

Abstract
Macrophage foam cells are a defining pathologic feature of atherosclerotic lesions. Recent studies have demonstrated that at high concentrations associated with hypercholesterolemia, native LDL induces macrophage lipid accumulation. LDL particles are taken up by macrophages as part of bulk fluid pinocytosis. However, the uptake and metabolism of cholesterol from native LDL during foam cell formation has not been clearly defined. Previous reports have suggested that selective cholesteryl ester (CE) uptake might contribute to cholesterol uptake from LDL independently of particle endocytosis. In this study we demonstrate that the majority of macrophage LDL-derived cholesterol is acquired by selective CE uptake in excess of LDL pinocytosis and degradation. Macrophage selective CE uptake does not saturate at high LDL concentrations and is not down-regulated during cholesterol accumulation. In contrast to CE uptake, macrophages exhibit little selective uptake of free cholesterol (FC) from LDL. Following selective uptake from LDL, CE is rapidly hydrolyzed by a novel chloroquine-sensitive pathway. FC released from LDL-derived CE hydrolysis is largely effluxed from cells but also is subject to ACAT-mediated reesterification. These results indicate that selective CE uptake plays a major role in macrophage metabolism of LDL.
Keywords: selective CE uptake, lipoprotein uptake, LDL endocytosis, CE hydrolysis, macrophage cholesterol metabolism
Macrophage foam cells are a defining pathologic feature of atherosclerotic lesions (1). These cells are characterized by cholesteryl ester (CE) accumulation within intracellular lipid droplets. LDL may play a major role in promoting foam cell formation by acting as a cholesterol donor during lipid accumulation. Indeed, high serum LDL is correlated with atherosclerosis in humans and experimental animals, and treatments that reduce serum LDL can halt progression of the disease and reduce complications such as myocardial infarction (2–4). However, there are currently no medications in use that target the macrophage foam cell directly, and further work is needed to determine the mechanisms responsible for macrophage LDL uptake and cholesterol accumulation in vivo (5).
Several pathways have been shown to induce cholesterol accumulation in cultured macrophages. Classically, scavenger receptor-mediated uptake of oxidized or enzyme-modified LDL is cited as a mechanism for foam cell formation (6, 7). Scavenger receptor-independent mechanisms are also involved in the uptake of some forms of enzyme-modified LDL including secretory phospholipase A2-modified LDL (8). In addition to chemically modified LDL, aggregated or matrix-associated LDL promotes cholesterol accumulation by stimulating LDL degradation in surface-connected compartments (9). There is evidence that various modified LDLs are present in atherosclerotic lesions (10–13) but macrophage uptake of these ligands has not been shown to be required for foam cell formation in vivo. More recently, a series of studies have demonstrated that native, unmodified LDL induces macrophage lipid accumulation at high concentrations (14–16). This accumulation is dependent on the induction of bulk fluid uptake through macropinocytosis and micropinocytosis and is independent of LDL binding to the LDL receptor and other cell surface receptors (17, 18). Pinocytosis of LDL-sized nanoparticles was demonstrated in atherosclerotic lesions of mice, supporting the potential significance of pinocytosis of native LDL during atherogenesis (19).
All of the current proposed mechanisms for macrophage foam cell formation involve intact (whole-particle) LDL uptake. However, CE and free cholesterol (FC) can also be taken up from lipoproteins independently of apolipoprotein by a process called selective lipid uptake. The HDL receptor scavenger receptor class B type I (SR-BI) functions as the major mediator of selective uptake from HDL in hepatocytes and steroidogenic cells (20). However, other mechanisms have been shown to be involved in selective CE uptake in other cell types (21–23). Importantly, macrophages exhibit SR-BI-independent selective CE uptake from both HDL and LDL (24–28). Selective lipid uptake is enhanced in atherosclerotic lesions of mice, indicating that macrophage selective uptake may be important during foam cell formation in vivo (29). However, a role for selective CE uptake in macrophage cholesterol accumulation from LDL has not been established. In this study, we investigate the uptake and metabolism of CE from native LDL during macrophage foam cell formation.
MATERIALS AND METHODS
Animals and reagents
Mice were housed in the Veterans Affairs Medical Center (Lexington, KY) and all experiments were approved by the Institutional Animal Care and Use Committee. Animals were maintained in a pathogen-free facility with a 12 h light/dark cycle and free access to food and water. SR-BI-null mice were obtained from Monty L. Krieger (MIT; Cambridge, MA) (30), and were bred together with wild-type control mice from a common mating pair of SR-BI heterozygous (SR-BI+/−) mice. CD36-null mice were obtained from Roy L. Silverstein (Cleveland Clinic; Cleveland, OH) (31). Iodide-125 radionuclide (125I), 1, 2, 3H(N)cholesterol ([3H]FC) and cholesteryl-1,2-3H(N)]hexadecyl ether ([3H]CEt) were obtained from Perkin Elmer. Cholesteryl 1, 2, 6, 7 H(N) oleate ([3H]CE) was purchased from American Radiolabeled Chemicals. Silica-coated polyvinyl chloride (PVC) TLC plates were purchased from Macherey-Nagel. Total cholesterol (TC) and FC E-kits were purchased from Wako. Cytochalasin D, latrunculin B, the ACAT inhibitor CI-976, chloroquine, and diethylumbelliferyl phosphate (UBP) were purchased from Sigma.
Lipoprotein isolation, characterization, radiolabeling, and modification
LDL (ρ = 1.019–1.063 g/ml) and HDL (ρ = 1.063–1.21 g/ml) were isolated from human plasma by density gradient ultracentrifugation (32). Human HDL was subfractionated to obtain HDL3 (ρ = 1.13–1.18 g/ml). Protein concentrations were determined by the method of Lowry et al. (33), and TC, FC, and CE content using Cholesterol E-kits. Lipoproteins were analyzed by SDS-PAGE and nondenaturing gradient gel electrophoresis. LDL was 125iodinated by the iodine monochloride method (34). To measure LDL CE uptake, [3H]CEt or [3H]CE were incorporated into 125I-LDL using the CETP method as previously described (35). Alternately, to measure LDL FC uptake, [3H]FC was incorporated into 125I-LDL as described (36). After labeling, 125I- and 3H-specific activities were determined as dpm/nmol CE (for [3H]CEt/125I-LDL and [3H]CE/125I-LDL) or dpm/nmol FC (for [3H]FC/125I-LDL). Specific activity with respect to the CE or FC content of the radiolabeled LDL was used so that uptake of cholesterol from LDL could be compared with changes in the measured cholesterol content of cells. Specific activities were between 2,000–10,000 and 500–1,000 dpm/nmol for 125I and 3H, respectively. LDL was aggregated by vortexing at maximum speed for 30 s, resulting in a milky suspension, or was acetylated by repeated addition of acetic anhydride to LDL as previously described (37).
Bone marrow-derived macrophage isolation and culture
Murine bone marrow-derived macrophages (BMMs) were obtained from wild-type or class B scavenger receptor-deficient mice and cultured by standard procedures (38). Isolated cells were suspended in medium A [RPMI 1640 plus 15% (v/v) L-cell-conditioned medium (LCM), 10% (v/v) heat-inactivated FBS, 50 IU/ml penicillin G, and 50 µg/ml streptomycin] and plated in 12-well plates (with or without glass cover slips for microscopy and cholesterol content determination, respectively) or in 48-well plates (for radiolabeled LDL uptake experiments). BMMs were cultured in medium A for 7 days before being used for experiments, and culture media was changed on days 3, 5, 6, and 7.
Microscopy and cellular lipid and protein content determinations
For microscopy, BMMs were treated as indicated in the figure legends then fixed for 5 min in freshly prepared 1% paraformaldehyde and stained with Oil Red O. Images were obtained from at least five randomly chosen fields for each condition. For cellular lipid determination, lipid was extracted from cells with hexane-isopropanol (3:2), and cells were dried and resuspended in aqueous solution with 1% Triton X-100-H2O. TC and FC content (nmol cholesterol/mg cell protein) were determined using Cholesterol E-kits. CE content was calculated as the difference between TC and FC content. Cell protein was determined using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific).
