

Abstract
The asymmetric unit of the title compound, C4H5N7, comprises two independent but virtually superimposable molecules. Each molecule is planar with the dihedral angles between the five-membered rings being 2.8 (3) and 2.1 (3)°. The crystal structure is formed by an extensive network of relatively strong N—H⋯N hydrogen-bond interactions. Individual molecules are arranged into supramolecular zigzag chains running parallel to [001] by way of the strongest N—H⋯N interactions. Adjacent chains are interconnected by rather long (D⋯A distances range from ca 3.00 to 3.03 Å) but highly directional (interaction angles above ca 173°) hydrogen bonds forming a supramolecular layer in the bc plane.
Related literature
For the synthesis of 1-butyl-3-methylimidazolium bromide and 1,2-diformylhydrazine, see: Liu et al. (2007 ▶); Parnham & Morris (2006 ▶). For the use of triazole molecules, see: Wang et al. (2012 ▶); Zhang et al. (2009 ▶). For previous research studies on crystal engineering approaches, see: Fernandes et al. (2011 ▶); Silva et al. (2011 ▶); Amarante et al. (2009 ▶); Paz & Klinowski (2007 ▶). For graph-set notation, see: Grell et al. (1999 ▶). For a description of the Cambridge Structural Database, see: Allen (2002 ▶).
Experimental
Crystal data
C4H5N7
M r = 151.15
Monoclinic,
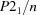
a = 13.765 (3) Å
b = 5.9378 (12) Å
c = 16.635 (4) Å
β = 112.914 (12)°
V = 1252.4 (5) Å3
Z = 8
Mo Kα radiation
μ = 0.12 mm−1
T = 293 K
0.10 × 0.05 × 0.03 mm
Data collection
Bruker X8 Kappa CCD APEXII diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1998 ▶) T min = 0.988, T max = 0.996
8003 measured reflections
2192 independent reflections
975 reflections with I > 2σ(I)
R int = 0.079
Refinement
R[F 2 > 2σ(F 2)] = 0.073
wR(F 2) = 0.226
S = 0.96
2192 reflections
217 parameters
8 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.52 e Å−3
Δρmin = −0.36 e Å−3
Data collection: APEX2 (Bruker, 2006 ▶); cell refinement: SAINT-Plus (Bruker, 2005 ▶); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: DIAMOND (Brandenburg, 2009 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812034691/tk5136sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812034691/tk5136Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812034691/tk5136Isup3.cdx
Supplementary material file. DOI: 10.1107/S1600536812034691/tk5136Isup4.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1A⋯N14i | 0.90 (1) | 2.43 (2) | 3.239 (6) | 150 (4) |
| N1—H1B⋯N4ii | 0.90 (1) | 2.11 (1) | 3.002 (6) | 173 (5) |
| N2—H2⋯N13i | 0.90 (1) | 1.93 (2) | 2.772 (6) | 155 (5) |
| N8—H8A⋯N7 | 0.90 (1) | 2.43 (2) | 3.264 (6) | 154 (4) |
| N8—H8B⋯N11iii | 0.90 (1) | 2.14 (1) | 3.034 (6) | 176 (5) |
| N10—H10⋯N6 | 0.90 (1) | 1.91 (2) | 2.777 (6) | 161 (5) |
Symmetry codes: (i) 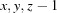 ; (ii)
; (ii)  ; (iii)
; (iii) 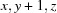 .
.
Acknowledgments
The authors gratefully acknowledge the Fundação para a Ciência e a Tecnologia (FCT, MEC, Portugal), the European Union, QREN, FEDER, COMPETE for financial support by the strategic projects PEst-C/CTM/LA0011/2011 (to CICECO) and Pest-C/EQB/LA0006/2011 (to REQUIMTE), the R&D projects PTDC/CTM/100357/2008 and PTDC/QUI-QUI/098098/2008 (FCOMP-01–0124-FEDER-010785), as well as the post-doctoral research grant SFRH/BPD/47566/2008 (to BL). They also wish to thank FCT for the specific funding towards the purchase of the single-crystal X-ray diffractometer.
