

Abstract
The human pregnane X receptor (PXR) is a ligand-regulated transcription factor belonging to the nuclear receptor superfamily. PXR is activated by a large, structurally diverse, set of endogenous and xenobiotic compounds, and coordinates the expression of genes central to metabolism and excretion of potentially harmful chemicals and therapeutic drugs in humans. Walrycin A is a novel antibacterial compound targeting the WalK/WalR two-component signal transduction system of Gram (+) bacteria. Here we report that, in hepatoma cells, walrycin A potently activates a gene set known to be regulated by the xenobiotic sensor PXR. Walrycin A was as efficient as the reference PXR agonist rifampicin to activate PXR in a transactivation assay at non cytoxic concentrations. Using a limited proteolysis assay, we show that walrycin A induces conformational changes at a concentration which correlates with walrycin A ability to enhance the expression of prototypic target genes, suggesting that walrycin A interacts with PXR. The activation of the canonical human PXR target gene CYP3A4 by walrycin A is dose- and PXR-dependent. Finally, in silico docking experiments suggest that the walrycin A oxidation product Russig’s blue is the actual a ligand for PXR. Taken together, these results identify walrycin A as novel human PXR activator.
Keywords: Anti-Bacterial Agents; toxicity; Cell Line, Transformed; Cell Survival; drug effects; Computational Biology; Computer Simulation; Cytochrome P-450 CYP3A; biosynthesis; genetics; Gene Expression; drug effects; Hepatocytes; drug effects; metabolism; Humans; Naphthols; toxicity; Oligonucleotide Array Sequence Analysis; Protein Binding; Quantitative Structure-Activity Relationship; RNA, Small Interfering; administration & dosage; genetics; Real-Time Polymerase Chain Reaction; Receptors, Steroid; drug effects; genetics; Rifampin; pharmacology; Transfection
Keywords: Walrycin A, Pregnane X Receptor, Nuclear Receptor, CYP3A4, Ligand Binding Domain, Xenobiotic
Introduction
Xenobiotics, such as drugs and environmental chemicals, exert a profound influence on human health. Xenobiotics can alter homeostasis and induce deleterious metabolic perturbations. In order to promote the metabolic inactivation and excretion of these compounds, multiple signalling pathways are activated to trigger hepatic biotransformation, biliary excretion and renal elimination. Part of these clearance mechanisms are coordinately controlled by nuclear receptors such as Pregnane X Receptor (PXR/NR1I2) and Constitutive Androstane Receptor (CAR/NR1I3). Being important transcription factors controlling xenobiotic detoxification, CAR and PXR display a strong expression in the primarily exposed organs, the liver and the intestine (Lamba et al. 2004; Savkur et al. 2003).
As many nuclear receptors, CAR and PXR possess a conserved DNA Binding Domain (DBD) and a variable C terminal Ligand Binding Domain (LBD). Within the LBD, the ligand-binding pocket of CAR and PXR accommodate a wide range of structurally unrelated endogenous and exogenous ligands (di Masi A. et al. 2009). For instance, human PXR and human CAR are both activated by endogenous ligands such as bile acids and steroid hormones (Guo et al. 2003; Timsit and Negishi 2007; Xie et al. 2003), xenobiotics such as drugs (e.g. rifampicin, dexamethasone and phenobarbital), endocrine disrupters (bisphenol A, phthalates) and natural plant compounds (hyperforine, zearalenone) (Ayed-Boussema et al. 2011; DeKeyser et al. 2011; Lehmann et al. 1998; Moore et al. 2000; Sueyoshi et al. 1999).
Through their DBD, CAR and PXR bind to various response elements (direct repeats DR3, DR4 and DR5 as well as everted repeats ER6 and ER8), thereby controlling the expression of a large set of target genes involved in energy metabolism and hormone homeostasis, inflammation, cell differentiation, bile acids and bilirubin detoxification (Moreau et al. 2008; Pascussi and Vilarem 2008; Wada et al. 2009). Moreover, this versatile DNA binding property allows cross talks between CAR and PXR, and also with other nuclear receptors such as FXR, LXR, VDR, PPAR, ER, GR, COUP-TFI and II (Breuker et al. 2010; di Masi A. et al. 2009; Faucette et al. 2006; Ihunnah et al. 2011; Istrate et al. 2010).
PXR and CAR have been initially described as xenobiotic sensors modulating the expression of several hepatic target genes driven by a so-called “xenobiotics response element” and involved in detoxification pathways, including drug-metabolizing enzymes and transporters (Omiecinski et al. 2011; Wada et al. 2009). For instance, human cytochrome P450 2B6 (CYP2B6) and 3A4 (CYP3A4) expression is under the control of CAR and PXR respectively (Kliewer et al. 2002; Lehmann et al. 1998; Maglich et al. 2003; Mo et al. 2009; Sueyoshi et al. 1999). The CYP3A sub-family member CYP3A4 is a key player in detoxification pathways, since about 50% of therapeutically used drugs are metabolized by this enzyme (Istrate et al. 2010; Kliewer et al. 2002). Moreover, the PXR/CYP3A4 pathway is involved in 60% of known drug-drug interactions (Evans 2005). Rifampicin, an antibiotic used to treat tuberculosis as well as nosocomial pneumonia caused by methicillin-resistant Staphylococcus aureus (MRSA) is a human PXR agonist inducing CYP3A4 expression. CYP3A4 metabolizes more than 100 drugs including oral contraceptives, anti-HIV protease inhibitors (Baciewicz et al. 2008; Ivanovic et al. 2008; Ma et al. 2008) and antibiotics, (Jung et al. 2010). Thus, activation of the PXR signalling pathway leads to a diminished therapeutic efficacy of many drugs and also potentially produces toxic metabolites. There is therefore a need to determine the effects of each novel therapeutic compound on PXR activity.
Recently, a novel antibacterial compound called walrycin A (4-methoxy-1-naphthol) has been identified through a high throughput screening approach and shown to target the WalK/WalR two-component signal transduction system of Gram (+) bacteria such as Staphylococcus aureus and Bacillus subtilis (Gotoh et al. 2010). Given that walrycin A belongs to a potential novel class of antibacterial compounds, effects on human xenobiotics metabolism and hepaotoxicity remain to be studied. Here we report that walrycin A modulates human PXR activity and impacts on hepatic cell viability.
