1 protein expression at the cell surface by more than 80% compared to non‐infected or shLuc‐LV‐transduced MLEC. The lack of effect of shLuc control LV on PV1 protein levels is shown in Figure 1E. The efficiency of shLuc‐LV control virus to decrease luciferase activity in 293 cells constitutively expressing luciferase was previously validated 25. The two most efficient mouse shRNA‐LVs, namely shPV1‐1 and shPV1‐5, were used in all subsequent experiments.
Intratumoural delivery of PV1‐shRNA inhibits the growth of established pancreatic tumours
To determine whether the LV‐mediated PV1 down‐regulation is able to inhibit PDAC growth, we employed a model system consisting of heterotopic tumours derived from two different human PDAC cell lines (AsPC‐1 and BxPC‐3) grown subcutaneously in nude mice.
Following subcutaneous injections of AsPC‐1 or BxPC‐3 cells, tumour growth was monitored and mice with established tumours (volume of 50 mm3) were enrolled in four different groups, one to be left untreated and three treated with one injection either of shPV1‐1‐LV, of shPV1‐5‐LV or shLuc‐LV. Tumour growth was determined over a period of several weeks. A statistically significant difference in tumour volumes between shLuc‐LV‐injected tumours and shPV1‐LV‐injected tumours was seen as early as 24 and 33 days following viral injection of AsPC‐1‐ and BxPC‐3‐derived tumours respectively. The subsequent growth of all shPV1‐LV‐injected tumours remained significantly attenuated. By the end of the experiment, AsPC‐1 tumours that were infected with shPV1‐1‐LV exhibited a 35% (P < 0.05) decrease in volume and 65% (P < 0.05) with shPV1‐5 (Fig. 2A) in comparison with tumours infected with shLuc‐LV or control tumours left untreated. No statistically significant differences were detected between the growth of tumours infected with shLuc‐LV and control tumours left untreated. With respect to BxPC‐3, shPV1‐1 inhibited tumour growth by 57% (P < 0.01) and shPV1‐5 one of 49% (P < 0.01) (Fig. 2B). Taken together, these data demonstrate that PV1 down‐regulation by two different shRNAs are able to inhibit pancreatic tumour growth in two different human‐PDAC xenograft models.
Figure 2.
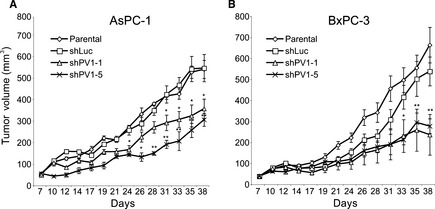
Intratumoural delivery of PV1 shRNA inhibits pancreatic tumour growth. Growth curves of AsPc‐1 (A) and BxPC‐3 (B) tumours either non‐treated (open diamonds) or injected with shLuc‐LV (open squares), shPV1‐1‐LV (open triangles) and shPV1‐5‐LV (x). (n = 8 per group, P < 0.01, error bars – SEM).
PV1 is expressed specifically in the EC of tumour xenografts and is not expressed in tumour cells
The sequence mismatches between mouse PV1 shRNAs and human PV1 strongly argued that tumour growth inhibition occurred via PV1 down‐regulation in the tumour microenvironment (i.e. ECs, stromal cells and tumour‐infiltrating leucocytes), which is of murine origin. To strengthen this observation, we defined the precise cellular type(s) expressing PV1 in the AsPC‐1‐ and BxPC‐3‐derived tumours. Although PV1 is expressed in tumour ECs in many solid tumours including pancreatic cancer, there is no previous information regarding PV1 expression in AsPC‐1‐ and BxPC‐3‐derived tumours. To address this, the vascular space of tumour‐bearing mice was labelled with fluorescent microspheres, tumours harvested and sections stained with anti‐PV1 antibodies directly labelled to fluorophores. We used the MECA‐32 mAb that recognizes only mouse PV1 24 16, and the anti‐hPV1C pAb that specifically recognizes human PV1 6. In agreement with data published for other tumours 31, 32, 33, 34, mouse‐PV1 expression was detected solely in the pancreatic tumour ECs, as shown by its co‐localization with the fluorescent microspheres as luminal markers of the functional blood vessels (Fig. 3A and B) and EC markers PECAM‐1/CD31 (Fig. 3A and B), ICAM‐2/CD102 (Fig. 3E), endoglin/CD105 and CD34 (data not shown). Similar results were obtained by staining of serial sections with anti‐PV1 mAb MECA‐32 and anti‐CD31 (Fig. 3C). We also found that both AsPC‐1 and BxPC‐3 cells in culture lack expression of PV1 mRNA as shown by real‐time PCR (Fig. 3D). Positive controls for real‐time PCR were human lung microvascular endothelial cells (HLMVEC) and HLMVEC treated with phorbol myristate acetate, expressing low and high levels of PV1 respectively 6. As a control for differences between cancer cells in culture and tumours in vivo, tumour sections were also probed with human PV1 antibodies. No expression of human PV1 was detected in the AsPC‐1 or BxPC‐3 tumour sections.