supplementary crystallographic information
Comment
In the context of our interest in Crystal Engineering approaches (Wang et al. 2012; Fernandes et al.; 2011; Silva et al., 2011; Amarante, Gonçalves et al., 2009; Amarante et al., 2009; Paz & Klinowski, 2007), we are currently designing new triazole molecules which could be simultaneously employed in the construction of novel Metal-Organic Frameworks (MOFs) or organic crystals. We note that this type of molecule has received considerable interest in the synthesis of polynuclear complexes and in the preparation of MOFs (Zhang et al., 2009). A survey in the Cambridge structural Database (Allen, 2002) and in the literature revealed that the use of the asymmetrical bridging bitriazole molecule 5-amino-3-(1,2,4-triazol-4-yl)-1H-1,2,4-triazole (HAtrtr) has not been reported to date, either in MOFs nor in organic crystals. Furthermore, to the best of our knowledge, its crystal structure has not been reported. Following our recent efforts we were able to isolate good-quality single-crystals of the title compound as a minor secondary phase and here we wish to report its crystal structure at ambient temperature.
The asymmetric unit of the title compound (I) comprises two whole molecules (i.e., Z'=2) of HAtrtr as depicted in Fig. 1. We note that these two individual moieties are almost perfectly overlaid by assuming a combination of both inversion and molecular flexibility (maximum distance of ca 0.021 Å with RMS of ca 0.014 Å). Nevertheless, this feature is not described by crystal symmetry as the "head-to-tail" orientation of the molecules can not be described by the screw-axis parallel to the b-axis of the unit cell. Both molecules are almost planar with the dihedral angles between the 1,2,4-triazole rings being only of ca 2.8 and 2.1°. In addition, the medium planes of all non-hydrogen atoms of each molecule subtend an angle of just ca 7.1°, indicating that the molecules can also be envisaged as coplanar.
HAtrtr is rich in groups capable of forming strong N—H···N hydrogen bonding interactions (see Table 1 for further geometrical details), which direct the crystal packing features of the title compound. The most striking intermolecular interactions concern the double donation of hydrogen atoms from each 5-amino-3-(1,2,4-triazole moiety to the 1,2,4-triazole group of an adjacent molecule (N1—H1A···N14, N2—H2···N13, N8—H8A···N7 and N10—H10···N6), forming a R22(7) graph set motif (dashed yellow lines in Fig. 2) (Grell et al., 1999). This supramolecular motif constitutes the basis of the formation of a supramolecular zigzag tape parallel to the c axis, for which the repeating motif are the two molecules composing the asymmetric unit. Tapes are interconnected by additional N—H···N interactions (N1—H1B···N4 and N8—H8B···N11) which, despite being slightly long (dD···A ranging from 3.002 (6) to 3.034 (6) Å), are nevertheless highly directional with the interaction angles approaching linearity (of ca 173 and 176°). Noteworthy, these inter-chain connections are further strengthened by the presence of two weak C—H···N interactions (not represented): C3—H3···N3i with dC···N=3.389 (6) Å and <(CHN)=174°; C8—H8···N9ii with dC···N=3.435 (6) Å and <(CHN)=167° (symmetry codes: (i) x, 1 + y, z; (ii) x, -1 + y, z). The combination of these N—H···N and C—H···N contacts can be described by the R22(8) graph set motif.
The hydrogen bonding interactions connecting adjacent molecular units and summarized in the previous paragraph lead to the formation of a two-dimensional supramolecular layer placed in the bc plane as shown in Fig. 2. Individual layers close pack parallel to the a-axis of the unit cell mediated by offset π-π contacts between HAtrtr molecules (Fig. 3 and Table 2 for intercentroid distances). Noteworthy, the inter-layer distance along the a axis alternates: layers are disposed into pairs so to promote the aforementioned strong offset π-π contacts within one pair; connections between adjacent pairs of supramolecular layers are weaker (i.e. longer inter-centroid distances - not shown).
Experimental
3,5-Di(amino)-1,2,4-triazole (98% purity) and Mn(OAc)2.4H2O (99%+ purity) were purchased from Sigma-Aldrich and were used as received without further purification. 5-Amino-3-(1,2,4-triazol-4-yl)-1H-1,2,4-triazole and 1,2-diformylhydrazine were prepared by methods reported in the literature (Liu et al., 2007). 1-Butyl-3-methylimidazolium bromide ([BMI]Br) was also prepared according to literature procedures (Parnham & Morris, 2006) (pale-yellow oil; yield: 78°).