Materials and Methods
Materials
Rifampicin, 6-methoxy-1-naphtol (6MNol) and 4-methoxy-1-naphtol (walrycin A), purchased from Sigma-Aldrich (St-Louis, MO, USA) were dissolved in dimethylsulfoxyde (DMSO). The housekeeping gene ribosomal protein large P0 (RPLP0, NM_001002.3) forward (CATGCTCAACATCTCCCCCTTCTCC) and reverse (ATGCAGCCCCGAATGCT CCTCATCGTGGCC) primers, CYP3A4 (NM_017460.5) forward (CATTCCTCATCCCAA TTCTTGAGGT) and reverse (CCACTCGGTGCTTTTGTGTATCT) primers and PXR isoforms 1 (NM_003889.3) and 2 (NM_022002.2) forward (ACCTTTGACACTACCTTCT CCCAT) and reverse (CGCAGCCACTGCTAAGCA) primers were purchased from Sigma-Aldrich (St Quentin-Fallavier, France)
Cell culture and treatment
The immortalized human hepatocyte (IHH) cell line was established by F. Kuipers (University Medical Center, Groningen, Netherlands) from primary human hepatocytes. Cells were routinely maintained as previously described (Schippers et al. 1997). Twenty-four hours before treatment, cells were seeded (3.105 cells per well) in 12-well plates in seeding medium (phenol-red free Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 2 g/L glucose, 2 mM glutamine (Invitrogen Life Technologies, Carlsbad, CA, USA), 7 μg/mL bovine insulin (Sigma-Aldrich), 100 U/mL penicillin (Invitrogen), 100 μg/mL streptomycin (Invitrogen) and 10 % charcoal dextran-stripped fetal calf serum (CD-FCS). Cells were washed once before treatment with stimulating medium (seeding medium with CD-FCS reduced to 1%) and treated for 24 h by compounds at indicated concentrations (0.1% DMSO final concentration).
qPCR-based array
First Strand cDNA Synthesis Kit and Human Drug Metabolism RT2Profiler™ PCR Arrays were purchased from SABiosciences (SABiosciences, Frederick, MD, USA). Both reverse transcription and qPCR (Stratagen Mx3005P QPCR System) were performed following manufacturer’s instructions. Five endogenous control genes – β-2-microglobulin (B2M), hypoxanthine phosphoribosyltransferase (HPRT1), ribosomal protein L13a (RPL13A), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and β-actin (ACTB) - displayed on the PCR array were used for normalization. Cycle threshold (Ct) was normalized to the average Ct of these 5 endogenous controls. The comparative Ct method was used to calculate the relative quantification of gene expression (Livak and Schmittgen 2001). The following formula was used to calculate the relative amount of the transcripts in the walrycin A and vehicle (DMSO) treated sample, both of which were normalized to the endogenous controls: ΔΔCt = ΔCt (walrycin A) − ΔCt (DMSO). ΔCt is the log2 difference in Ct between the target gene and the average Ct of the five endogenous controls. The fold change for walrycin A-treated sample is expressed relative to the control (DMSO) sample = 2−ΔΔCt.
Cell transfection
IHH cells were seeded 24 hours before transfection in 6-well plates (106 cells per well) in maintenance medium (Schippers et al. 1997). Cells were transfected using 0.1 nmol of ON-TARGETplus SMARTpool hPXR siRNA or non-targeting siRNA using the Dharmafect1 transfection reagent according to the manufacturer’s protocol (Thermo Fisher Scientific, Lafayette, CO, USA). After a 24 h incubation, cells were washed with seeding medium and incubated further for 24 h. Cells were then washed with stimulating medium and treated as described above.
RNA extraction, reverse transcription and real time qPCR
At indicated times, cells were washed with 1x phosphate buffer saline (1x PBS) and total RNA was extracted using the Extract-all reagent (Eurobio, Courtabeuf, France) according to the manufacturer’s protocol. One μg of total RNA was reverse-transcribed using the High Capacity Reverse Transcription Kit (Applied Biosystem, Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. A 1:20 dilution of cDNA was then amplified by real time qPCR using Brilliant II Fast SybR Green Master Mix (Agilent Technologies, Santa Clara, CA, USA) and specific primers in a Stratagen Mx3005P QPCR System (Agilent Technologies). Gene expression levels were normalized using the RPLP0 housekeeping gene expression level as internal control. Fold induction were expressed as the ratio of the gene induced expression level to that of the basal level arbitrarily set to one.
Western blotting
At indicated times, transfected cells were washed with 1x PBS followed by total protein extraction using 100 μL of Cell Lysis Buffer (Cell Signaling Technology, Beverly, MA, USA) according to the manufacturer’s protocol. Western blotting was performed using 40 μg of total proteins. hPXR protein expression was monitored using an anti-hPXR mouse monoclonal antibody (Perseus Proteomics Inc, Tokyo, Japan) and a HRP-conjugated goat anti-mouse secondary antibody (Sigma-Aldrich). The immune complexes were detected by chemiluminescence using Pierce ECL-plus Western Blotting Substrate (Thermo Fisher Scientific) according to the manufacturer’s protocol and visualized with a G:Box gel dock system (Syngene, Cambridge, UK). HSP90 protein was used as internal standard for equal loading using a rabbit anti HSP90 antibody (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA) and a HRP-coupled goat anti rabbit as secondary antibody (Sigma-Aldrich).
Limited proteolysis assay
In vitro translation of hPXR was performed using the TNT T7 Quick Coupled Transcription/Translation System (Promega, Madison, WI, USA) and the [35S]-containing Protein Labelling Mix Easy Tag (Perkin Elmer, Waltham, MA, USA), in the presence of vehicle (0.1% DMSO), 50 μM walrycin A, 50 μM rifampicin or 50 μM 6MNol following the supplier’s instructions. Limited proteolysis of the radiolabeled receptor (5 μL of TnT mix) was carried out using increasing concentrations of chymotrypsin ranging from 1 to 5 μg/mL. After a 10 min. incubation, digestion was stopped by the addition of 6X Laemmli buffer. Samples were boiled for 5 min. and separated by 12% SDS-PAGE. After gel drying, radiolabeled digestion products were visualized using a STORM Phosphorimager (GE Healthcare, Orsay, France).
Gene reporter assay
The stable reporter cell line HGPXR stably expressing a GAL4DBD-hPXR LBD chimeric fusion protein was described previously (Lemaire et al. 2006).
Methods for computer-simulated ligand binding (docking)
Protein input files preparation
Ligand-free PXR (1ILH.pdb and 3HVL.pdb) input files were generated using the protein preparation wizard from the Maestro software (Maestro 8.5, Academic Campaign, http://www.schrodinger.com). The bond orders were assigned and hydrogen atoms were added. The resulting receptor coordinates were saved as a pdb file.
Ligand input files preparation
Ligand input structures were generated and 3D-optimized with the MarvinSketch Academic Package (MarvinSketch 5.4.1.1, 2011, ChemAxon http://www.chemaxon.com). Ligand structures were saved as mol2 files.