Determination of CE and FC uptake
LDL CE and FC uptake were determined by treating BMMs with radiolabeled LDL, as indicated in the figure legends, in medium B [RPMI 1640 plus 15% (v/v) LCM, 0.5% (w/v) FA-free BSA (Sigma), 20 mM HEPES, pH 7.4, 50 IU/ml penicillin G, and 50 µg/ml streptomycin]. After the treatment period, 125I-LDL uptake, [3H]CEt uptake and selective [3H]CEt uptake were quantified as previously described for HDL CE selective uptake (28). Briefly, cells were lysed by the addition of 0.1 M NaOH, collected, and counted for cell-associated 125I-LDL. Cell lysates were also used to determine cell protein content (mg/well) by BCA. Culture medium was precipitated with TCA, chloroform was extracted, and the aqueous phase was counted for degraded 125I-LDL. Lipid was extracted from cell lysates using Dole reagent (isopropanol-heptane-1M sulfuric acid, 40:10:1) and counted for 3H by scintillation counting. Cellular 3H (dpm/mg cell protein) was used to quantify CE uptake (nmol CE/mg cell protein). 125I uptake (dpm/mg cell protein, the sum of cell-associated and degraded 125I) was used to calculate CE equivalents (nmol/mg cell protein) assuming whole-particle uptake with concomitant CE delivery, as previously validated (6). Selective CE uptake was calculated as the difference between total [3H]CEt uptake and the CE uptake equivalents contributed by 125I-LDL uptake and represents CE uptake that cannot be accounted for by whole-particle uptake. The same calculation was used to quantify CE and FC uptake in cells treated with [3H]CE/125I-LDL or [3H]FC/125I-LDL, respectively. In some experiments in which the primary purpose of the 125I measurement was to analyze LDL protein uptake (rather than CE or FC uptake), 125I data were expressed as µg 125I-LDL protein/mg cell protein (see Figs. 4, 8).
Fig. 4.
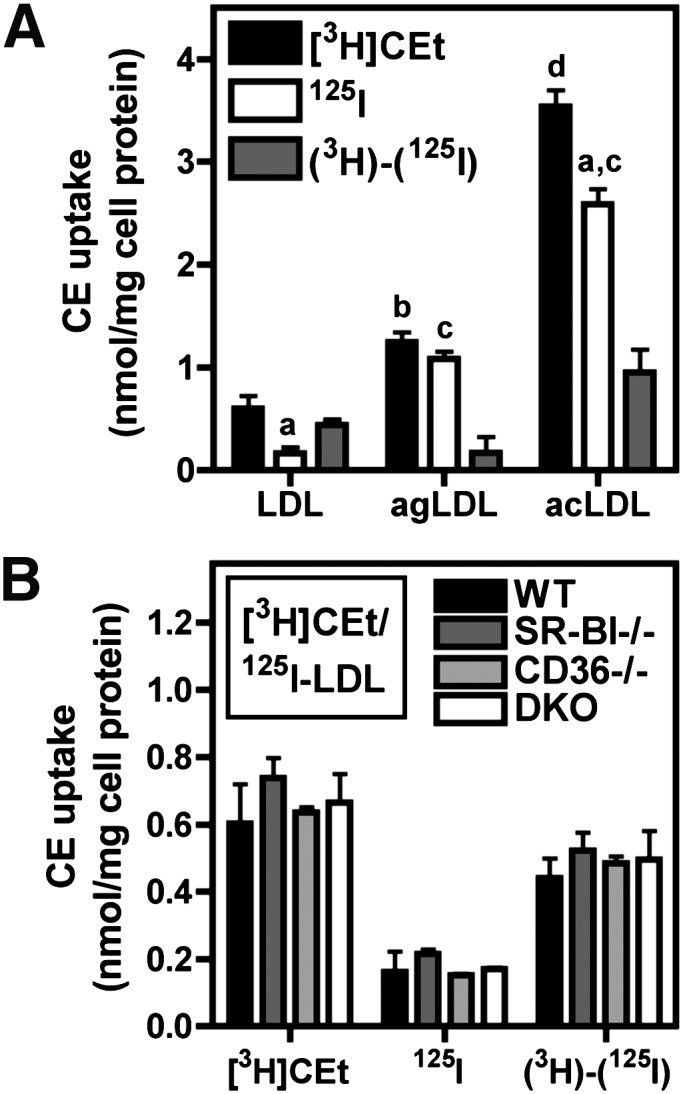
Effect of LDL modification or scavenger receptor deficiency on selective CE uptake. A: BMMs were treated with 10 µg/ml [3H]CEt/125I-labeled LDL, aggregated LDL (agLDL), or acetylated LDL (acLDL) for 4 h. a, P < 0.05 compared with the corresponding [3H]CEt from the same ligand; b, P < 0.05 compared with LDL [3H]CEt; c, P < 0.001 compared with LDL 125I; d, P < 0.001 compared with LDL [3H]CEt. B: BMMs from wild type, SR-BI-null, CD36-null, or DKO mice were treated with 10 µg/ml [3H]CEt/125I-LDL for 4 h. CE uptake was determined from [3H]CEt and 125I-LDL uptake, and selective CE uptake [(3H)-(125I)] was calculated as the difference between these two values, as described in Materials and Methods. Values are the mean ± SEM (n = 3).
Fig. 8.
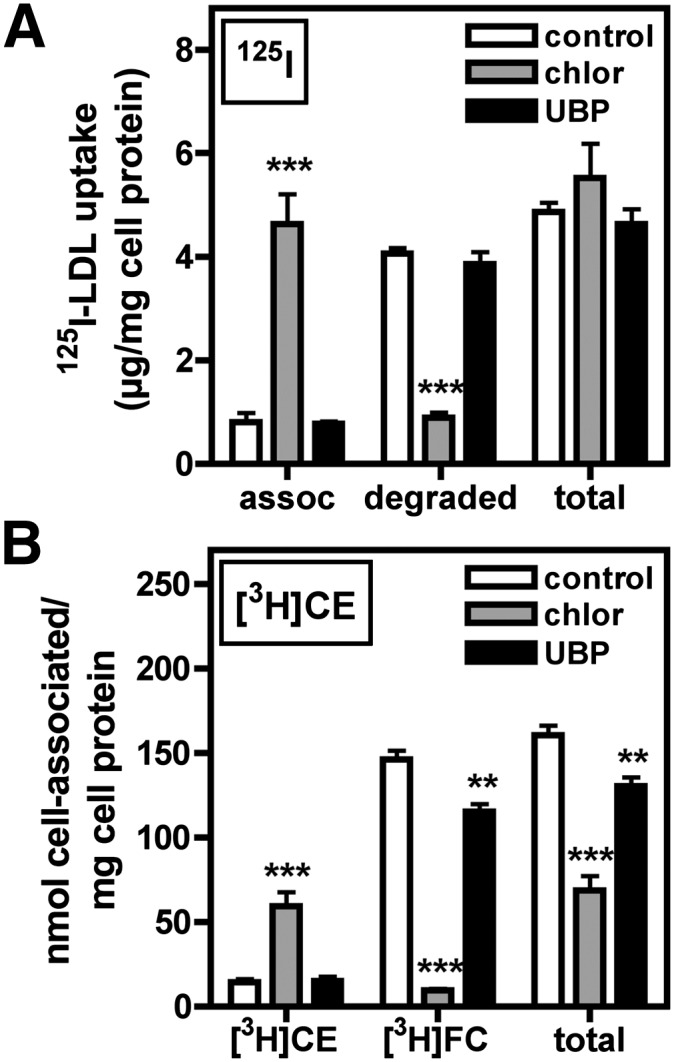
Effect of chloroquine and UBP on CE uptake and hydrolysis. BMMs were pretreated for 2 h in medium A with or without 200 µM chloroquine (chlor) or 400 µM UBP, then washed and treated under the same conditions in medium B plus 250 µg/ml [3H]CE/125I-LDL for 4 h. Cells were then washed thoroughly, and media and cell lysates were collected and used for determination of 125I-LDL uptake, association, and degradation as described in Materials and Methods (A). Cell lipid was extracted and separated by TLC, and [3H]CE and [3H]FC were quantified as described in Materials and Methods (B). Values are the mean ± SEM (n = 4). Results are representative of three independent experiments. ** P < 0.01 compared with control; *** P < 0.001.
TLC and CE hydrolysis assay
After treatment in medium B, as indicated in the figure legends, BMMs were washed and lipid was extracted from cells with hexane-isopropanol (3:2) and culture media (with Dole reagent) and dried overnight. Cell lysates and aliquots from culture media were used for determination of 125I association, degradation, and uptake. The next day, lipid extracts were resuspended in chloroform containing FC and CE (cholesteryl oleate) standards. An aliquot of this solution was removed and counted to determine total 3H for each sample. The remainder was spotted onto silica-coated PVC TLC plates and developed for 15 min with hexane-diethyl ether-acetic acid (60:40:1). CE and FC spots were visualized by iodine, cut out, and counted in scintillation fluid. The fraction of 3H dpm in the CE and FC spots was determined for each sample and multiplied by the total 3H to determine the amount of [3H]CE and [3H]FC, respectively. The amount of [3H]FC already present in the [3H]CE/125I-LDL before incubating with cells was determined in each experiment and was subtracted as a background during determination of [3H]FC accumulation in the culture medium. This background was <5% of total 3H and did not change during 37°C incubation in empty wells. Total CE uptake was calculated as the sum of [3H]FC and [3H]CE in cells plus the quantity of [3H]FC in the culture medium. The TLC procedural loss was less than 20% in each experiment.