1,2-Diformylhydrazine (0.1 mol; 8.8095 g) and 3,5-di(amino)-1,2,4-triazole (0.05 mol; 4.9564 g) were mixed in a 25 mL Teflon-lined stainless-steel reaction vessel and heated at 170 °C for 3 days in a furnace. After this time, the vessel was allowed to cool slowly to ambient temperature yielding 5-amino-3-(1,2,4-triazol-4-yl)-1H-1,2,4-triazole (HAtrtr) as a white microcrystalline powder. The solid was then washed with copious amounts of water and ethanol and dried at ca 60 °C for 24 h. Yield: 6.85 g, 67.4%.
HAtrtr (0.2 mmol, 0.0407 g) and Mn(OAc)2.4H2O (1.0 mmol, 0.2451 g) were mixed with ca 0.50 g of [BMI]Br in a 25 mL teflon-lined stainless-steel reaction vessel. The resulting mixture was heated to 110 °C for 7 days. The vessel was then allowed to cool to ambient temperature at a rate of ca 1 °C/h. Small colourless platelets of HAtrtr were formed as a minor secondary product, whose crystal structure is reported in this manuscript.
Refinement
Hydrogen atoms bound to carbon were placed at their idealized positions with C—H = 0.93 Å and included in the final structural model in the riding-motion approximation with isotropic displacement parameters fixed at 1.2×Ueq(C).
Hydrogen atoms associated with nitrogen have been directly located from difference Fourier maps and were included in the structural model with the N—H and H···H (only for the –NH2 moieties) distances restrained to 0.900 (5) and 1.560 (5) Å, respectively, in order to ensure a chemically reasonable environment. These hydrogen atoms were refined using a riding-motion approximation with isotropic displacement parameters fixed at 1.5×Ueq(N).
Figures
Fig. 1.

Asymmetric unit of the title compound showing all non-hydrogen atoms represented as anisotropic displacement ellipsoids drawn at the 50% probability level and hydrogen atoms as small spheres with arbitrary radius. The labeling scheme for all non-hydrogen atoms is also provided.
Fig. 2.

Two-dimensional supramolecular network placed in the bc plane formed by the N—H···N hydrogen bonding interactions between adjacent molecular units. Zigzag supramolecular tapes running parallel to the c axis of the unit cell are based on R22(7) graph set motifs (yellow dashed lines) formed between the 5-amino-3-(1,2,4-triazole) moiety of one molecule and the 1,2,4-triazole group from the adjacent one. Connections between adjacent tapes (dashed green lines) are ensured by additional N—H···N interactions along [010]. For geometrical details on the represented hydrogen bonding interactions see Table 1.
Fig. 3.

(a) Schematic representation of the supramolecular π-π interactions between HAtrtr molecules belonging to adjacent supramolecular layers. For clarity one layer is represented as pink bonds and all hydrogen atoms have omitted. For inter-centroid distances see Table 2. (b) Crystal packing of the title compound viewed along [001] direction, clearly showing the planar nature of the two-dimensional supramolecular layer.