Figure 3.
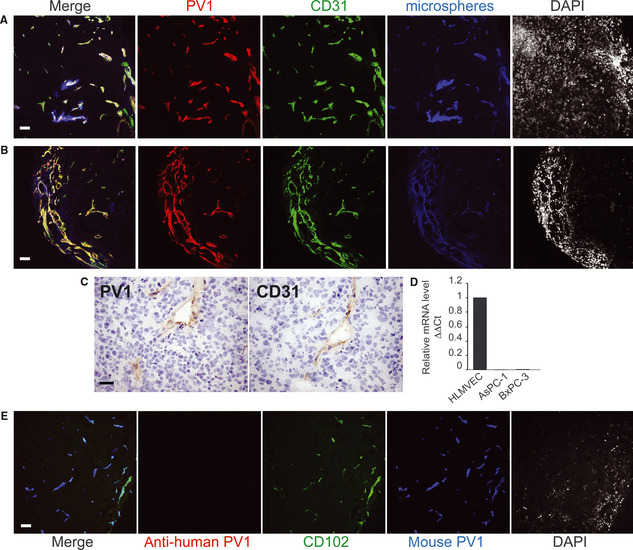
In both AsPC‐1 and BxPc‐3 pancreatic tumours PV1 is expressed only in tumour endothelial cells (A–B) Confocal microscopy of frozen sections from AsPC‐1 pancreatic tumours grown in nude mice, with anti‐PV1 (red) and anti‐CD31 (green) and antibodies directly coupled to fluorophores. The vascular lumen (blue) was labelled by the introduction of dark red microspheres in the circulation. In the merged field, white means triple co‐localization. Last panel in each shows nuclei stained with DAPI. (A) is a representative field showing vessels inside the tumour whereas (B) shows vessels at the tumour periphery. Magnification 20×. Bars – 20 μm. (C) Immunohistochemistry of serial frozen sections from human pancreatic xenograft tumours grown in nude mice, with anti‐PV1 (PV1) and anti‐CD31 (CD31) antibodies. The nuclei are counter stained with haematoxylin. Bars – 20 μm. (D) PV1‐mRNA levels in HLMVEC and human pancreatic tumour cell lines AsPC‐1 and BxPC‐3 measured by real‐time quantitative PCR. The data were obtained from triplicate samples and normalized to β‐actin levels (ΔΔCt). Bars – SEM (E) Confocal microscopy of frozen sections from AsPC‐1 pancreatic tumours grown in nude mice, stained with anti‐human PV1 (red) showing lack of expression of PV1 by human cancer cells. In the same specimen, mouse PV1 (blue) is present and colocalizes with the vascular marker CD102 (green), both detected by antibodies directly coupled to fluorophores. Magnification 20×. Bars – 10 μm.
Thus, in both AsPC‐1 and BxPC‐3 tumours, PV1 is specifically expressed in the tumour ECs and not in the tumour cells per se, in good agreement with data in the literature obtained in other tumours.