[14C]oleate incorporation into cellular CE
[14C]oleic acid was mixed with unlabeled oleic acid to a final specific activity of 1 µCi/mmol, saponified, and complexed to BSA as previously described (6). To measure incorporation of [14C]oleate into cellular CE, cells were treated with [3H]CE-LDL in medium C [RPMI 1640 plus 15% (v/v) LCM, 0.2 mM [14C]oleate complexed to 0.5% w/v BSA, 20 mM HEPES, pH 7.4, 50 IU/ml penicillin G, and 50 µg/ml streptomycin) as indicated in the figure legends. After treatment, lipid was extracted from cells, dried and separated by TLC as described for CE hydrolysis determinations. CE, trigylceride, FA, and FC spots were visualized by iodine, cut out, and counted for both 3H and 14C.
Statistics and data analysis
Results are expressed as the mean ± SEM as indicated in the figure legends. Where not visible, error bars are contained within the symbols for results with n ≥ 3. Statistical analysis was done when appropriate using unpaired, two-tailed t-tests. Best-fit curves were generated using GraphPad Prism4 software.
RESULTS
Native LDL and macrophage foam cell formation
Native LDL has been shown to induce cholesterol accumulation in human monocyte-derived macrophages (14). In the current study, murine BMMs also accumulated cholesterol during treatment with native LDL (Fig. 1). Oil Red O-positive lipid droplets were observed when macrophages were treated for 24 h with 250 µg/ml LDL (Fig. 1A). Higher concentrations resulted in large increases in the number of droplets. Cholesterol quantification showed that cellular TC increased 1.5- to 3-fold over baseline in these conditions (Fig. 1B). CE was not detected in untreated cells, but accumulated in cells treated with LDL in a nonsaturating, linear manner with respect to LDL concentration. FC accumulation was less pronounced and exhibited saturation, with a near-maximal level of FC observed in cells treated with 250 µg/ml LDL.
Fig. 1.

Foam cell formation with native LDL. BMMs were treated for 24 h with the indicated concentration of LDL in medium B. Cells were then washed and stained with Oil Red O (A), or cell lipids were extracted and used for determination of TC, FC, and CE content (B, shown as the mean ± SEM, n = 3). Results are representative of four independent experiments.
Macrophage selective CE uptake from native LDL
To investigate the pathway involved in LDL-induced cholesterol accumulation, the rates of uptake of LDL CE and LDL protein were quantified (Fig. 2). This was carried out using [3H]CEt/125I double-labeled LDL (tracers for the CE and protein components of LDL, respectively), as previously reported (25). [3H]CEt is a nonhydrolyzable analog of CE that is intracellularly trapped and commonly used to measure selective CE uptake. BMMs were treated with 250, 500, or 1,000 µg/ml double-radiolabeled LDL, conditions that induced foam cell formation (see Fig. 1). Following treatment of macrophages with radiolabeled LDL, cellular 3H dpm was used to determine CE uptake (expressed as nmol CE/mg cell protein), whereas 125I was used to calculate CE uptake equivalents assuming whole-particle LDL uptake as described in Materials and Methods.
Fig. 2.

Macrophage selective LDL CE uptake. BMMs were treated in medium B with the indicated concentration of [3H]CEt/125I-LDL for 4 h (A) or with 250 µg/ml [3H]CEt/125I-LDL for the indicated time periods (B, C: panel C is a magnification of panel B for clarification). D: BMMs were pretreated for 24 h in medium A with or without 500 µg/ml LDL or 25 µg/ml acetylated LDL and were then washed and treated for 4 h in medium B with 250 µg/ml [3H]CEt/125I-LDL. CE uptake was determined from [3H]CEt and 125I-LDL uptake, and selective CE uptake [(3H)-(125I)] was calculated as the difference between these two values, as described in Materials and Methods. Values in panels A–C are the mean ± SEM (n = 3), and values in panel D are the mean ± SEM (n = 4) expressed as the percent of the control value (not treated with LDL or acLDL). * P < 0.05 compared with 125I or control; ** P < 0.01; *** P < 0.001. Results are representative of three independent experiments.
Total CE uptake in BMMs ([3H]CEt) was more than 3.5 times greater than CE contributed by whole-particle uptake (i.e., CE uptake equivalents determined from cell-associated and degraded 125I-LDL), indicating significant selective CE uptake (Fig. 2A–C). The basic finding of macrophage-selective CE uptake from LDL was consistently observed in multiple experiments using six separate LDL preparations. Selective CE uptake from LDL was also observed in cultured mouse peritoneal macrophages (see supplementary Fig. I). Macrophage CE uptake was linearly proportional to the LDL concentration and did not exhibit saturability at high concentrations (Fig. 2A). As LDL concentration increased, the ratio of 3H to 125I uptake increased from 3.5:1 to more than 8:1, due to partial saturation of the 125I-LDL uptake process. When macrophages were treated with 250 µg/ml LDL, the time course of [3H]CEt uptake was characterized by a rapid initial rate that decreased with time to reach a constant rate within 4 h (Fig. 2B). Thereafter, accumulation of 3H was constant with time for up to 24 h with no indication of down-regulation. In contrast, 125I-LDL uptake exhibited a single-phase, linear uptake. The majority (>90%) of 125I taken up was degraded and released from cells within 24 h (data not shown), consistent with a process of whole-particle endocytosis and delivery to lysosomes. The initial rate of [3H]CEt uptake, also shown in Fig. 2C (a magnification of Fig. 2B), was more than 12-fold higher than 125I-LDL CE uptake (42.41 versus 3.49 nmol CE/mg cell protein/h, respectively), suggesting that selective CE uptake was not subsequent to rapid LDL particle uptake and retro-endocytosis. Total CE uptake after 24 h was approximately 175 and 50 nmol CE/mg cell protein/24 h when determined from [3H]CEt and 125I-LDL, respectively.
Effect of cholesterol loading on selective CE uptake
Cellular cholesterol content is controlled by the coordinate regulation of lipoprotein uptake, cholesterol synthesis, and cholesterol efflux. Therefore we tested whether prior cholesterol loading might affect CE uptake from LDL (Fig. 2D). Cholesterol accumulation was induced by pretreating cells for 24 h with 500 µg/ml LDL (shown in Fig. 1 to cause cholesterol accumulation) or 25 µg/ml acetylated LDL (acLDL). At the end of this treatment, lipid droplets were visible in LDL- and acLDL-pretreated cells when viewed by phase-contrast microscopy (data not shown). However, cholesterol loading did not lead to a reduction in [3H]CEt or 125I-LDL uptake, both of which increased in cholesterol-loaded cells.
Comparison of CE uptake and FC uptake from LDL
Uptake of LDL [3H]CEt was also compared with [3H]CE and [3H]FC to further characterize macrophage LDL metabolism (Fig. 3). [3H]CE uptake was similar to uptake of [3H]CEt, suggesting that CE was not hydrolyzed prior to uptake and indicating that [3H]CEt is a valid marker for LDL CE uptake (Fig. 3A). In contrast, [3H]FC uptake was much lower than [3H]CEt or [3H]CE uptake (6-fold, Fig. 3A). In addition, macrophage [3H]FC accumulation was almost completely accounted for by whole-particle 125I-LDL uptake, with little selective FC uptake (Fig. 3B). Selective uptake of CE but not FC from LDL was observed in repeated experiments at LDL concentrations ranging from 250 to 1000 µg/ml LDL (data not shown). Together, these results demonstrated that selective CE uptake is responsible for the majority of cholesterol uptake during foam cell formation from LDL.
Fig. 3.