Crystal data
| C4H5N7 | F(000) = 624 |
| Mr = 151.15 | Dx = 1.603 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 823 reflections |
| a = 13.765 (3) Å | θ = 2.7–21.7° |
| b = 5.9378 (12) Å | µ = 0.12 mm−1 |
| c = 16.635 (4) Å | T = 293 K |
| β = 112.914 (12)° | Prism, yellow |
| V = 1252.4 (5) Å3 | 0.10 × 0.05 × 0.03 mm |
| Z = 8 |
Data collection
| Bruker X8 Kappa CCD APEXII diffractometer | 2192 independent reflections |
| Radiation source: fine-focus sealed tube | 975 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.079 |
| ω / φ scans | θmax = 25.0°, θmin = 3.7° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1998) | h = −16→16 |
| Tmin = 0.988, Tmax = 0.996 | k = −7→5 |
| 8003 measured reflections | l = −19→19 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.073 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.226 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.96 | w = 1/[σ2(Fo2) + (0.1189P)2] where P = (Fo2 + 2Fc2)/3 |
| 2192 reflections | (Δ/σ)max < 0.001 |
| 217 parameters | Δρmax = 0.52 e Å−3 |
| 8 restraints | Δρmin = −0.36 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.6176 (4) | −0.3457 (6) | −0.1686 (3) | 0.0465 (13) | |
| H1B | 0.625 (4) | −0.477 (4) | −0.140 (3) | 0.070* | |
| H1A | 0.625 (4) | −0.341 (7) | −0.2198 (16) | 0.070* | |
| N2 | 0.6244 (3) | 0.0473 (7) | −0.1487 (3) | 0.0431 (12) | |
| H2 | 0.624 (4) | 0.106 (9) | −0.1987 (18) | 0.065* | |
| N4 | 0.6274 (3) | 0.2015 (6) | −0.0855 (3) | 0.0443 (12) | |
| C2 | 0.6268 (4) | 0.0647 (8) | −0.0238 (3) | 0.0346 (12) | |
| N3 | 0.6228 (3) | −0.1582 (6) | −0.0393 (2) | 0.0326 (10) | |
| C1 | 0.6217 (4) | −0.1618 (8) | −0.1201 (3) | 0.0368 (12) | |
| N6 | 0.6346 (3) | 0.1685 (7) | 0.1884 (3) | 0.0477 (12) | |
| N7 | 0.6317 (3) | 0.3877 (7) | 0.1576 (3) | 0.0493 (12) | |
| C3 | 0.6290 (4) | 0.3720 (8) | 0.0783 (3) | 0.0449 (14) | |
| H3 | 0.6272 | 0.4936 | 0.0426 | 0.054* | |
| N5 | 0.6292 (3) | 0.1502 (6) | 0.0556 (2) | 0.0339 (10) | |
| C4 | 0.6328 (4) | 0.0325 (9) | 0.1264 (3) | 0.0420 (13) | |
| H4 | 0.6338 | −0.1238 | 0.1302 | 0.050* | |
| N10 | 0.6263 (3) | 0.2101 (6) | 0.3519 (3) | 0.0417 (12) | |
| H10 | 0.636 (4) | 0.166 (9) | 0.3037 (19) | 0.063* | |
| N11 | 0.6323 (3) | 0.0579 (6) | 0.4167 (3) | 0.0383 (11) | |
| C6 | 0.6227 (3) | 0.1927 (7) | 0.4747 (3) | 0.0312 (11) | |
| N9 | 0.6124 (3) | 0.4162 (6) | 0.4575 (3) | 0.0350 (10) | |
| C5 | 0.6144 (4) | 0.4198 (8) | 0.3774 (3) | 0.0341 (12) | |
| N14 | 0.6344 (3) | −0.1263 (7) | 0.6590 (3) | 0.0441 (11) | |
| N13 | 0.6262 (3) | 0.0938 (7) | 0.6861 (3) | 0.0487 (12) | |
| C7 | 0.6202 (4) | 0.2269 (8) | 0.6221 (3) | 0.0440 (14) | |