GOLD and FRED docking protocol
Docking was performed using chemscore fitness function under standard default settings in the GOLD software (Cambridge Crystallographic Data Centre 12 Union Road, Cambridge, CB2 1EZ, UK, http://www.ccdc.cam.ac.uk): search efficiency: 200%, population size 100, number of islands 5, number of operations 100,000, niche size 2, migrate 10, mutate 95, cross over 95 and a selection pressure of 1.1. Early termination was allowed if 4 solutions were within 1.5 angstroms of root mean square deviation (RMSD). Docking with FRED 2.2.5, (OpenEye Scientific Software, Inc., Santa Fe, NM, USA, www.eyesopen.com, 2011) was performed using Chemgauss3 scoring for exhaustive search and Chemgauss3 scoring for optimization.
Viability assays
HepG2 cells were seeded in 96-well plates (104 cells per well) in maintenance medium and incubated for 24 hours. Cells were treated for 24 h with indicated concentrations of compounds. Cell viability was quantified using the CellTiter 96 Non-Radioactive Cell Proliferation Assay (Promega) according to the manufacturer’s protocol. Absorbance was monitored on MRX spectrophotometer (Thermo Labsystems, Issy Les Moulineaux, France). Results were calculated as follows: (A570 -A630)compound/(A570 - A630)DMSO. Curve fitting was performed using GraphPad Prism 4.0 software (San Diego, CA, USA). Each concentration was tested in quadruplicate and data are displayed as means ± SEM.
Statistical Analysis
Histograms represent means ± SEM (n=2–3). Statistical analyses were performed using GraphPad Prism 4.0. Statistical significance was determined using a one-way ANOVA followed by a Dunnett’s multiple comparison post-hoc test (p-values<0.01 were considered as significant). For knock-down assays, significant differences were determined using a two-way ANOVA followed by a Bonferroni post-hoc test.
Results
Walrycin A regulates mRNA expression of genes involved in phase I drug metabolism
Walrycin A harbors potent antibacterial activity against the methycillin-resistant S. aureus (MRSA) strain N315 (Gotoh et al. 2010). Since rifampicin displays a similar efficacy against MRSA (Perlroth et al. 2008) and induce drug metabolizing enzymes in human hepatocytes at a concentration of ~30–50 μM (Rae et al. 2001), we first assessed whether walrycin A could modulate the expression of enzymes involved in drug/xenobiotic metabolism pathway at a similar concentration. Since drug metabolism is initiated upon the activation of phase I enzymes, we focused our analysis on this class of enzymes using qPCR macroarrays. Human immortalized hepatocytes [IHH cell line, (Schippers et al. 1997)] were treated for 24 hours by 50 μM walrycin A and 0.1% DMSO as a control. The differential mRNA expression analysis of 83 phase I enzymes was monitored and analyzed using a fold change cut-off of 4. This allowed the identification of 31 upregulated (36.9 %), and of only 2 downregulated (2.4 %) genes (Figure 1B). The expression of 50 genes were not affected (59.5 %) (Supplementary figure S1). Walrycin A-modulated genes could be classified into 3 main classes of enzymes (alcool dehydrogenases, aldehyde dehydrogenases and cytochrome p450s). Genes encoding proteins with other functions, such as FMO1, are displayed as “Others” (Figure 1C). Very interestingly, the PXR target genes CYP1A1 and CYP3A4 were strongly upregulated (30.9 and 5.2 fold respectively) in response to walrycin A. Given that both drug-activated CAR and PXR may equally regulate the expression of genes in human hepatocytes such as CYP1A1 and CYP3A4 (Auerbach et al. 2007; Faucette et al. 2006), we determined the relative expression of mRNAs coding for these two nuclear receptors in IHH cells. As PXR mRNA was strongly expressed whereas CAR mRNA was not detectable (see Supplementary figure S2), this indicated that the observed altered gene expression pattern upon walrycin A treatment could result form PXR activation. In addition, this indicated that IHH cells provide a valid experimental model to evaluate specific PXR response upon exposure to xenobiotics.
Figure 1. Walrycin A regulates mRNA expression of a large set of drug metabolism phase I enzymes.

A: Structure of the tested compounds. B: Scatter plot of mRNA expression upon walrycin A treatment (2-ΔCt (walrycin A)) compared to vehicle treatment (2−ΔCt (DMSO)). Upregulated (>4 fold change), not deregulated (-4<fold change<4) and downregulated (>-4 fold change) gene expression are displayed within dark gray, central uncolored and light gray areas, respectively. C: Phase I enzymes differentially expressed in response to walrycin A treatment (fold-change>4). The gene list was clustered in 3 main families (alcool deshydrogenase, aldehyde dehydrogenase and cytochrome p450). Enzymes not belonging to these 3 main families are labeled as “others”.
Dose-dependent activation of CYP3A4 by walrycin A is PXR-dependent
We further assessed the contribution of PXR to the observed alteration of the gene expression pattern. IHH cells were treated in parallel for 24 hours with the PXR reference agonist rifampicin or walrycin A or the walrycin A structural analogue 6MNol. As expected, rifampicin induced CYP3A4 mRNA expression in a dose-dependent manner (Figure 2A) with an EC50~1 μM reaching a plateau starting at 10 μM (4-fold maximal induction). Interestingly, CYP3A4 mRNA expression was also enhanced in a dose-dependent manner by walrycin A, with a maximal induction reached at 100 μM and an EC50~30 μM (Figure 2B). 6MNol was inactive in this assay. Taken together, these results indicate that walrycin A activates signalling pathway(s) controlling CYP3A4 mRNA expression.
Figure 2. Dose-dependence activation of CYP3A4 by walrycin A is PXR-dependent.

A: IHH cells were treated for 24h with indicated concentrations of rifampicin (black bar). Expression of CYP3A4 was determined by qPCR, normalized to RPLP0 expression level and compared to DMSO control (white bar). Histograms represent means ± SEM of a representative experiment performed in triplicate. B: Cells were treated for 24h with indicated concentrations of walrycin A (black bar) or 6MNol (hatched bar). Expression of CYP3A4 was determined by qPCR and analyzed as described above. Histograms represent means ± SEM of 3 independent experiments performed with triplicates. Statistical significances were determined with a Dunnett’s Multiple Comparison Test (***p<0.001). C: Cells were transfected with control siRNA (white bars) or by PXR-targeting siRNA (black bars) and knock-down expression of PXR gene (mRNA and protein) was assessed by qPCR and western blot analysis. D: Control and PXR-targeting siRNA transfected cells were treated 24h by vehicle (0.1% DMSO), rifampicin or walrycin A. The expression of PXR and CYP3A4 was determined by qPCR, normalized to RPLP0 expression level and expressed relative to that detected in cells transfected by control siRNA. Histograms represent means ± SEM of a representative experiment performed in triplicate. Statistical significances were determined using a Bonferroni post-hoc test (***p<0.001 and **p<0.01).