PV1 down‐regulation does not affect vascular density of tumours
Next, we investigated whether the tumour growth inhibition induced by PV1 down‐regulation is because of inhibition of tumour angiogenesis. Morphometric analyses of tumour sections stained with antibodies against vascular markers such as CD31 were performed to determine the vascular area density, which is a measure of angiogenesis 35. Representative images of CD31 staining are shown in Figure 4A. Quantitation data show that there are no statistically significant differences in vascular area density between the control and PV1 shRNAs‐treated AsPC‐1‐ and BxPC‐3‐derived tumours (Fig. 4B).
Figure 4.
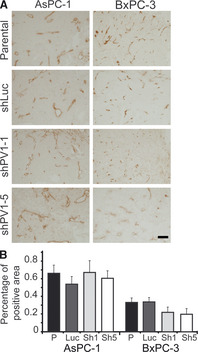
Down‐regulation of PV1 does not decrease tumour vascular density (A) Immunohistochemistry with anti‐CD31 antibodies of AsPC‐1 (left panels) or BxPC‐3 (right panels) tumours untreated (parental) or treated with shLuc‐LVs (shLuc), shPV1‐1‐LV (shPV1‐1) and shPV1‐5‐LV (shPV1‐5). Bars – 20 μm. (B) Morphometric analysis of vascular density in AsPC‐1 and BxPC‐3 tumours [shLuc‐LVs (Luc), shPV1‐1‐LV (sh1) and shPV1‐5‐LV (sh5)]. Error bars – SEM.
This result raises doubts that tumour growth inhibition was the result of decreased angiogenesis in this model system suggesting that endothelial PV1 is necessary for tumour growth via other mechanisms than promoting angiogenesis.
PV1 down‐regulation does not modify endothelial cell proliferation and migration in vitro
Angiogenesis, defined as the formation of new vessels from pre‐existing ones is based on the ability of ECs to proliferate and migrate 36 37. To strengthen the above observations we have next investigated whether the modulation of PV1 expression level influences any of these EC cellular functions in vitro. To determine the effect of PV1 gain of function, we used ECs that do not express PV1 (such as HUVEC and BAEC) either not transduced or transduced with AdPV1HA, AdNull or AdGFP. To determine the effect of PV1 loss of function we have compared MLEC, which constitutively express PV1 and with MLEC stably expressing different shRNAs.
De novo expression of PV1 does not modify proliferation rate of HUVEC (Fig. 5A) or BAEC (not shown), as determined using an assay measuring DNA content. Controls consisted of non‐transduced cells and cells transduced with the same MOI of AdGFP or AdNull. Flow cytometric analyses of PV1 and GFP expression shows that the cells are efficiently transduced by the adenovirus (Fig. 5B). Reciprocally, in MLEC that constitutively express PV1, PV1 down‐regulation by transduction with lentiviruses encoding PV1 shRNAs does not modify PV1 proliferation, as measured by population doubling time (Fig. 5C) or DNA content (Fig. 5D).
Figure 5.
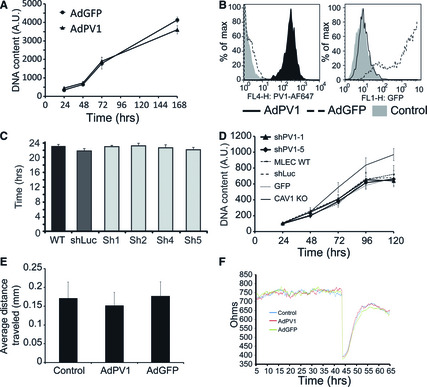
PV1 levels do not modify EC proliferation and migration (A) Proliferation of HUVEC transduced with adenoviruses encoding for PV1 (AdPV1) or GFP (AdGFP) measured with the DNA content assay. Data were obtained from n = 8 sample replicates per condition. Error bars – SEM. (B) Flow cytometric analysis of PV1 (left) and GFP expression (right) in HUVEC transduced with AdPV1HA (AdPV1) or AdGFP (AdGFP). Control cells were either non‐transduced or AdNull transduced HUVEC. Representative samples (of n = 4) are shown in each panel. Control cells (grey histogram), AdGFP‐transduced cells (dashed line histogram), AdPV1HA‐transduced cells (solid line histogram). (C) Average doubling time of MLEC parental or stably expressing shLuc (Luc), shPV1‐1 (sh1) or shPV1‐5 (sh5). Data were obtained from triplicate samples. Error bars –SEM. (D) Proliferation of MLEC parental or stably expressing shLuc (Luc), shPV1‐1 (sh1) or shPV1‐5 (sh5) measured with the DNA content assay. Data were obtained from n = 8 samples per condition. Error bars – SEM. (E–F) Migration of HUVEC transduced with adenoviruses encoding for PV1 (AdPV1) or GFP (AdGFP), as measured by the monolayer wound assay at 8 hrs after monolayer wounding (E) or by the ECIS method (F). For the latter, the cells were plated onto electrodes at near confluence and the monolayer allowed to mature for 44 hrs when the cells over the electrodes were eliminated with electric pulses and the monolayer recovery was monitored for 11 hrs. The traces are representative samples. Data were obtained from n = 4 sample replicates per condition. Error bars in E) – STDEV.