| H7 | 0.6136 | 0.3827 | 0.6229 | 0.053* | |
| N12 | 0.6250 (3) | 0.1088 (6) | 0.5547 (2) | 0.0342 (10) | |
| C8 | 0.6339 (4) | −0.1094 (8) | 0.5814 (3) | 0.0411 (13) | |
| H8 | 0.6390 | −0.2311 | 0.5480 | 0.049* | |
| N8 | 0.6036 (4) | 0.6025 (7) | 0.3278 (3) | 0.0509 (13) | |
| H8A | 0.605 (4) | 0.589 (7) | 0.2746 (15) | 0.076* | |
| H8B | 0.615 (4) | 0.737 (4) | 0.355 (3) | 0.076* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.083 (3) | 0.031 (2) | 0.035 (3) | 0.001 (2) | 0.033 (3) | −0.005 (2) |
| N2 | 0.084 (3) | 0.030 (2) | 0.026 (3) | −0.003 (2) | 0.032 (3) | −0.001 (2) |
| N4 | 0.083 (3) | 0.033 (2) | 0.028 (3) | −0.001 (2) | 0.033 (2) | 0.000 (2) |
| C2 | 0.045 (3) | 0.036 (3) | 0.026 (3) | 0.004 (2) | 0.017 (3) | −0.002 (2) |
| N3 | 0.051 (3) | 0.030 (2) | 0.022 (2) | 0.0005 (17) | 0.020 (2) | 0.0000 (17) |
| C1 | 0.050 (3) | 0.036 (3) | 0.030 (3) | 0.001 (2) | 0.022 (3) | −0.001 (2) |
| N6 | 0.075 (3) | 0.051 (3) | 0.025 (3) | 0.007 (2) | 0.028 (2) | −0.002 (2) |
| N7 | 0.066 (3) | 0.044 (3) | 0.044 (3) | −0.008 (2) | 0.028 (3) | −0.008 (2) |
| C3 | 0.067 (4) | 0.042 (3) | 0.030 (3) | −0.005 (3) | 0.022 (3) | −0.011 (2) |
| N5 | 0.052 (3) | 0.035 (2) | 0.021 (3) | 0.0012 (19) | 0.022 (2) | −0.0004 (18) |
| C4 | 0.059 (4) | 0.045 (3) | 0.027 (3) | 0.005 (2) | 0.022 (3) | 0.003 (3) |
| N10 | 0.073 (3) | 0.032 (3) | 0.029 (3) | 0.004 (2) | 0.029 (3) | 0.004 (2) |
| N11 | 0.067 (3) | 0.031 (2) | 0.023 (2) | −0.0016 (19) | 0.024 (2) | −0.0013 (19) |
| C6 | 0.043 (3) | 0.033 (3) | 0.022 (3) | 0.000 (2) | 0.017 (2) | 0.002 (2) |
| N9 | 0.055 (3) | 0.029 (2) | 0.026 (2) | 0.0003 (18) | 0.021 (2) | −0.0082 (18) |
| C5 | 0.050 (3) | 0.028 (3) | 0.028 (3) | 0.001 (2) | 0.020 (3) | 0.000 (2) |
| N14 | 0.061 (3) | 0.045 (3) | 0.030 (3) | 0.002 (2) | 0.022 (2) | 0.006 (2) |
| N13 | 0.073 (3) | 0.048 (3) | 0.029 (3) | −0.002 (2) | 0.024 (2) | 0.003 (2) |
| C7 | 0.069 (4) | 0.038 (3) | 0.033 (3) | −0.001 (2) | 0.029 (3) | 0.000 (3) |
| N12 | 0.052 (3) | 0.030 (2) | 0.024 (3) | −0.0001 (18) | 0.020 (2) | −0.0002 (18) |
| C8 | 0.059 (4) | 0.039 (3) | 0.032 (3) | 0.003 (2) | 0.026 (3) | 0.004 (2) |
| N8 | 0.097 (4) | 0.030 (2) | 0.036 (3) | 0.001 (2) | 0.037 (3) | 0.002 (2) |
Geometric parameters (Å, º)
| N1—C1 | 1.345 (6) | N10—C5 | 1.346 (6) |
| N1—H1B | 0.900 (5) | N10—N11 | 1.385 (5) |
| N1—H1A | 0.899 (5) | N10—H10 | 0.901 (5) |
| N2—C1 | 1.335 (6) | N11—C6 | 1.299 (5) |
| N2—N4 | 1.382 (5) | C6—N9 | 1.353 (5) |
| N2—H2 | 0.899 (5) | C6—N12 | 1.410 (5) |
| N4—C2 | 1.312 (6) | N9—C5 | 1.344 (6) |
| C2—N3 | 1.346 (5) | C5—N8 | 1.336 (6) |
| C2—N5 | 1.403 (5) | N14—C8 | 1.293 (6) |