We then hypothesized that walrycin A-induced CYP3A4 mRNA expression was PXR-dependent. To assess this possibility, PXR knock-down was performed in IHH cells using siRNAs (Figure 2C and 2D). Both PXR mRNA and protein were significantly downregulated upon anti-PXR siRNA treatment (Figure 2C), whereas a non-specific siRNA did not affect PXR expression. CYP3A4 mRNA was induced upon rifampicin treatment, whereas PXR knock-down significantly blunted this response (Figure 2D). Importantly, walrycin A-induced CYP3A4 mRNA expression was decreased upon PXR knock-down, but not affected by the non-specific siRNA (Figure 2D). Thus, induction of CYP3A4 expression in response to walrycin A is a PXR-dependent process.
Walrycin A interacts with PXR and modifies its conformation
As we demonstrated that the induction of CYP3A4 by walrycin A is PXR-dependent, we next hypothesized that walrycin A could directly interact with PXR. To assess this possibility, limited proteolysis experiments were performed to probe potential structural modifications of the PXR polypeptide upon walrycin A binding. Full-length PXR was translated in vitro using 35S-labelled methionine in the presence of either DMSO (negative control), rifampicin (positive control), walrycin A or the related compound 6MNol. Unliganded and liganded PXR were then submitted to limited proteolysis using increasing concentrations of chymotrypsin ranging from 0.5 μg/mL to 5 μg/mL (Figure 3). Full-length radiolabeled PXR protein was more amenable to degradation by chymotrypsin in the presence of rifampicin compared to negative control (1% DMSO), leading to several proteolysis resistant peptides (Figure 3). The PXR proteolysis pattern in the presence of walrycin A was totally different to that induced by DMSO, suggesting that walrycin A, like rifampicin, interacts with PXR and modifies its conformation, hence exposing chymotrypsin cleavage sequences. The digestion pattern generated in presence of 6MNol was comparable to that observed in the presence of DMSO, indicating that this compound is unlikely to bind PXR. These results are in line with the (lack of) effect of these compounds on PXR-mediated induction of CYP3A4 expression.
Figure 3. Walrycin A induces conformational changes in the human PXR receptor polypeptide.

In vitro translation of hPXR was carried out in the presence of 0.1% DMSO, 50μM 6MNol, walrycin A or rifampicin. Upon completion of protein synthesis, limited proteolysis assay was performed as described in materials and methods using varying concentrations of chymotrypsin (up to 5 μg/mL). Radio-labeled peptides were detected using a Phosphorimager system. Arrow indicates the short fragments corresponding to LBD of PXR partially protected from digestion in presence of rifampicin or walrycin A.
Walrycin A activates PXR through its ligand binding domain
Since protease-resistant fragments observed in limited proteolysis assays can be attributed to structural alterations occurring in the LBD of nuclear receptors (Benkoussa et al. 1997), our limited proteolysis assays strongly suggested that walrycin A modulates PXR activity through interaction with the PXR LBD. To verify this hypothesis, a luciferase gene reporter assay was performed using a modified HeLa cell line (HGPXR cells) in which a chimeric GAL4 DBD-hPXR LBD fusion protein is stably expressed (Lemaire et al. 2006; Lemaire et al. 2007). HeLa cells were treated with concentrations of walrycin A ranging from 0.5 μM to 500 μM, and the luciferase activity was monitored (Figure 4A). This system was not sensitive to walrycin A concentrations below 5 μM. Nonetheless, a dose-dependent increase in the luciferase activity was observed starting at 5μM and reached a maximum at approximately 150 μM, with a calculated EC50~10 μM. To evaluate PXR selectivity for walrycin A or 6MNol, an independent experiment was performed in the same system using 150 μM of walrycin A or of 6MNol. As shown in Figure 4B, walrycin A significantly enhanced luciferase transactivation when used at 150 μM when compared to vehicle (DMSO), up to a level close to 80% of the maximal response obtained with the reference PXR ligand SR12813 (Moore et al. 2000), whereas 6MNol was again inactive. Taken together, these results show that walrycin A activates PXR through its LBD and is likely to be a novel ligand for this nuclear receptor.
Figure 4. Walrycin A activates PXR through its ligand binding domain.
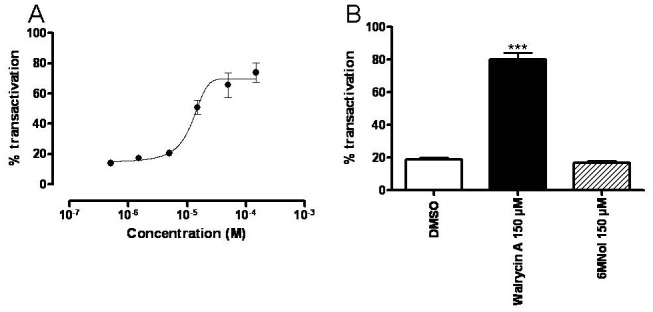
HeLa cell line in which a chimeric GAL4-DBD-PXR LBD fusion protein was stably expressed was treated with DMSO (negative control), SR12813 (positive control), walrycin A or 6MNol. A: Cells were treated with increasing concentrations of walrycin A ranging from 0.5 μM to 200 μM. B: Cells were treated with 150 μM of walrycin A or 6MNol. All experiments were performed in duplicates and results were plotted compared to maximal luciferase activity obtained using 1 μM of SR12813 (100% of transactivation).
The walrycin A oxidation product Russig’s blue is efficiently docked into the PXR LBD
Having shown that walrycin A can alter PXR conformation in vitro and activates PXR through its LBD in cellular assays, we next analyzed the ability of walrycin A, of the closely related molecule 6MNol, and of Russig’s blue, an oxidation product of walrycin A (Shoji et al. 2010), to fit in the ligand-binding pocket of PXR by in silico docking experiments. Docking was first implemented using PXR LBD coordinates extracted from the 3hvl.pdb file, corresponding to a human PXR LBD crystal obtained in the presence of a short SRC-1 coactivator peptide and of the bound synthetic PXR agonist SR12813 (Watkins et al. 2001). As shown in Figure 5A, SR12813 was efficiently docked by our procedure with solutions mostly superposable to the 3HVL coordinates (Watkins et al. 2001). Walrycin A and 6MNol docked perpendicular to the phenyl ring of SR12813, leaving most of the ligand-binding pocket empty (Figure 5A). In contrast, Russig’s blue positioned similarly to SR12813 (Figure 5B). Ranking ligand fit in 3HVL showed that Russig’s blue has the highest score compared to walrycin A and 6MNol, using either GOLD or FRED softwares (Figure 5B). Similar conclusions were drawn when using other PXR LBD coordinates (1ILH), corresponding to the human PXR LBD co-crystallized with SR12813 but without a coactivator peptide (data not shown). These data suggest that walrycin A may dock into the PXR LBD in its oxidized form Russig’s blue.