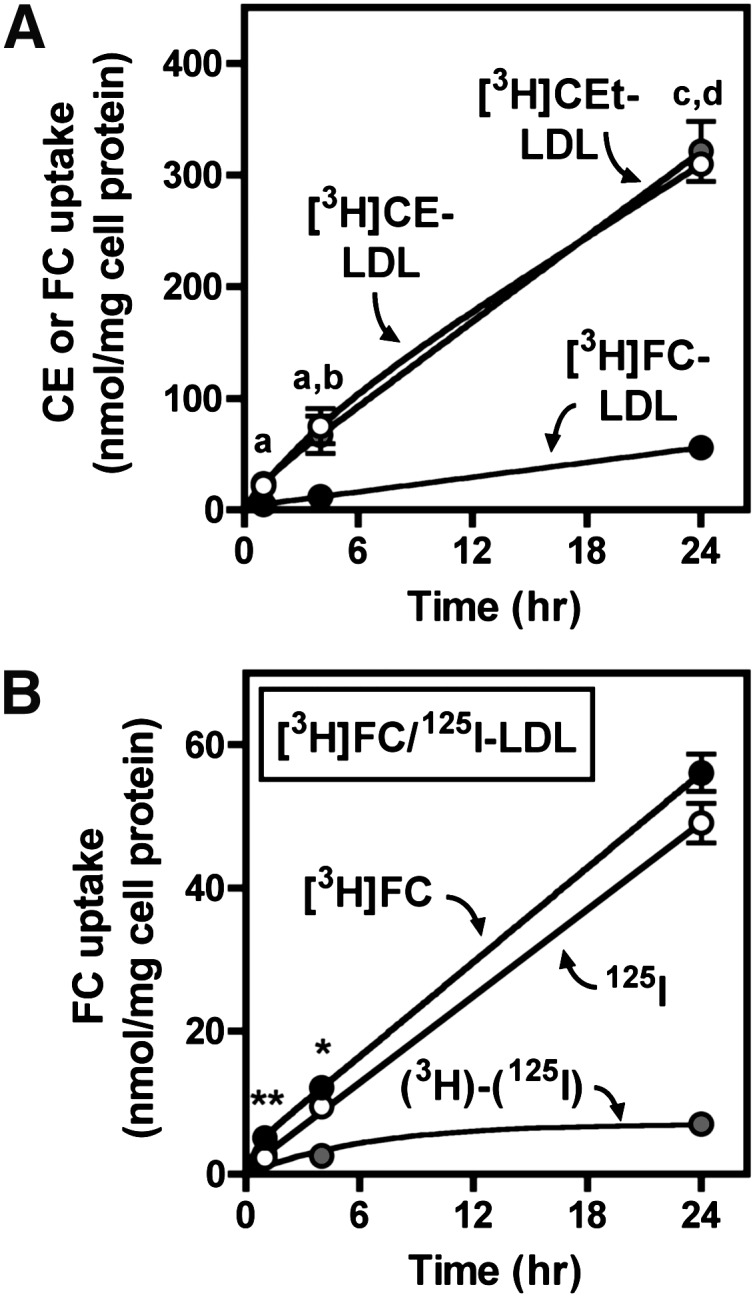
Comparison of CEt, CE, and FC uptake from LDL. BMMs were treated in medium B with 250 µg/ml of 125I-LDL labeled with [3H]CEt, [3H]CE, or [3H]FC for the indicated time periods. A: LDL CE or FC uptake was determined from cell-associated 3H after treatment with the indicated ligand. a, P < 0.05, [3H]CEt versus [3H]FC; b, P < 0.05, [3H]CE versus [3H]FC; c, P < 0.001, [3H]CEt versus [3H]FC; d, P < 0.001, [3H]CE versus [3H]FC. B: FC uptake from [3H]FC/125I-LDL was determined from [3H]FC and 125I-LDL uptake, and selective FC uptake [(3H)-(125I)] was calculated as the difference between these two values, as described in Materials and Methods. Values are the mean ± SEM (n = 3). Results are representative of three independent experiments.
Effect of LDL modification and scavenger receptor deficiency on selective CE uptake
Physical or chemical modification of LDL enhances its binding and degradation by macrophages (39, 40). To study the effect of LDL modification on selective CE uptake, BMMs were treated with native or modified LDL as previously described (7) using low concentrations (10 µg/ml) to study the high-affinity interactions known to be involved in macrophage uptake of modified LDL (Fig. 4). Consistent with previous reports, we found that endocytosis of acLDL or agLDL, respectively, as measured by 125I, was greatly increased compared with native LDL (Fig. 4A). 125I-LDL uptake was enhanced approximately 7- and 16-fold by aggregation or acetylation of LDL, respectively. However, compared with native LDL, the relative contribution of selective CE uptake from modified LDL was either largely abolished (agLDL) or significantly reduced (acLDL). Thus, selective CE uptake represents only a minor component of high-affinity macrophage cholesterol uptake from modified LDL. SR-BI and CD36 both bind native LDL as well as modified LDL and have been shown to mediate selective CE uptake in other cell types (20, 41–43). However, CE uptake was not impaired in BMMs from SR-BI-deficient, CD36-deficient, or SR-BI/CD36 double-deficient macrophages (Fig. 4B).
Role of actin polymerization-dependent pinocytosis in selective CE uptake
Macrophage LDL endocytosis during foam cell formation was previously shown to be dependent on fluid-phase pinocytosis and was inhibited by cytochalasin D, an inhibitor of actin polymerization (17). Consistent with this, we found that cytochalasin D blocked 125I-LDL uptake by >75% (Fig. 5A). Cytochalasin D also blocked 125I-BSA uptake (a marker of fluid-phase pinocytosis) by >50% (Fig. 5B). In contrast, selective [3H]CEt uptake was only modestly reduced (<35%) by cytochalasin D (Fig. 5C). In three independent experiments, cytochalasin D reduced 125I-LDL uptake by 62.5 ± 13.5% and [3H]CEt uptake by 27.6 ± 8.6%, a difference that was statistically significant (P < 0.05). This distinct pattern of inhibition was also conserved at a range of LDL concentrations (250–2,000 µg/ml) and during a time course of LDL CE uptake up to 24 h (data not shown). A similar result was obtained with latrunculin B, another inhibitor of actin polymerization (see supplementary Fig. II). These data indicated that selective CE uptake from LDL is largely independent of actin polymerization and fluid-phase pinocytosis.
Fig. 5.
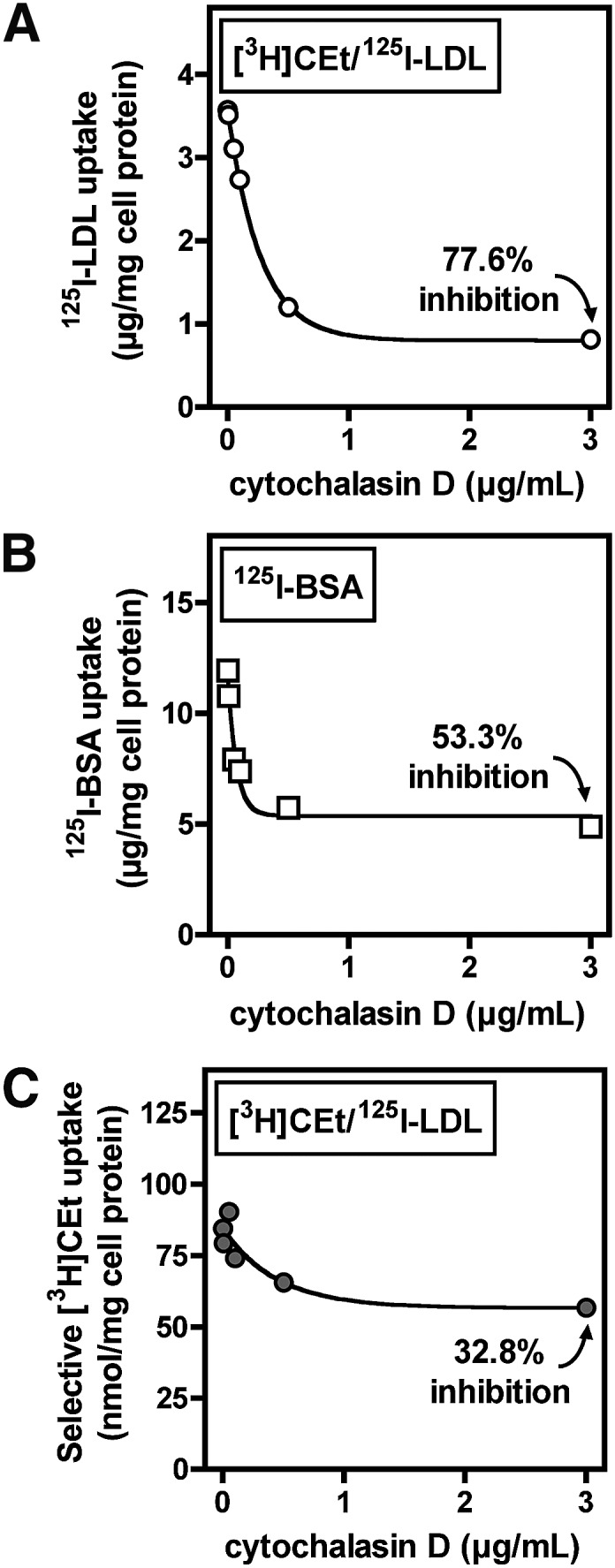
Role of actin polymerization in selective LDL CE uptake. BMMs were pretreated for 30 min with the indicated concentration of cytochalasin D in medium A, then washed and treated for 4 h with the same addition of cytochalasin D and 500 µg/ml [3H]CEt/125I-LDL or 100 µg/ml 125I-BSA in medium B. 125I-LDL uptake (A), 125I-BSA uptake (B), and selective [3H]CEt uptake (C) were determined as described in Materials and Methods. Values are the mean of duplicate determinations, and results are representative of three independent experiments.