| N3—C1 | 1.337 (6) | N14—N13 | 1.401 (5) |
| N6—C4 | 1.303 (6) | N13—C7 | 1.302 (6) |
| N6—N7 | 1.394 (6) | C7—N12 | 1.346 (6) |
| N7—C3 | 1.307 (6) | C7—H7 | 0.9300 |
| C3—N5 | 1.370 (6) | N12—C8 | 1.360 (6) |
| C3—H3 | 0.9300 | C8—H8 | 0.9300 |
| N5—C4 | 1.353 (6) | N8—H8A | 0.899 (5) |
| C4—H4 | 0.9300 | N8—H8B | 0.900 (5) |
| C1—N1—H1B | 114 (3) | C5—N10—N11 | 109.6 (4) |
| C1—N1—H1A | 123 (3) | C5—N10—H10 | 129 (4) |
| H1B—N1—H1A | 120 (4) | N11—N10—H10 | 121 (4) |
| C1—N2—N4 | 110.0 (4) | C6—N11—N10 | 100.6 (4) |
| C1—N2—H2 | 134 (4) | N11—C6—N9 | 118.7 (4) |
| N4—N2—H2 | 116 (4) | N11—C6—N12 | 120.8 (4) |
| C2—N4—N2 | 100.2 (4) | N9—C6—N12 | 120.5 (4) |
| N4—C2—N3 | 118.1 (4) | C5—N9—C6 | 100.6 (4) |
| N4—C2—N5 | 120.5 (4) | N8—C5—N9 | 125.8 (4) |
| N3—C2—N5 | 121.4 (4) | N8—C5—N10 | 123.7 (4) |
| C1—N3—C2 | 101.1 (4) | N9—C5—N10 | 110.5 (4) |
| N2—C1—N3 | 110.6 (4) | C8—N14—N13 | 106.2 (4) |
| N2—C1—N1 | 122.8 (5) | C7—N13—N14 | 106.9 (4) |
| N3—C1—N1 | 126.6 (5) | N13—C7—N12 | 110.9 (4) |
| C4—N6—N7 | 107.4 (4) | N13—C7—H7 | 124.5 |
| C3—N7—N6 | 106.8 (4) | N12—C7—H7 | 124.5 |
| N7—C3—N5 | 110.1 (5) | C7—N12—C8 | 104.6 (4) |
| N7—C3—H3 | 124.9 | C7—N12—C6 | 127.8 (4) |
| N5—C3—H3 | 124.9 | C8—N12—C6 | 127.6 (4) |
| C4—N5—C3 | 105.1 (4) | N14—C8—N12 | 111.4 (4) |
| C4—N5—C2 | 127.7 (4) | N14—C8—H8 | 124.3 |
| C3—N5—C2 | 127.2 (4) | N12—C8—H8 | 124.3 |
| N6—C4—N5 | 110.6 (4) | C5—N8—H8A | 120 (3) |
| N6—C4—H4 | 124.7 | C5—N8—H8B | 117 (3) |
| N5—C4—H4 | 124.7 | H8A—N8—H8B | 120 (4) |
| C1—N2—N4—C2 | 0.4 (5) | C5—N10—N11—C6 | 0.3 (5) |
| N2—N4—C2—N3 | −0.7 (6) | N10—N11—C6—N9 | −0.9 (6) |
| N2—N4—C2—N5 | −179.9 (4) | N10—N11—C6—N12 | −179.6 (4) |
| N4—C2—N3—C1 | 0.7 (6) | N11—C6—N9—C5 | 1.0 (6) |
| N5—C2—N3—C1 | 180.0 (4) | N12—C6—N9—C5 | 179.8 (4) |
| N4—N2—C1—N3 | 0.0 (6) | C6—N9—C5—N8 | 177.7 (5) |
| N4—N2—C1—N1 | 179.4 (4) | C6—N9—C5—N10 | −0.7 (5) |
| C2—N3—C1—N2 | −0.4 (5) | N11—N10—C5—N8 | −178.2 (5) |
| C2—N3—C1—N1 | −179.8 (5) | N11—N10—C5—N9 | 0.3 (5) |
| C4—N6—N7—C3 | −0.4 (5) | C8—N14—N13—C7 | 0.7 (5) |
| N6—N7—C3—N5 | 0.4 (6) | N14—N13—C7—N12 | −0.6 (6) |
| N7—C3—N5—C4 | −0.3 (5) | N13—C7—N12—C8 | 0.3 (6) |
| N7—C3—N5—C2 | 180.0 (4) | N13—C7—N12—C6 | −178.9 (4) |
| N4—C2—N5—C4 | −177.5 (5) | N11—C6—N12—C7 | 177.2 (4) |
| N3—C2—N5—C4 | 3.3 (7) | N9—C6—N12—C7 | −1.5 (7) |
| N4—C2—N5—C3 | 2.1 (7) | N11—C6—N12—C8 | −1.8 (7) |
| N3—C2—N5—C3 | −177.1 (4) | N9—C6—N12—C8 | 179.4 (4) |
| N7—N6—C4—N5 | 0.2 (5) | N13—N14—C8—N12 | −0.5 (5) |
| C3—N5—C4—N6 | 0.0 (5) | C7—N12—C8—N14 | 0.2 (5) |