Figure 5. The walrycin A oxidation product Russig’s blue fits in PXR ligand-binding pocket.

A: Images of docking solutions for SR12813, Russig’s blue, Walrycin A and 6MNol in PXR ligand-binding domain from 3HVL. Endogenous 3HVL SR12813 ligand is shown in magenta and three solutions for each ligand are shown in gold. Oxygen atoms are shown in red except in the reference ligand (magenta). B: Ligand fitting in 3HVL and 1ILH ligand-binding pockets as assessed with GOLD (Chemscore, good fitting results in high score) and FRED (FRED score, good fitting results in low score). Russig’s blue and SR12813 grouped as best fitting compounds compared to walrycin A and 6MNol.
Walrycin A and hepatic cells viability
Rifampicin and walrycin A share common bactericidal properties, raising the possibility of a combined antibacterial therapy. We therefore investigated the effect of walrycin A alone or in combination with rifampicin on the viability of human hepatoma cell lines IHH and HepG2. Cell viability assays were performed using MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) as a substrate for mitochondrial reductase. To assess walrycin A toxicity, cells were independently exposed for 24 h to walrycin A and 6MNol concentrations increasing from 2 μM up to 5 mM, whereas rifampicin concentrations were from 0.5μM to 1 mM. Walrycin A concentrations up to 156 μM did not affect HepG2 viability (Figure 6A). Walrycin A IC50 was calculated to be 240 μM, indicating that this compound impacted cell viability at a concentration lower than that of rifampicin [IC50~1 mM, Figure 6A and (Nakajima et al. 2011)] and 6MNol (IC50~1.7 mM, data not shown). Similar results were obtained using IHH cell line (IC50 walrycin A~350 μM, IC50 rifampicin~4.5 mM, IC50 6MNol~1.8 mM, data not shown).
Figure 6. Impacts of rifampicin and Walrycin A on hepatoma cell viability.

A: HepG2 cells were treated for 24h using compounds concentrations ranging from 2 μM to 5 mM for walrycin A and from 0.5 μM to 1mM for rifampicin. Data were normalized using DMSO as internal control. Rifampicin: black curve, walrycin A: gray curve. B: Cells were treated for 24h and 48h using 100 μM rifampicin (black bars), 50 μM walrycin A (gray bars) or treated in combination with rifampicin 100 μM and walrycin A 50 μM (white bars). Data were normalized using DMSO as internal control. Histograms represent means ± SEM of a representative experiment performed in quadruplicate. Statistical significances were determined with a Dunnett’s Multiple Comparison Test (**p<0.01).
The potential synergy between walrycin A and rifampicin was then assessed in the MTS assay. HepG2 cells were exposed to increasing concentrations of rifampicin (from 3 μM to 100 μM) with or without 50 μM walrycin A for 24 or 48 hours (Supplementary figure S4). Highest rifampicin and walrycin A concentrations (100 μM and 50 μM respectively) did not impact cell viability after the 24 h exposure, whereas the combined walrycin A/rifampicin treatment significantly decreased cell viability by approximately 35 % (Figure 6B, left panel). After a 48 h exposure, HepG2 cell viability was significantly decreased upon rifampicin (100 μM) treatment when compared to walrycin A (50 μM) treatment, corroborating data from (Singh et al. 2011). The combined walrycin A/rifampicin exposure for 48 hours significantly decreased cell viability by approximately 50 % when compared to rifampicin alone (Figure 6B, right panel). Similar results were obtained by exposing HepG2 cells to increasing concentrations of walrycin A (from 3 μM to 100 μM) together with rifampicin 10 μM (Supplementary figure S4). Taken together, these results indicated that rifampicin and walrycin A synergistically impacted human hepatoma cell viability in a dose- and time-dependent manner.
Discussion
Bacterial infections, despite intense efforts to thwart them, remain a major public health problem not only in developing but also in occidental countries, as highlighted by the reminiscence of nosocomial infections. Methicillin-resistant Staphylococcus aureus (MRSA) is a widespread nosocomial pathogen (Diekema et al. 2001) resistant to β-lactam antibiotics, cephalosporins and to the last resort antibiotic vancomycin, to which 40% of MRSA infected patients are resistant (Jeffres et al. 2006; Rello et al. 1994). Therefore, given the high mortality rates caused by these drug-resistant bacteria and the difficulty to develop novel potent and specific antibiotics targeting these bacterial pathogens, combined antibactericidal treatments are currently used to kill increasingly common antibiotic resistant-strains. Rifampicin harbors valuable properties in combination with first-line antibiotics, and the combination of rifampicin and vancomycin is an effective treatment against nosocomial MRSA-induced pneumoniae (Jung et al. 2010). The novel antibacterial compound walrycin A is effective against the MRSA S. aureus N315 strain (Gotoh et al. 2010; Kuroda et al. 2001), suggesting that it could be successfully used in combination with other antibiotics such as rifampicin, since its high minimal inhibitory concentration on MRSA (734μM) precludes its use as a standalone therapy.
However, drug-drug interactions induced upon antibiotics (co)treatment may lead to increased cytotoxicity as exemplified by liver damage, and to reduced efficiency due to rapid drug inactivation resulting from the activation of the nuclear xenobiotic sensors CAR and PXR (Singh et al. 2011). Evaluating the activity of walrycin A on hepatic functions is therefore critical to evaluate its potential usefulness as an anti-MRSA drug.
The structure of walrycin A is based on a naphthalen scaffold, conferring hydrophobic properties, substituted by alcool and methoxy functions in position 1 and 4 respectively. As described by Gotoh et al., walrycin A targets the WalK/WalR two-component signal transduction system of Gram (+) bacteria, indicating that this compound gets effectively through the bacterial peptidoglycanic wall and cytoplasmic membrane of prokaryotes (Gotoh et al. 2010). Therefore, walrycin A could pass through the eukaryote plasma membrane to activate various signalling pathways, including those controlled by PXR. Indeed, walrycin A was found to induce the expression of several PXR target genes such as the drug metabolism phase I enzymes CYP1A1/1A2/2B6/2C8/2C19/3A4/11A1/11B1/11B2 (di Masi A. et al. 2009) and CYP4A11(Siest G. et al. 2008).Walrycin A also deregulated the expression of several enzymes not identified as PXR target genes, suggesting that walrycin A modulates other non identified signalling pathways.