LDL CE hydrolysis
CE derived from lipoproteins through endocytosis is typically hydrolyzed and incorporated into cellular cholesterol pools before it can be reesterified in lipid droplets (6). However, it is not known whether CE derived selectively from LDL is hydrolyzed in macrophages. Results from other experiments (Figs. 2–5) characterized the uptake of CE from LDL but did not discriminate between [3H]CE and [3H]FC that may have been formed by hydrolysis or esterification within cells. TLC revealed that during treatment of BMMs with 1,000 µg/ml [3H]CE/125I-LDL, the majority of cell-associated 3H accumulated as [3H]FC (Fig. 6A). This indicated that macrophages hydrolyzed the majority of CE taken up from LDL. CE hydrolysis was cell-mediated, inasmuch as [3H]FC was not generated when media containing [3H]CE-LDL was incubated in empty wells without BMMs (data not shown). [3H]FC accumulated in cells at a nearly constant rate for 8 h before reaching a plateau. [3H]FC was also detected in the treatment medium (Fig. 6B) and accumulated in a linear pattern with time. After 24 h, the amount of [3H]FC in the culture medium was greater than the amount present in cells, indicating that more than 50% of the formed [3H]FC was effluxed from cells. The total uptake of [3H]CE, calculated as the sum of cell-associated [3H]CE/[3H]FC and released [3H]FC, was much greater than what could be accounted for by uptake of 125I-LDL particles (Fig. 6C), consistent with substantial selective CE uptake.
Fig. 6.
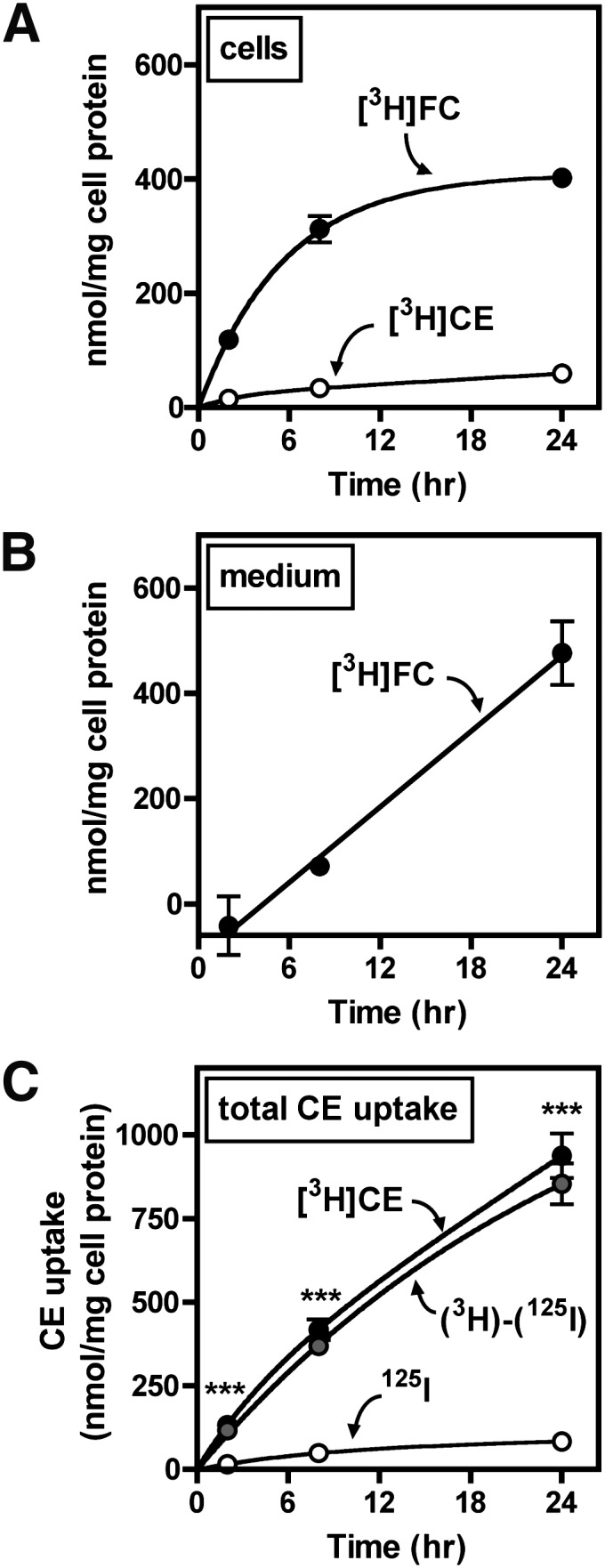
LDL CE uptake and hydrolysis. BMMs were treated with 1,000 µg/ml [3H]CE/125I-LDL for the indicated time period, then lipid was extracted from cells (A) and media (B) and separated by TLC. [3H]CE and [3H]FC were quantified as described in Materials and Methods. C: In the same experiment, CE uptake was determined from [3H]CEt and 125I-LDL uptake, and selective CE uptake [(3H)-(125I)] was calculated as the difference between these two values, as described in Materials and Methods. ***, P < 0.001, [3H]CE compared to 125I. Values are the mean ± SEM (n = 3). Results are representative of two independent experiments.
Because [3H]CEt uptake was not significantly less than [3H]CE uptake in previous experiments (see Fig. 3A), it was expected that CE hydrolysis and efflux occurred after uptake into cells. To confirm this, macrophages were pulsed for 30 min with 250 µg/ml [3H]CE/125I-LDL, washed thoroughly, and chased with unlabeled LDL (Fig. 7). LDL-derived [3H]CE diminished rapidly during the chase period, with a half-time of about 30 min, and this was accompanied by an increase in [3H]FC in both cells (Fig. 7A) and culture medium (Fig. 7B). The estimated rate of efflux of [3H]FC was ∼13.5% of total cellular 3H in 4 h, similar to the rate observed in cells treated continuously with [3H]CE/125I-LDL (Fig. 6B, >50% in 24 h). In a separate experiment using a standard cholesterol efflux assay, efflux of cellular [3H]FC to LDL was found to be comparable to efflux to apo-equivalent amounts of HDL (see supplementary Fig. III), confirming previous reports of significant macrophage cholesterol efflux to LDL (44). [3H]CE release was not detected in any experiment. These data indicated that selective CE uptake into macrophages is followed by rapid CE hydrolysis and significant release of newly generated FC from cells.
Fig. 7.

LDL-derived CE hydrolysis and efflux. BMMs were treated for 30 min in medium B with 250 µg/ml [3H]CE/125I-LDL and then washed thoroughly and chased with 250 µg/ml unlabeled LDL in medium B for the indicated time period, after which lipid was extracted from cells (A) and media (B), and [3H]CE and [3H]FC were quantified as described in Materials and Methods. Values are the mean ± SEM (n = 3). Results are representative of two independent experiments.
Involvement of acid and neutral lipase activity in LDL CE hydrolysis
The contributions of acidic lipase and neutral cholesteryl esterase to LDL-derived CE hydrolysis were investigated using the inhibitors chloroquine and UBP, respectively (45, 46). Macrophage LDL apolipoprotein degradation (measured as µg 125I-LDL protein/mg cell protein) involves acid proteases and is inhibited by chloroquine (47). Consistent with this, chloroquine significantly reduced 125I-LDL degradation and concomitantly increased cell-associated 125I-LDL within BMMs (Fig. 8A). Currently, there are no reports on the type of hydrolase involved in macrophage hydrolysis of LDL-derived CE. Similar to the effect on 125I-LDL degradation, chloroquine almost completely inhibited the generation of [3H]FC in cells treated with [3H]CE/125I-LDL (Fig. 8B). Unlike 125I-LDL uptake, total [3H]CE uptake was also significantly reduced by chloroquine. This indicated that chloroquine inhibited CE uptake but not LDL particle uptake, supporting the hypothesis that a portion of CE is taken up from LDL by a mechanism that is distinct from the 125I-LDL endocytosis and lysosomal delivery pathway. In contrast to chloroquine, the neutral cholesteryl esterase inhibitor UBP had no effect on LDL degradation and only a modest effect on [3H]CE uptake and hydrolysis (Fig. 8A, B, respectively). UBP was shown to block the hydrolysis of intracellular CE from lipid-loaded cells (see supplementary Fig. IV), confirming that UBP was an effective inhibitor of neutral cholesteryl esterase activity in BMMs. These data, therefore, implicated the involvement of acid but not neutral hydrolase activity in LDL-derived CE hydrolysis during foam cell formation.
ACAT-dependent reesterification of cholesterol derived from LDL CE
[3H]CE detected in cells treated with [3H]CE-LDL (Figs. 6–8) could represent nonhydrolyzed [3H]CE or hydrolyzed and reesterified [3H]CE. To distinguish these pools of CE, cells were treated with the ACAT inhibitor CI-976 to block intracellular esterification of [3H]FC (Fig. 9A). At the same LDL concentration, LDL CE contributed more to cellular [3H]CE accumulation than did LDL FC (Fig. 9A, left panel). As expected, CI-976 almost completely inhibited formation of [3H]CE in cells treated with [3H]FC-LDL. CI-976 also partially inhibited accumulation of [3H]CE during treatment with [3H]CE-LDL, indicating that a portion of [3H]CE was generated by ACAT-mediated reesterification. This effect did not reach statistical significance, but a similar magnitude of inhibition with statistical significance was observed in a second experiment which did not include the comparison to [3H]FC-LDL (data not shown). As observed in Fig. 3, total [3H]CE uptake from LDL was greater than [3H]FC uptake, and uptake was not affected by ACAT inhibition (Fig. 9A, right panel).