| C2—N5—C4—N6 | 179.7 (4) | C6—N12—C8—N14 | 179.4 (4) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···N14i | 0.90 (1) | 2.43 (2) | 3.239 (6) | 150 (4) |
| N1—H1B···N4ii | 0.90 (1) | 2.11 (1) | 3.002 (6) | 173 (5) |
| N2—H2···N13i | 0.90 (1) | 1.93 (2) | 2.772 (6) | 155 (5) |
| N8—H8A···N7 | 0.90 (1) | 2.43 (2) | 3.264 (6) | 154 (4) |
| N8—H8B···N11iii | 0.90 (1) | 2.14 (1) | 3.034 (6) | 176 (5) |
| N10—H10···N6 | 0.90 (1) | 1.91 (2) | 2.777 (6) | 161 (5) |
Symmetry codes: (i) x, y, z−1; (ii) x, y−1, z; (iii) x, y+1, z.
Selected π-π contacts
| π-π interaction | d(Cg···Cg) |
| Cg(1)···Cg(2)i | 3.580 (3) |
| Cg(3)···Cg(4)ii | 3.700 (3) |
Symmetry codes: (i) 1-x, -y, -z; (ii) 1-x, -y, 1-z; Cg(1): centroid of the ring formed by N2, N3, N4, C1 and C2; Cg(2): centroid of the ring formed by N5, N6, N7, C3 and C4; Cg(3): centroid of the ring formed by N9, N10, N11, C5 and C6; Cg(4): centroid of the ring formed by N12, N13, N14, C7 and C8.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: TK5136).
References
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Amarante, T. R., Gonçalves, I. S. & Almeida Paz, F. A. (2009). Acta Cryst. E65, o1962–o1963. [DOI] [PMC free article] [PubMed]
- Brandenburg, K. (2009). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Bruker (2005). SAINT-Plus Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2006). APEX2 Bruker AXS Inc., Madison, Wisconsin, USA.
- Fernandes, J. A., Liu, B., Tomé, J. C., Cunha-Silva, L. & Almeida Paz, F. A. (2011). Acta Cryst. E67, o2073–o2074. [DOI] [PMC free article] [PubMed]
- Grell, J., Bernstein, J. & Tinhofer, G. (1999). Acta Cryst. B55, 1030–1043. [DOI] [PubMed]
- Liu, B., Zhang, X.-C. & Wang, Y.-F. (2007). Inorg. Chem. Commun. 10, 199–203.
- Parnham, E. R. & Morris, R. E. (2006). Chem. Mater. 18, 4882–4887.
- Paz, F. A. A. & Klinowski, J. (2007). Pure Appl. Chem. 79, 1097–1110.
- Sheldrick, G. M. (1998). SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Silva, P., Vieira, F., Gomes, A. C., Ananias, D., Fernandes, J. A., Bruno, S. M., Soares, R., Valente, A. A., Rocha, J. & Paz, F. A. A. (2011). J. Am. Chem. Soc. 133, 15120–15138. [DOI] [PubMed]
- Wang, N., Feng, Y.-C., Shi, W., Zhao, B., Cheng, P., Liao, D.-Z. & Yan, S.-P. (2012). CrystEngComm, 14, 2769–2278.
- Zhang, X.-C. & Liu, B. (2009). Inorg. Chem. Commun. 12, 808–810.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812034691/tk5136sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812034691/tk5136Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812034691/tk5136Isup3.cdx
Supplementary material file. DOI: 10.1107/S1600536812034691/tk5136Isup4.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report