The PXR-CYP3A4 pathway is involved in approximately 60% of reported drug-drug interactions (Evans 2005). CYP3A4 mRNA expression was enhanced in a PXR-dependent manner upon walrycin A treatment. Owing to its structure, walrycin A can potentially activate PXR through direct binding or by triggering post-translational modifications of this nuclear receptor. To sort out these non-exclusive hypotheses, we investigated whether walrycin A is a direct activator of PXR. Using in vitro limited proteolysis and gene reporter assays, we demonstrated that walrycin A is likely to act through the PXR LBD. However, in silico docking of walrycin A into the PXR ligand binding pocket revealed that the walrycin A oxidation product, Russig’s blue, is more likely to behave as a bona-fide PXR ligand. This is consistent with the fact that walrycin A spontaneously oxidizes in various aqueous solutions (Shoji et al. 2010 and our unpublished observations), and with the strict stereoselectivity of PXR activation, on which the structurally related 6MNol is inactive and unable to convert into a spectrally detectable compound (our unpublished observations).
Activation of PXR is known to be species-specific, and several reports document the irrelevance of rodent models for assessing the ability of xenobiotics to regulate human PXR activity (Jones et al. 2000; Lecluyse 2001; Ma et al. 2007). Importantly, the expression of mouse Cyp3a family members is not altered upon administration of rifampicin (Ma et al. 2007) in contrast to dexamethasone, a known mouse PXR activator (Scheer et al. 2010). Our in vitro transactivation assays established that walrycin A is an activator of human, but not of mouse PXR (Supplementary figures S3A and S3B). In line with these results, orally administrated walrycin A (200 mg/kg) for eight days to C57Bl6 mice neither induced the expression of Cyp3a11, the mouse orthologue of human CYP3A4, nor caused significant macroscopic liver damage (data not shown). This clearly suggested that walrycin A is unable to activate mouse PXR. Our data and others (Ma et al. 2007) thus underline the need for humanized-PXR mouse models to study the in vivo effects of walrycin A and other antibiotics rather than wild-type mice.
CAR also plays a major role in drug biotransformation pathways by regulating the expression of, among others, CYP2B, CYP2C, CYP3A, UGTs, GSTs and membrane transporters MRP2&4 (di Masi A. et al. 2009; Omiecinski et al. 2011). It is therefore important to investigate the effects of walrycin A on CAR activity. Our data indicates that walrycin A behaves, like many CAR modulators, as an inverse agonist of CAR1 (Supplementary figure S5). Up to 15 CAR isoforms are expressed in human liver (Lamba et al. 2004) and some compounds, such as clotrimazole, display opposite modulatory properties on CAR1 and CAR3 isoforms (Auerbach et al. 2005; Moore et al. 2000). This highly complex signalling system, which does not occur in IHH and HepG2 hepatoma cell lines, does not allow an easy prediction of biological outcomes following CAR ctivation in the liver, and certainly deserves further investigation.
In addition to its PXR and CAR modulatory effects, walrycin A exhibited weak cytotoxic properties on human hepatoma cells, but synergistically increased rifampicin toxicity and vice-versa. This suggests that a combination of rifampicin and walrycin A could induce deleterious hepatic effects in vivo. This drawback has also been reported for the vancomycin/ rifampicin combined treatment (Jung et al. 2010), which is likely due at least in part to drug-drug interactions. Finally, we noted that some walrycin A-exposed mice exhibited a marked splenomegaly, in agreement with a study in rats (Eastman Kodak Co. et al. 1992), and which might stem also from a portal hypertension secondary to liver disease. The design of efficient antibiotics thus comes up against important problems of hepatotoxicity and of hepatic xenobiotic sensors regulation.
Finally, to the best of our knowledge, walrycin A is not yet validated as a therapeutically usable drug and there is therefore no information on human exposure level. Nevertheless, as this compound and/or its derivative Russig’s blue activate PXR and CAR, hepatic effects of a simultaneous exposure to walrycin A and to other drugs should be considered in the future. As walrycin A is also a widely used compound in chemical synthesis (Talaat and Nelson 1986), the effects of a fortuitous acute or chronic exposition should be monitored in workers exposed to this compound, especially since combined, unwanted exposure to other environmental pollutants such as phthalates and bisphenol A, also known to activate human PXR and CAR (DeKeyser et al. 2011), can dangerously impact health (Howdeshell et al. 2007).
Supplementary Material
Acknowledgments
We thank Dr. Jérome Eeckhoute for help with the writing of the manuscript.
References
- Auerbach SS, DeKeyser JG, Stoner MA, Omiecinski CJ. CAR2 displays unique ligand binding and RXRalpha heterodimerization characteristics. Drug Metab Dispos. 2007;35 (3):428–439. doi: 10.1124/dmd.106.012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach SS, Stoner MA, Su S, Omiecinski CJ. Retinoid X receptor-alpha-dependent transactivation by a naturally occurring structural variant of human constitutive androstane receptor (NR1I3) Mol Pharmacol. 2005;68(5):1239–1253. doi: 10.1124/mol.105.013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayed-Boussema I, Pascussi JM, Maurel P, Bacha H, Hassen W. Zearalenone activates pregnane X receptor, constitutive androstane receptor and aryl hydrocarbon receptor and corresponding phase I target genes mRNA in primary cultures of human hepatocytes. Environ Toxicol Pharmacol. 2011;31(1):79–87. doi: 10.1016/j.etap.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Baciewicz AM, Chrisman CR, Finch CK, Self TH. Update on rifampin and rifabutin drug interactions. Am J Med Sci. 2008;335(2):126–136. doi: 10.1097/MAJ.0b013e31814a586a. [DOI] [PubMed] [Google Scholar]
- Benkoussa M, Nomine B, Mouchon A, Lefebvre B, Bernardon JM, Formstecher P, Lefebvre P. Limited proteolysis for assaying ligand binding affinities of nuclear receptors. Recept Signal Transduct. 1997;7(4):257–267. [PubMed] [Google Scholar]
- Breuker C, Moreau A, Lakhal L, Tamasi V, Parmentier Y, Meyer U, Maurel P, Lumbroso S, Vilarem MJ, Pascussi JM. Hepatic expression of thyroid hormone-responsive spot 14 protein is regulated by constitutive androstane receptor (NR1I3) Endocrinology. 2010;151(4):1653–1661. doi: 10.1210/en.2009-1435. [DOI] [PubMed] [Google Scholar]
- DeKeyser JG, Laurenzana EM, Peterson EC, Chen T, Omiecinski CJ. Selective phthalate activation of naturally occurring human constitutive androstane receptor splice variants and the pregnane X receptor. Toxicol Sci. 2011;120(2):381–391. doi: 10.1093/toxsci/kfq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Masi A, De Marinis E, Ascenzi P, Marino M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol Aspects Med. 2009;30(5):297–343. doi: 10.1016/j.mam.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, Jones RN, Beach M SENTRY Participants Group. Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997–1999. Clin Infect Dis. 2001;32(Suppl 2):S114–S132. doi: 10.1086/320184. [DOI] [PubMed] [Google Scholar]
- Rochester N. Eastman Kodak Co. Environmental Protection Agency, and Office of Toxic Substances. Basic Toxicity of 4-Methoxy-1-Naphthol with Cover Letter. Sep 8, 1992. 8EHQ-0992-10994. http://cfpub.epa.gov.