Cell migration was assessed using wound assays by either the classical scratch method or by ECIS (electric cell impedance sensing). By the two methods, we found no statistically significant differences between ECs expressing PV1 and those that did not (Fig. 5E and F). Taken together, these data argued that either PV1 overexpression or PV1 down‐regulation did not modify EC proliferation and migration.
Discussion
PV1 is an endothelial protein with important roles in formation of diaphragms of caveolae, fenestrae, transendothelial channels and, possibly, vesiculo‐vacuolar organelles. The diaphragms are structures with unknown functions. PV1 is specifically expressed on normal ECs in a subset of vascular beds. PV1 is also specifically expressed in ECs of many human and mouse tumours, pancreatic cancer included. The strong expression of PV1 in the tumour ECs raises the question as to its function, if any, especially related to tumour initiation and progression.
On the basis of its specific location, PV1 was hypothesized to play an active role in tumour angiogenesis, which is sustained by the published in vitro data where PV1 down‐regulation by siRNA was shown to block the formation of capillary tubes by human ECs in Matrigel and migration towards tumour‐secreted angiogenic growth factors 12. These in vitro data, supporting a role of PV1 in EC migration, have not yet been validated in vivo prompting us to ask this question in a heterotopic xenograft tumour model.
As a model system, we have chosen heterotopic human tumour xenografts in athymic nude mice treated with LV encoded PV1 shRNA delivered by intratumoural injection. Tumours were derived from AsPC‐1 and BxPC‐3 cells, two established human PDAC cell lines isolated from metastatic and primary PDAC tumours, respectively. The model is simple to execute and the intratumoural injection of lentiviruses expressing various shRNAs was previously validated to be an effective means in down‐regulating target molecules in this model 25. Because the tumour microenvironment (i.e. tumour ECs, stromal cells and tumour‐infiltrating leucocytes) is all derived from the mouse host, the model also allows the study of the down‐regulation of PV1 expressed in the stroma versus tumour cells using species‐specific shRNAs.
The salient finding of our work is the efficacy of PV1 down‐regulation by shRNA in inhibiting the growth of pre‐existing tumours in both PDAC models. This study demonstrates, for the first time, the potential therapeutic effect of targeting PV1 for the treatment of PDAC. As shown before, no inhibitory effect of the empty virus or shLuc‐LV was observed when compared to the untreated tumours, demonstrating that tumour inhibition is not because of non‐specific effects of the LVs or the shRNA. Moreover, this also shows that shRNAs do not inhibit angiogenesis non‐specifically, as was suggested by other studies with targeted and non‐targeted siRNAs acting via toll‐like receptor 3 (TLR3) 38, 39. The specificity we observe could be because of the fact that in our model, shRNAs are expressed inside the LV‐infected cells and as such, may not engage TLR3. Finally, the inhibition of tumour growth by two different PV1 shRNAs in two different models rules out (or makes highly unlikely) off‐target effects of individual shRNAs.