Fig. 9.

ACAT-dependent reesterification of LDL CE-derived cholesterol. A: BMMs were treated for 24 h in medium B with 250 µg/ml [3H]CE-LDL or [3H]FC-LDL with or without 10 µg/ml of the ACAT inhibitor CI-976. Lipid from cells was extracted and separated by TLC, and [3H]CE (left panel) and CE or FC uptake (right panel) were quantified as described in Materials and Methods. * P < 0.05 compared with DMSO control. B: BMMs were treated for the indicated time with 1,000 µg/ml [3H]CE-LDL in medium C (containing 0.2 mM [14C]oleate, left panel) or in medium C with the addition of 10 µg/ml CI-976 (right panel), then washed, and cellular [3H]CE and [14C]CE were extracted and quantified as described in Materials and Methods. C: BMMs were treated for 24 h in medium B with 500 µg/ml [3H]CE-LDL (left panel) or 25 µg/ml [3H]CE-acLDL (right panel). Cells were then washed, and [3H]CE and [3H]FC were quantified as described in Materials and Methods. Aliquots of lipid extracts from the same cells were also used to determine the CE and FC content, as described in Materials and Methods. Values are the mean ± SEM (n = 3). Results are representative of two independent experiments.
ACAT-mediated esterification was also investigated by quantifying incorporation of [14C]oleate into cellular CE as described in Materials and Methods (Fig. 9B). Treatment with either 1,000 µg/ml [3H]CE-LDL or 50 µg/ml [3H]CE-acLDL significantly increased [14C]CE generation in BMMs compared with cells not treated with lipoproteins (data not shown). As expected, [14C]CE formation was almost completely blocked by CI-976 (Fig. 9B). In the same cells, the quantity of [3H]CE accumulated was larger than [14C]CE and was only partially blocked by CI-976, supporting the hypothesis that in addition to ACAT-dependent [3H]CE generation, a portion of cellular [3H]CE represents incoming LDL [3H]CE prior to hydrolysis. Similar results were obtained with [3H]CE-acLDL (data not shown).
Finally, ACAT-mediated reesterification was examined by comparing the accumulation of [3H]CE and [3H]FC in cells treated with [3H]CE-LDL to the CE and FC mass measured in the same cells (Fig. 9C). After 24 h, BMM CE and FC mass could be accounted for by cell-associated [3H]CE and [3H]FC, respectively (Fig. 9C, left panel). A similar result was obtained with cells treated with [3H]CE-acLDL (Fig. 9C, right panel). These results suggested that the quantity of cholesterol derived from selective CE uptake is sufficient to explain FC and CE accumulation during macrophage foam cell formation.
DISCUSSION
Atherosclerosis is associated with elevated plasma LDL cholesterol levels, often more than 150 mg/dl LDL-C (greater than 1,000 µg/ml LDL). LDL accumulates at even higher levels in the arterial intima, where foam cells develop (48, 49). Therefore, studies of lipoprotein receptors carried out at very low lipoprotein concentrations may not accurately reflect disease conditions related to atherosclerosis and could have led to an underestimation of the role of native LDL in foam cell formation (6). In the current investigation, we observed that high-concentration LDL induces foam cell formation in murine BMMs, similar to previous reports in human macrophages (14). As described in the earlier studies, lipid accumulation from native LDL is a low-affinity process, with lipid droplet formation occurring at 250–1,000 µg/ml LDL or higher (Fig. 1). These results support the hypothesis that during hyperlipidemia, native LDL may contribute to macrophage cholesterol accumulation.
The initial goal of the current study was to define macrophage CE uptake during lipid accumulation from native LDL. Previous investigations demonstrated that LDL particles are taken up and degraded by macrophages as part of receptor-independent pinocytosis (17), but these reports did not quantify CE uptake and therefore did not address the possible contribution of selective CE uptake to cholesterol accumulation induced by native LDL. Our results demonstrate that the majority of cholesterol taken up from LDL is acquired by selective CE uptake in excess of LDL pinocytosis and degradation. Specifically, a traditional endocytic model cannot explain the following observations: 1) the quantity of [3H]CEt or [3H]CE uptake was not accounted for by 125I-LDL whole-particle uptake; 2) [3H]CEt and [3H]CE uptake were more than six times greater than [3H]FC uptake; 3) the kinetics and concentration dependence of [3H]CEt uptake differed from 125I-LDL uptake; and 4) [3H]CEt uptake was largely resistant to inhibition by cytochalasin D in comparison to 125I-LDL uptake. Together, these results provide strong evidence that a distinct selective CE uptake pathway contributes to macrophage cholesterol uptake from LDL.
This is the first report demonstrating selective CE uptake during macrophage foam cell formation. Results therefore extend previous observations of macrophage selective CE uptake from LDL (25, 26) and emphasize that macrophage selective CE uptake is predominantly a low-affinity, high-capacity process. The current results also demonstrate that macrophage selective CE uptake from LDL is independent of scavenger receptors, as we and others demonstrated for HDL selective CE uptake (27, 28). Macrophage selective CE uptake was previously reported to be down-regulated by cholesterol loading (26); however, we found that prior cholesterol loading did not reduce selective CE uptake. Rather, [3H]CEt accumulated with time in a nonsaturating, linear manner during foam cell formation. This difference could be because cholesterol accumulation was induced in the previous study by ACAT inhibition or cholesterol-saturated methyl-β-cyclodextrin, whereas the current study used native or acetylated LDL. Cholesterol accumulation from lipoproteins including native LDL is likely to be more relevant to the regulation of macrophage selective uptake than the treatments used previously. In addition, several reports indicated that selective CE uptake may be a bidirectional exchange process in macrophages and other cell types (50, 51), but the current results demonstrate that after transfer to macrophages, CE is efficiently hydrolyzed and not released into the medium as CE. This difference could be due to the much lower LDL concentrations used in previous studies reporting bidirectional selective CE uptake, or to differences in the reversibility of [3H]CEt (used in a previous study (51)) and [3H]CE.
To further characterize the selective CE uptake pathway in macrophages, we studied subsequent metabolism of LDL-derived CE. Our results demonstrate that after uptake from LDL, CE is rapidly hydrolyzed by macrophages, similar to a previous description of macrophage metabolism of acetylated LDL (6), but not to oxidized (52) or aggregated (53) LDL, which were reported to be resistant to CE hydrolysis. As already mentioned, cell-associated [3H]CE was not directly released into the chase medium in pulse-chase experiments. These findings are important because they demonstrate that [3H]CE uptake is a unidirectional process and not a reflection of simple radioisotope exchange resulting from a reversible CE uptake process. Irreversible, unidirectional CE uptake suggests cellular internalization. Although endocytosis of LDL particles could not account for CE uptake in this study, it is possible that one or more endocytic processes are involved in the selective CE uptake mechanism. One possibility is that CE is first transferred from LDL to the macrophage plasma membrane and subsequently sequestered and internalized within the membranes of endocytic vesicles. In support of this, LDL [3H]CE hydrolysis was almost completely inhibited by chloroquine (>90%). On the other hand, the neutral lipase inhibitor UBP did not substantially reduce [3H]CE hydrolysis, in contrast to a previous report investigating SR-BI-directed HDL CE hydrolysis (54).
ACAT-mediated cholesterol esterification is responsible for the development of cellular lipid droplets during macrophage foam cell formation. The low ratio of [3H]CE to [3H]FC in some experiments (see Fig. 6) suggested relatively inefficient reesterification of cholesterol derived from LDL CE. However, additional studies, in which ACAT-dependent [3H]CE accumulation was directly compared with [14C]CE generation and cellular cholesterol mass, provided strong evidence that cholesterol from LDL CE uptake contributed to FC and CE accumulation in BMMs. One limitation of the current study was that it was not possible to distinguish cholesterol derived from LDL endocytosis from that acquired by selective CE uptake. While providing less cholesterol to macrophages than selective CE uptake, whole-particle uptake of LDL could theoretically contribute enough cholesterol to account for the observed CE accumulation induced by LDL, assuming markedly different trafficking of cholesterol compared with the selective CE uptake pathway. Therefore, our results do not rule out the possibility that cholesterol derived from pinocytic uptake of LDL particles is responsible for foam cell formation during treatment with LDL as previously proposed (17). Strategies for specific pharmacologic and/or genetic inhibition of selective LDL CE uptake are needed to confirm the contribution of this pathway to macrophage foam cell formation.