- Evans RM. The nuclear receptor superfamily: a rosetta stone for physiology. Mol Endocrinol. 2005;19(6):1429–1438. doi: 10.1210/me.2005-0046. [DOI] [PubMed] [Google Scholar]
- Faucette SR, Sueyoshi T, Smith CM, Negishi M, Lecluyse EL, Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. J Pharmacol Exp Ther. 2006;317(3):1200–1209. doi: 10.1124/jpet.105.098160. [DOI] [PubMed] [Google Scholar]
- Gotoh Y, Doi A, Furuta E, Dubrac S, Ishizaki Y, Okada M, Igarashi M, Misawa N, Yoshikawa H, Okajima T, Msadek T, Utsumi R. Novel antibacterial compounds specifically targeting the essential WalR response regulator. J Antibiot (Tokyo) 2010;63 (3):127–134. doi: 10.1038/ja.2010.4. [DOI] [PubMed] [Google Scholar]
- Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Jr, Kliewer SA, Gonzalez FJ, Sinal CJ. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem. 2003;278(46):45062–45071. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- Howdeshell KL, Furr J, Lambright CR, Rider CV, Wilson VS, Gray LE., Jr Cumulative effects of dibutyl phthalate and diethylhexyl phthalate on male rat reproductive tract development: altered fetal steroid hormones and genes. Toxicol Sci. 2007;99(1):190–202. doi: 10.1093/toxsci/kfm069. [DOI] [PubMed] [Google Scholar]
- Ihunnah CA, Jiang M, Xie W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim Biophys Acta. 2011;1812(8):956–963. doi: 10.1016/j.bbadis.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istrate MA, Nussler AK, Eichelbaum M, Burk O. Regulation of CYP3A4 by pregnane X receptor: The role of nuclear receptors competing for response element binding. Biochem Biophys Res Commun. 2010;393(4):688–693. doi: 10.1016/j.bbrc.2010.02.058. [DOI] [PubMed] [Google Scholar]
- Ivanovic J, Nicastri E, Ascenzi P, Bellagamba R, De ME, Notari S, Pucillo LP, Tozzi V, Ippolito G, Narciso P. Therapeutic drug monitoring in the management of HIV-infected patients. Curr Med Chem. 2008;15(19):1925–1939. doi: 10.2174/092986708785132960. [DOI] [PubMed] [Google Scholar]
- Jeffres MN, Isakow W, Doherty JA, McKinnon PS, Ritchie DJ, Micek ST, Kollef MH. Predictors of mortality for methicillin-resistant Staphylococcus aureus health-care-associated pneumonia: specific evaluation of vancomycin pharmacokinetic indices. Chest. 2006;130(4):947–955. doi: 10.1378/chest.130.4.947. [DOI] [PubMed] [Google Scholar]
- Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NC, Lecluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14(1):27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- Jung YJ, Koh Y, Hong SB, Chung JW, Ho Choi S, Kim MN, Choi IS, Han SY, Kim WD, Yun SC, Lim CM. Effect of vancomycin plus rifampicin in the treatment of nosocomial methicillin-resistant Staphylococcus aureus pneumonia. Crit Care Med. 2010;38(1):175–180. doi: 10.1097/ccm.0b013e3181b9ecea. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23(5):687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet. 2001;357(9264):1225–1240. doi: 10.1016/s0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- Lamba JK, Lamba V, Yasuda K, Lin YS, Assem M, Thompson E, Strom S, Schuetz E. Expression of constitutive androstane receptor splice variants in human tissues and their functional consequences. J Pharmacol Exp Ther. 2004;311(2):811–821. doi: 10.1124/jpet.104.069310. [DOI] [PubMed] [Google Scholar]
- Lecluyse EL. Pregnane X receptor: molecular basis for species differences in CYP3A induction by xenobiotics. Chem Biol Interact. 2001;134(3):283–289. doi: 10.1016/s0009-2797(01)00163-6. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102(5):1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire G, Benod C, Nahoum V, Pillon A, Boussioux AM, Guichou JF, Subra G, Pascussi JM, Bourguet W, Chavanieu A, Balaguer P. Discovery of a highly active ligand of human pregnane x receptor: a case study from pharmacophore modeling and virtual screening to “in vivo” biological activity. Mol Pharmacol. 2007;72(3):572–581. doi: 10.1124/mol.106.033415. [DOI] [PubMed] [Google Scholar]
- Lemaire G, Mnif W, Pascussi JM, Pillon A, Rabenoelina F, Fenet H, Gomez E, Casellas C, Nicolas JC, Cavailles V, Duchesne MJ, Balaguer P. Identification of new human pregnane X receptor ligands among pesticides using a stable reporter cell system. Toxicol Sci. 2006;91(2):501–509. doi: 10.1093/toxsci/kfj173. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real- time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma X, Idle JR, Gonzalez FJ. The pregnane X receptor: from bench to bedside. Expert Opin Drug Metab Toxicol. 2008;4(7):895–908. doi: 10.1517/17425255.4.7.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Shah Y, Cheung C, Guo GL, Feigenbaum L, Krausz KW, Idle JR, Gonzalez FJ. The Pregnane X Receptor gene-humanized mouse: a model for investigating drug-drug interactions mediated by cytochromes P450 3A. Drug Metab Dispos. 2007;35(2):194–200. doi: 10.1124/dmd.106.012831. [DOI] [PubMed] [Google Scholar]
- Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, Stoltz CA, Kliewer SA, Lambert MH, Willson TM, Moore JT. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J Biol Chem. 2003;278(19):17277–17283. doi: 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- Mo SL, Liu YH, Duan W, Wei MQ, Kanwar JR, Zhou SF. Substrate specificity, regulation, and polymorphism of human cytochrome P450 2B6. Curr Drug Metab. 2009;10(7):730–753. doi: 10.2174/138920009789895534. [DOI] [PubMed] [Google Scholar]
- Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, Goodwin B, Liddle C, Blanchard SG, Willson TM, Collins JL, Kliewer SA. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275(20):15122–15127. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- Moreau A, Vilarem MJ, Maurel P, Pascussi JM. Xenoreceptors CAR and PXR activation and consequences on lipid metabolism, glucose homeostasis, and inflammatory response. Mol Pharm. 2008;5(1):35–41. doi: 10.1021/mp700103m. [DOI] [PubMed] [Google Scholar]
- Nakajima A, Fukami T, Kobayashi Y, Watanabe A, Nakajima M, Yokoi T. Human arylacetamide deacetylase is responsible for deacetylation of rifamycins: rifampicin, rifabutin, and rifapentine. Biochem Pharmacol. 2011;82(11):1747–1756. doi: 10.1016/j.bcp.2011.08.003. [DOI] [PubMed] [Google Scholar]
- Omiecinski CJ, Vanden Heuvel JP, Perdew GH, Peters JM. Xenobiotic Metabolism, Disposition, and Regulation by Receptors: From Biochemical Phenomenon to Predictors of Major Toxicities. Toxicol Sci. 2011;120(Suppl 1):S49–S75. doi: 10.1093/toxsci/kfq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascussi JM, Vilarem MJ. Inflammation and drug metabolism: NF-kappB and the CAR and PXR xeno-receptors. Med Sci (Paris) 2008;24(3):301–305. doi: 10.1051/medsci/2008243301. [DOI] [PubMed] [Google Scholar]
- Perlroth J, Kuo M, Tan J, Bayer AS, Miller LG. Adjunctive use of rifampin for the treatment of Staphylococcus aureus infections: a systematic review of the literature. Arch Intern Med. 2008;168(8):805–819. doi: 10.1001/archinte.168.8.805. [DOI] [PubMed] [Google Scholar]
- Rae JM, Johnson MD, Lippman ME, Flockhart DA. Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: studies with cDNA and oligonucleotide expression arrays. J Pharmacol Exp Ther. 2001;299(3):849–857. [PubMed] [Google Scholar]
- Rello J, Torres A, Ricart M, Valles J, Gonzalez J, Artigas A, Rodriguez-Roisin R. Ventilator-associated pneumonia by Staphylococcus aureus. Comparison of methicillin-resistant and methicillin-sensitive episodes. Am J Respir Crit Care Med. 1994;150(6 Pt 1):1545–1549. doi: 10.1164/ajrccm.150.6.7952612. [DOI] [PubMed] [Google Scholar]
- Savkur RS, Wu Y, Bramlett KS, Wang M, Yao S, Perkins D, Totten M, Searfoss G, III, Ryan TP, Su EW, Burris TP. Alternative splicing within the ligand binding domain of the human constitutive androstane receptor. Mol Genet Metab. 2003;80(1–2):216–226. doi: 10.1016/j.ymgme.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Scheer N, Ross J, Kapelyukh Y, Rode A, Wolf WC. In vivo responses of the human and murine pregnane X receptor to dexamethasone in mice. Drug Metab Dispos. 2010;38 (7):1046–1053. doi: 10.1124/dmd.109.031872. [DOI] [PubMed] [Google Scholar]
- Schippers IJ, Moshage H, Roelofsen H, Muller M, Heymans HS, Ruiters M, Kuipers F. Immortalized human hepatocytes as a tool for the study of hepatocytic (de-)differentiation. Cell Biol Toxicol. 1997;13(4–5):375–386. doi: 10.1023/a:1007404028681. [DOI] [PubMed] [Google Scholar]
- Shoji O, Wiese C, Fujishiro T, Shirataki C, Wunsch B, Watanabe Y. Aromatic C-H bond hydroxylation by P450 peroxygenases: a facile colorimetric assay for monooxygenation activities of enzymes based on Russig’s blue formation. J Biol Inorg Chem. 2010;15(7):1109–1115. doi: 10.1007/s00775-010-0671-9. [DOI] [PubMed] [Google Scholar]
- Siest G, Jeannesson E, Marteau JB, Samara A, Marie B, Pfister M, Visvikis-Siest S. Transcription factor and drug-metabolizing enzyme gene expression in lymphocytes from healthy human subjects. Drug Metab Dispos. 2008;36(1):182–189. doi: 10.1124/dmd.107.017228. [DOI] [PubMed] [Google Scholar]
- Siest G, Jeannesson E, Marteau JB, Samara A, Marie B, Pfister M, Visvikis-Siest S. Transcription factor and drug-metabolizing enzyme gene expression in lymphocytes from healthy human subjects. Drug Metab Dispos. 2008;36(1):182–189. doi: 10.1124/dmd.107.017228. [DOI] [PubMed] [Google Scholar]
- Singh M, Sasi P, Rai G, Gupta VH, Amarapurkar D, Wangikar PP. Studies on toxicity of antitubercular drugs namely isoniazid, rifampicin, and pyrazinamide in an in vitro model of HepG2 cell line. Med Chem Res. 2011;20:1611–1615. [Google Scholar]
- Sueyoshi T, Kawamoto T, Zelko I, Honkakoski P, Negishi M. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J Biol Chem. 1999;274(10):6043–6046. doi: 10.1074/jbc.274.10.6043. [DOI] [PubMed] [Google Scholar]
- Talaat RE, Nelson WL. Synthesis and identification of 3-(4-hydroxy-1-naphthoxy)lactic acid as a metabolite of propranolol in the rat, in man, and in the rat liver 9000 g supernatant fraction. Drug Metab Dispos. 1986;14(2):202–207. [PubMed] [Google Scholar]
- Timsit YE, Negishi M. CAR and PXR: the xenobiotic-sensing receptors. Steroids. 2007;72(3):231–246. doi: 10.1016/j.steroids.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Gao J, Xie W. PXR and CAR in energy metabolism. Trends Endocrinol Metab. 2009;20(6):273–279. doi: 10.1016/j.tem.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292(5525):2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- Xie W, Yeuh MF, Radominska-Pandya A, Saini SP, Negishi Y, Bottroff BS, Cabrera GY, Tukey RH, Evans RM. Control of steroid, heme, and carcinogen metabolism by nuclear pregnane X receptor and constitutive androstane receptor. Proc Natl Acad Sci U S A. 2003;100(7):4150–4155. doi: 10.1073/pnas.0438010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.