The LVs infect all cell types present in the tumour 25, therefore it was important to demonstrate the specificity of PV1 location in these tumours. Staining tumour sections with PV1 and various EC markers used to detect tumour vessels, we show that PV1 is specifically expressed in ECs and not tumour cells, as shown for many other tumours before 10, 11, 12, 15. We verified that tumour cells lack PV1 at both protein and mRNA level. Most importantly, the shRNA sequences used do not target human PV1. These data demonstrate that down‐regulation of endothelial PV1 is responsible for the anti‐tumour effect, opening the possibility to use PV1 as a therapeutic target in a broad range of cancers, where PV1 is expressed in the vessels.
The effect of PV1 down‐regulation on tumour growth can be reasonably explained by the effect of either inhibition of tumour angiogenesis resulting in decreased ratio of tumour vascular volume to tumour tissue volume and/or impaired function of the tumour vessels. We find that the vascular area counts inside and at the periphery of the tumour were similar in both shPV1 and control‐treated tumours, suggesting that angiogenesis may not be impaired in the PDAC models used. Although the value of density counts in measuring whether angiogenesis is inhibited or not has limits, as discussed in the past by Hlatky, et al., 40, these conclusions are supported by the lack of effect of PV1 gain and loss of function on both EC proliferation and migration in vitro. Moreover, analysis of PV1‐knockout mice demonstrates that PV1 is not required for both vasculogenesis and angiogenesis during mouse development, as these mice have normally formed vessels (D. Tse, S. Deharvengt, C. McGarry, D. S. Longnecker, C. Carriere and R. V. Stan, unpublished data).
Several laboratories have demonstrated PV1 to be expressed specifically in tumour ECs and we have recently published data strongly suggesting that, at cellular level, the function of PV1 in ECs is to form diaphragms 9. These diaphragms are associated with four different EC structures, of which caveolae 41 and vesiculo‐vacuolar organelles 42 were previously postulated as regulating tumour growth. Additional experiments will be needed to dissect which diaphragm on which organelle is important.
At the vascular physiology function level, to date, PV1 was implicated in the maintenance of basal permeability in normal vessels (Tse, et al., 2012, submitted) and in the recruitment of leucocytes into inflammation sites, by unknown mechanisms 17. Tumour vessels are fenestrated in most solid cancers and are quite disorganized and leaky; the significance of this increased leakiness for tumour growth is under debate 43. If we are to speculate, the deletion of PV1 will presumably increase the permeability of tumour vessels by deletion of the diaphragms of fenestrae, and possibly those of VVOs. Presently, it is not obvious what consequences this increased leakiness of already leaky vessels will have on tumour growth. Moreover, data in the literature argues that decreasing tumour vascular permeability inhibits tumour growth 44. Finally, PV1 promotion of diapedesis of leucocytes in inflammation sites 17 raises the possibility of PV1 being important for recruitment of tumour‐infiltrating leucocytes with roles in promoting tumour growth 45, which is currently under study in immunocompetent models of cancer.
Finally, by far the most exciting, is the translational potential of PV1 function to be harnessed in devising new drugs against cancer. Here too, experiments employing disease‐relevant genetically engineered mouse models will be needed to further explore the role of PV1 in tumour growth.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported from National Institutes of Health grants HL83249, HL092085 and 5P30RR032136‐02/8P30GM103415‐02 (PI Green) to RVS and CA127095 to CC. SD was the recipient of a postdoctoral fellowship award from the SASS Foundation for Medical Research. SJL, CC, RVS designed the research study. SJL, DT, CLM, OS, JRG, CC, RVS performed the research. SJL, DT, DSL, CC, RVS analysed the data. SJL, DSL, CC, RVS wrote the paper. We would like to thank C. Tomlinson for cells, BW Pogue for cells and help with fluorescent microspheres experiments, A. Eastman for advice on proliferation assays and M. Israel for the use of the microscope.