In addition to ACAT-mediated reesterification, our results demonstrate rapid efflux of cholesterol generated from hydrolysis of LDL-derived CE. Substantial LDL-dependent cholesterol efflux was observed in time-course and pulse-chase experiments using [3H]CE/125I-LDL and in a standard [3H]FC efflux assay. Efflux of cholesterol during treatment with LDL provides an explanation for our observation that net cholesterol accumulation was less than predicted from [3H]CEt uptake (compare Figs. 1, 2). These results demonstrate that an accurate determination of the rate of CE uptake requires consideration of the magnitude of FC efflux, especially at relatively high LDL concentrations. In addition to controlling cellular cholesterol content, macrophage cholesterol efflux is recognized as a key step in reverse cholesterol transport and may impact the removal of cholesterol from atherosclerotic lesions (55, 56). It was previously reported that LDL-derived cholesterol is more efficiently released from macrophages than acetylated LDL-derived cholesterol, a finding attributed to distinct intracellular trafficking of internalized lipoproteins (57). The current results suggest that the greater efflux of cholesterol derived from LDL CE is more likely to be related to differences between the selective CE uptake pathway and the classic endocytic pathway. Although selective CE uptake may promote macrophage cholesterol accumulation, it is interesting to consider that this process may also facilitate reverse cholesterol transport by linking LDL CE hydrolysis to cholesterol efflux to HDL and other extracellular acceptors.
In summary, we show that selective CE uptake contributes the majority of macrophage cholesterol uptake during treatment with high concentrations of native LDL. Following uptake, CE is rapidly hydrolyzed in a chloroquine-sensitive pathway and contributes to cellular FC and CE accumulation. These results indicate that selective CE uptake plays a major role in macrophage metabolism of LDL.
Supplementary Material
Acknowledgments
The authors thank Xin Shi for excellent technical support, and Dr. Monty Krieger and Dr. Roy Silverstein for kindly providing SR-BI-null mice and CD36-null mice, respectively. The authors acknowledge Dr. Nancy R. Webb and Dr. Maria C. de Beer for very helpful discussion and comments.
Footnotes
Abbreviations:
- BCA
- bicinchoninic acid
- BMM
- bone marrow-derived macrophage
- CE
- cholesteryl ester
- FC
- free cholesterol
- LCM
- l-cell-conditioned medium
- PVC
- polyvinyl chloride
- SR-BI
- scavenger receptor class B type I
- TC
- total cholesterol
- UBP
- diethylumbelliferyl phosphate
This work was supported by the Department of Veterans Affairs, Biomedical Laboratory Research and Development [VA Merit Award IO1BX000672, (D.R.v.d.W.), Research Chemist, Department of Veterans Affairs Medical Center, Lexington, KY], and by National Institutes of Health research Grants P01-HL-086670-04 (D.R.v.d.W.) and T32-HL-072743. Additional support was provided by grants from the National Center for Research Resources (5P20RR-021954-05) and the National Institute of General Medical Sciences (8P20GM-103527-05) from the National Institutes of Health. The contents of this study do not represent the views of the Department of Veterans Affairs or the United States Government.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of four figures.
REFERENCES
- 1.Moore K. J., Tabas I. 2011. Macrophages in the pathogenesis of atherosclerosis. Cell. 145: 341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.1984 The Lipid Research Clinics Coronary Primary Prevention Trial Results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. J. Am. Med. Assoc. 251: 365–374 [PubMed] [Google Scholar]
- 3.1994 Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 344: 1383–1389 [PubMed] [Google Scholar]
- 4.Nissen S. E., Nicholls S. J., Sipahi I., Libby P., Raichlen J. S., Ballantyne C. M., Davignon J., Erbel R., Fruchart J. C., Tardif J. C., et al. 2006. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. J. Am. Med. Assoc. 295: 1556–1565 [DOI] [PubMed] [Google Scholar]
- 5.Webb N. R., Moore K. J. 2007. Macrophage-derived foam cells in atherosclerosis: lessons from murine models and implications for therapy. Curr. Drug Targets. 8: 1249–1263 [DOI] [PubMed] [Google Scholar]
- 6.Brown M. S., Goldstein J. L., Krieger M., Ho Y. K., Anderson R. G. 1979. Reversible accumulation of cholesteryl esters in macrophages incubated with acetylated lipoproteins. J. Cell Biol. 82: 597–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunjathoor V. V., Febbraio M., Podrez E. A., Moore K. J., Andersson L., Koehn S., Rhee J. S., Silverstein R., Hoff H. F., Freeman M. W. 2002. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 277: 49982–49988 [DOI] [PubMed] [Google Scholar]
- 8.Boyanovsky B. B., van der Westhuyzen D. R., Webb N. R. 2005. Group V secretory phospholipase A2-modified low density lipoprotein promotes foam cell formation by a SR-A- and CD36-independent process that involves cellular proteoglycans. J. Biol. Chem. 280: 32746–32752 [DOI] [PubMed] [Google Scholar]
- 9.Haka A. S., Grosheva I., Chiang E., Buxbaum A. R., Baird B. A., Pierini L. M., Maxfield F. R. 2009. Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Mol. Biol. Cell. 20: 4932–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tamminen M., Mottino G., Qiao J. H., Breslow J. L., Frank J. S. 1999. Ultrastructure of early lipid accumulation in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 19: 847–853 [DOI] [PubMed] [Google Scholar]
- 11.Ylä-Herttuala S., Palinski W., Rosenfeld M. E., Parthasarathy S., Carew T. E., Butler S., Witztum J. L., Steinberg D. 1989. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Invest. 84: 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore K. J., Kunjathoor V. V., Koehn S. L., Manning J. J., Tseng A. A., Silver J. M., McKee M., Freeman M. W. 2005. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J. Clin. Invest. 115: 2192–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wooton-Kee C. R., Boyanovsky B. B., Nasser M. S., de Villiers W. J., Webb N. R. 2004. Group V sPLA2 hydrolysis of low-density lipoprotein results in spontaneous particle aggregation and promotes macrophage foam cell formation. Arterioscler. Thromb. Vasc. Biol. 24: 762–767 [DOI] [PubMed] [Google Scholar]
- 14.Kruth H. S., Huang W., Ishii I., Zhang W. Y. 2002. Macrophage foam cell formation with native low density lipoprotein. J. Biol. Chem. 277: 34573–34580 [DOI] [PubMed] [Google Scholar]
- 15.Zhao B., Li Y., Buono C., Waldo S. W., Jones N. L., Mori M., Kruth H. S. 2006. Constitutive receptor-independent low density lipoprotein uptake and cholesterol accumulation by macrophages differentiated from human monocytes with macrophage-colony-stimulating factor (M-CSF). J. Biol. Chem. 281: 15757–15762 [DOI] [PubMed] [Google Scholar]
- 16.Anzinger J. J., Chang J., Xu Q., Barthwal M. K., Bohnacker T., Wymann M. P., Kruth H. S. 2012. Murine bone marrow-derived macrophages differentiated with GM-CSF become foam cells by PI3Kγ-dependent fluid-phase pinocytosis of native LDL. J. Lipid Res. 53: 34–42 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kruth H. S., Jones N. L., Huang W., Zhao B., Ishii I., Chang J., Combs C. A., Malide D., Zhang W. Y. 2005. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J. Biol. Chem. 280: 2352–2360 [DOI] [PubMed] [Google Scholar]
- 18.Anzinger J. J., Chang J., Xu Q., Buono C., Li Y., Leyva F. J., Park B. C., Greene L. E., Kruth H. S. 2010. Native low-density lipoprotein uptake by macrophage colony-stimulating factor-differentiated human macrophages is mediated by macropinocytosis and micropinocytosis. Arterioscler. Thromb. Vasc. Biol. 30: 2022–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buono C., Anzinger J. J., Amar M., Kruth H. S. 2009. Fluorescent pegylated nanoparticles demonstrate fluid-phase pinocytosis by macrophages in mouse atherosclerotic lesions. J. Clin. Invest. 119: 1373–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Acton S., Rigotti A., Landschulz K. T., Xu S., Hobbs H. H., Krieger M. 1996. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 271: 518–520 [DOI] [PubMed] [Google Scholar]
- 21.Vassiliou G., McPherson R. 2004. A novel efflux-recapture process underlies the mechanism of high-density lipoprotein cholesteryl ester-selective uptake mediated by the low-density lipoprotein receptor-related protein. Arterioscler. Thromb. Vasc. Biol. 24: 1669–1675 [DOI] [PubMed] [Google Scholar]