References
- 1. Stan RV, Roberts WG, Predescu D, et al. Immunoisolation and partial characterization of endothelial plasmalemmal vesicles (caveolae). Mol Biol Cell. 1997; 8: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stan RV, Ghitescu L, Jacobson BS, et al. Isolation, cloning, and localization of rat PV‐1, a novel endothelial caveolar protein. J Cell Biol. 1999; 145: 1189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ghitescu LD, Crine P, Jacobson BS. Antibodies specific to the plasma membrane of rat lung microvascular endothelium. Exp Cell Res. 1997; 232: 47–55. [DOI] [PubMed] [Google Scholar]
- 4. Stan RV, Kubitza M, Palade GE. PV‐1 is a component of the fenestral and stomatal diaphragms in fenestrated endothelia. Proc Natl Acad Sci USA. 1999; 96: 13203–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tse D, Stan RV. Morphological heterogeneity of endothelium. Semin Thromb Hemost. 2010; 36: 236–45. [DOI] [PubMed] [Google Scholar]
- 6. Stan RV, Tkachenko E, Niesman IR. PV1 is a key structural component for the formation of the stomatal and fenestral diaphragms. Mol Biol Cell. 2004; 15: 3615–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stan RV. Multiple PV1 dimers reside in the same stomatal or fenestral diaphragm. Am J Physiol Heart Circ Physiol. 2004; 286: H1347–53. [DOI] [PubMed] [Google Scholar]
- 8. Ioannidou S, Deinhardt K, Miotla J, et al. An in vitro assay reveals a role for the diaphragm protein PV‐1 in endothelial fenestra morphogenesis. Proc Natl Acad Sci USA. 2006; 103: 16770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tkachenko E, Tse D, Sideleva O, et al. Caveolae, fenestrae and transendothelial channels retain PV1 on the surface of endothelial cells. PLoS One. 2012; 7: e32655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stan RV. Endothelial stomatal and fenestral diaphragms in normal vessels and angiogenesis. J Cell Mol Med. 2007; 11: 621–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Madden SL, Cook BP, Nacht M, et al. Vascular gene expression in nonneoplastic and malignant brain. Am J Pathol. 2004; 165: 601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carson‐Walter EB, Hampton J, Shue E, et al. Plasmalemmal vesicle associated protein‐1 is a novel marker implicated in brain tumour angiogenesis. Clin Cancer Res. 2005; 11: 7643–50. [DOI] [PubMed] [Google Scholar]
- 13. Shue EH, Carson‐Walter EB, Liu Y, et al. Plasmalemmal vesicle associated protein‐1 (PV‐1) is a marker of blood‐brain barrier disruption in rodent models. BMC Neurosci. 2008; 9: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Y, Carson‐Walter EB, Cooper A, et al. Vascular gene expression patterns are conserved in primary and metastatic brain tumours. J Neurooncol. 2010; 99: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Strickland LA, Jubb AM, Hongo JA, et al. Plasmalemmal vesicle‐associated protein (PLVAP) is expressed by tumour endothelium and is upregulated by vascular endothelial growth factor‐A (VEGF). J Pathol. 2005; 206: 466–75. [DOI] [PubMed] [Google Scholar]
- 16. Niemela H, Elima K, Henttinen T, et al. Molecular identification of PAL‐E, a widely used endothelial‐cell marker. Blood. 2005; 106: 3405–9. [DOI] [PubMed] [Google Scholar]
- 17. Keuschnigg J, Henttinen T, Auvinen K, et al. The prototype endothelial marker PAL‐E is a leukocyte trafficking molecule. Blood. 2009; 114: 478–84. [DOI] [PubMed] [Google Scholar]
- 18. Ades EW, Candal FJ, Swerlick RA, et al. HMEC‐1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992; 99: 683–90. [DOI] [PubMed] [Google Scholar]
- 19. Sanders Strickland LA, Koeppen H. Comments on plasmalemmal vesicle associated protein‐1 as a novel marker implicated in brain tumour angiogenesis. Clin Cancer Res. 2006; 12: 2649–50. [DOI] [PubMed] [Google Scholar]
- 20. ACS American Cancer Society . Cancer facts and figures 2012. American Cancer Society; 2012. Available at http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-031941.pdf. [Google Scholar]
- 21. Abbruzzese JL. Adjuvant therapy for surgically resected pancreatic adenocarcinoma. JAMA. 2008; 299: 1066–7. [DOI] [PubMed] [Google Scholar]
- 22. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011; 364: 1817–25. [DOI] [PubMed] [Google Scholar]
- 23. Campen CJ, Dragovich T, Baker AF. Management strategies in pancreatic cancer. Am J Health Syst Pharm. 2011; 68: 573–84. [DOI] [PubMed] [Google Scholar]
- 24. Hallmann R, Mayer DN, Berg EL, et al. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn. 1995; 202: 325–32. [DOI] [PubMed] [Google Scholar]
- 25. Deharvengt SJ, Gunn JR, Pickett SB, et al. Intratumoral delivery of shRNA targeting cyclin D1 attenuates pancreatic cancer growth. Cancer Gene Ther. 2009; 17: 325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Naldini L, Blomer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996; 272: 263–7. [DOI] [PubMed] [Google Scholar]
- 27. Falcon BL, Pietras K, Chou J, et al. Increased vascular delivery and efficacy of chemotherapy after inhibition of platelet‐derived growth factor‐B. Am J Pathol. 2011; 178: 2920–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moreira LR, Schenka AA, Latuf‐Filho P, et al. Immunohistochemical analysis of vascular density and area in colorectal carcinoma using different markers and comparison with clinicopathologic prognostic factors. Tumour Biol. 2011; 32: 527–34. [DOI] [PubMed] [Google Scholar]
- 29. Rao J, Otto WR. Fluorimetric DNA assay for cell growth estimation. Anal Biochem. 1992; 207: 186–92. [DOI] [PubMed] [Google Scholar]
- 30. Montano R, Chung I, Garner KM, et al. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA‐damaging agents and antimetabolites. Mol Cancer Ther. 2012; 11: 427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Montesano R, Pepper MS, Mohle‐Steinlein U, et al. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell. 1990; 62: 435–45. [DOI] [PubMed] [Google Scholar]
- 32. Dineen SP, Sullivan LA, Beck AW, et al. The Adnectin CT‐322 is a novel VEGF receptor 2 inhibitor that decreases tumour burden in an orthotopic mouse model of pancreatic cancer. BMC Cancer. 2008; 8: 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Okaji Y, Tsuno NH, Kitayama J, et al. A novel method for isolation of endothelial cells and macrophages from murine tumours based on Ac‐LDL uptake and CD16 expression. J Immunol Methods. 2004; 295: 183–93. [DOI] [PubMed] [Google Scholar]
- 34. Yanagawa J, Walser TC, Zhu LX, et al. Snail promotes CXCR2 ligand‐dependent tumour progression in non‐small cell lung carcinoma. Clin Cancer Res. 2009; 15: 6820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Falcon BL, Hashizume H, Koumoutsakos P, et al. Contrasting actions of selective inhibitors of angiopoietin‐1 and angiopoietin‐2 on the normalization of tumour blood vessels. Am J Pathol. 2009; 175: 2159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007; 6: 273–86. [DOI] [PubMed] [Google Scholar]
- 37. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011; 473: 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kalluri R, Kanasaki K. RNA interference: generic block on angiogenesis. Nature. 2008; 452: 543–5. [DOI] [PubMed] [Google Scholar]
- 39. Kleinman ME, Yamada K, Takeda A, et al. Sequence‐ and target‐independent angiogenesis suppression by siRNA via TLR3. Nature. 2008; 452: 591–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hlatky L, Hahnfeldt P, Folkman J. Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn't tell us. J Natl Cancer Inst. 2002; 94: 883–93. [DOI] [PubMed] [Google Scholar]
- 41. Goetz JG, Lajoie P, Wiseman SM, et al. Caveolin‐1 in tumour progression: the good, the bad and the ugly. Cancer Metastasis Rev. 2008; 27: 715–35. [DOI] [PubMed] [Google Scholar]
- 42. Chang SH, Feng D, Nagy JA, et al. Vascular permeability and pathological angiogenesis in caveolin‐1‐null mice. Am J Pathol. 2009; 175: 1768–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002; 62: 5381–5. [PubMed] [Google Scholar]
- 44. Gratton JP, Lin MI, Yu J, et al. Selective inhibition of tumour microvascular permeability by cavtratin blocks tumour progression in mice. Cancer Cell. 2003; 4: 31–9. [DOI] [PubMed] [Google Scholar]
- 45. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011; 331: 1565–70. [DOI] [PubMed] [Google Scholar]