- 22.Rinninger F., Brundert M., Brosch I., Donarski N., Budzinski R. M., Greten H. 2001. Lipoprotein lipase mediates an increase in selective uptake of HDL-associated cholesteryl esters by cells in culture independent of scavenger receptor BI. J. Lipid Res. 42: 1740–1751 [PubMed] [Google Scholar]
- 23.Morrison J. R., Silvestre M. J., Pittman R. C. 1994. Cholesteryl ester transfer between high density lipoprotein and phospholipid bilayers. J. Biol. Chem. 269: 13911–13918 [PubMed] [Google Scholar]
- 24.Rinninger F., Deichen J. T., Jackle S., Windler E., Greten H. 1994. Selective uptake of high-density lipoprotein-associated cholesteryl esters and high-density lipoprotein particle uptake by human monocyte-macrophages. Atherosclerosis. 105: 145–157 [DOI] [PubMed] [Google Scholar]
- 25.Rinninger F., Brundert M., Jackle S., Kaiser T., Greten H. 1995. Selective uptake of low-density lipoprotein-associated cholesteryl esters by human fibroblasts, human HepG2 hepatoma cells and J774 macrophages in culture. Biochim. Biophys. Acta. 1255: 141–153 [DOI] [PubMed] [Google Scholar]
- 26.Seo T., Velez-Carrasco W., Qi K., Hall M., Worgall T. S., Johnson R. A., Deckelbaum R. J. 2002. Selective uptake from LDL is stimulated by unsaturated fatty acids and modulated by cholesterol content in the plasma membrane: role of plasma membrane composition in regulating non-SR-BI-mediated selective lipid transfer. Biochemistry. 41: 7885–7894 [DOI] [PubMed] [Google Scholar]
- 27.Brundert M., Heeren J., Bahar-Bayansar M., Ewert A., Moore K. J., Rinninger F. 2006. Selective uptake of HDL cholesteryl esters and cholesterol efflux from mouse peritoneal macrophages independent of SR-BI. J. Lipid Res. 47: 2408–2421 [DOI] [PubMed] [Google Scholar]
- 28.Ji A., Meyer J. M., Cai L., Akinmusire A., de Beer M. C., Webb N. R., van der Westhuyzen D. R. 2011. Scavenger receptor SR-BI in macrophage lipid metabolism. Atherosclerosis. 217: 106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seo T., Qi K., Chang C., Liu Y., Worgall T. S., Ramakrishnan R., Deckelbaum R. J. 2005. Saturated fat-rich diet enhances selective uptake of LDL cholesteryl esters in the arterial wall. J. Clin. Invest. 115: 2214–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rigotti A., Trigatti B. L., Penman M., Rayburn H., Herz J., Krieger M. 1997. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc. Natl. Acad. Sci. USA. 94: 12610–12615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Febbraio M., Abumrad N. A., Hajjar D. P., Sharma K., Cheng W., Pearce S. F., Silverstein R. L. 1999. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J. Biol. Chem. 274: 19055–19062 [DOI] [PubMed] [Google Scholar]
- 32.Coetzee G. A., Strachan A. F., van der Westhuyzen D. R., Hoppe H. C., Jeenah M. S., de Beer F. C. 1986. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J. Biol. Chem. 261: 9644–9651 [PubMed] [Google Scholar]
- 33.Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193: 265–275 [PubMed] [Google Scholar]
- 34.Bilheimer D. W., Eisenberg S., Levy R. I. 1972. The metabolism of very low density lipoprotein proteins. I. Preliminary in vitro and in vivo observations. Biochim. Biophys. Acta. 260: 212–221 [DOI] [PubMed] [Google Scholar]
- 35.Eckhardt E. R., Cai L., Sun B., Webb N. R., van der Westhuyzen D. R. 2004. High density lipoprotein uptake by scavenger receptor SR-BII. J. Biol. Chem. 279: 14372–14381 [DOI] [PubMed] [Google Scholar]
- 36.Yancey P. G., de la Llera-Moya M., Swarnakar S., Monzo P., Klein S. M., Connelly M. A., Johnson W. J., Williams D. L., Rothblat G. H. 2000. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J. Biol. Chem. 275: 36596–36604 [DOI] [PubMed] [Google Scholar]
- 37.Basu S. K., Goldstein J. L., Anderson G. W., Brown M. S. 1976. Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc. Natl. Acad. Sci. USA. 73: 3178–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirsch S., Austyn J. M., Gordon S. 1981. Expression of the macrophage-specific antigen F4/80 during differentiation of mouse bone marrow cells in culture. J. Exp. Med. 154: 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein J. L., Ho Y. K., Basu S. K., Brown M. S. 1979. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA. 76: 333–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khoo J. C., Miller E., McLoughlin P., Steinberg D. 1988. Enhanced macrophage uptake of low density lipoprotein after self-aggregation. Arteriosclerosis. 8: 348–358 [DOI] [PubMed] [Google Scholar]
- 41.Acton S. L., Scherer P. E., Lodish H. F., Krieger M. 1994. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J. Biol. Chem. 269: 21003–21009 [PubMed] [Google Scholar]
- 42.Endemann G., Stanton L. W., Madden K. S., Bryant C. M., White R. T., Protter A. A. 1993. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 268: 11811–11816 [PubMed] [Google Scholar]
- 43.Connelly M. A., Klein S. M., Azhar S., Abumrad N. A., Williams D. L. 1999. Comparison of class B scavenger receptors, CD36 and scavenger receptor BI (SR-BI), shows that both receptors mediate high density lipoprotein-cholesteryl ester selective uptake but SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J. Biol. Chem. 274: 41–47 [DOI] [PubMed] [Google Scholar]
- 44.Zimetti F., Weibel G. K., Duong M., Rothblat G. H. 2006. Measurement of cholesterol bidirectional flux between cells and lipoproteins. J. Lipid Res. 47: 605–613 [DOI] [PubMed] [Google Scholar]
- 45.Goldstein J. L., Dana S. E., Faust J. R., Beaudet A. L., Brown M. S. 1975. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J. Biol. Chem. 250: 8487–8495 [PubMed] [Google Scholar]
- 46.Harrison E. H., Bernard D. W., Scholm P., Quinn D. M., Rothblat G. H., Glick J. M. 1990. Inhibitors of neutral cholesteryl ester hydrolase. J. Lipid Res. 31: 2187–2193 [PubMed] [Google Scholar]
- 47.Tabas I., Boykow G. C., Tall A. R. 1987. Foam cell-forming J774 macrophages have markedly elevated acyl coenzyme A:cholesterol acyl transferase activity compared with mouse peritoneal macrophages in the presence of low density lipoprotein (LDL) despite similar LDL receptor activity. J. Clin. Invest. 79: 418–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoff H. F., Gaubatz J. W., Gotto A. M., Jr 1978. Apo B concentration in the normal human aorta. Biochem. Biophys. Res. Commun. 85: 1424–1430 [DOI] [PubMed] [Google Scholar]
- 49.Hoff H. F., Heideman C. L., Gaubatz J. W., Titus J. L., Gotto A. M., Jr 1978. Quantitation of apo B in human aortic fatty streaks. A comparison with grossly normal intima and fibrous plaques. Atherosclerosis. 30: 263–272 [DOI] [PubMed] [Google Scholar]
- 50.Takahashi Y., Zhu H., Xu W., Murakami T., Iwasaki T., Hattori H., Yoshimoto T. 2005. Selective uptake and efflux of cholesteryl linoleate in LDL by macrophages expressing 12/15-lipoxygenase. Biochem. Biophys. Res. Commun. 338: 128–135 [DOI] [PubMed] [Google Scholar]
- 51.Graf G. A., Connell P. M., van der Westhuyzen D. R., Smart E. J. 1999. The class B, type I scavenger receptor promotes the selective uptake of high density lipoprotein cholesterol ethers into caveolae. J. Biol. Chem. 274: 12043–12048 [DOI] [PubMed] [Google Scholar]
- 52.Maor I., Aviram M. 1994. Oxidized low density lipoprotein leads to macrophage accumulation of unesterified cholesterol as a result of lysosomal trapping of the lipoprotein hydrolyzed cholesteryl ester. J. Lipid Res. 35: 803–819 [PubMed] [Google Scholar]
- 53.Griffin E. E., Ullery J. C., Cox B. E., Jerome W. G. 2005. Aggregated LDL and lipid dispersions induce lysosomal cholesteryl ester accumulation in macrophage foam cells. J. Lipid Res. 46: 2052–2060 [DOI] [PubMed] [Google Scholar]
- 54.Connelly M. A., Kellner-Weibel G., Rothblat G. H., Williams D. L. 2003. SR-BI-directed HDL-cholesteryl ester hydrolysis. J. Lipid Res. 44: 331–341 [DOI] [PubMed] [Google Scholar]
- 55.Wang X., Collins H. L., Ranalletta M., Fuki I. V., Billheimer J. T., Rothblat G. H., Tall A. R., Rader D. J. 2007. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Invest. 117: 2216–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang M. D., Franklin V., Marcel Y. L. 2007. In vivo reverse cholesterol transport from macrophages lacking ABCA1 expression is impaired. Arterioscler. Thromb. Vasc. Biol. 27: 1837–1842 [DOI] [PubMed] [Google Scholar]
- 57.Wang M. D., Kiss R. S., Franklin V., McBride H. M., Whitman S. C., Marcel Y. L. 2007. Different cellular traffic of LDL-cholesterol and acetylated LDL-cholesterol leads to distinct reverse cholesterol transport pathways. J. Lipid Res. 48: 633–645 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.