

Abstract
The title compound, C13H9ClN2O3, is almost planar, showing a dihedral angle of 4.63 (6)° between the aromatic ring planes. The nitro group also lies in the plane, the C—C—N—O torsion angle being 6.7 (2)°. There is an intamolecular C—H⋯O hydrogen bond. The crystal structure features N—H⋯O(nitro) hydrogen bonds that link the molecules into zigzag chains extending along [010].
Related literature
For background information on aromatic polyimides, see: Yang et al. (1999 ▶); More et al. (2010 ▶); Litvinov et al., (2010 ▶); Sheng et al. (2009 ▶); Choi et al. (1992 ▶); Hsiao & Lin (2004 ▶); Li et al. (2007 ▶); Liaw et al. (2005 ▶). For related structures, see Saeed et al. (2011 ▶); Wardell et al. (2006 ▶).
Experimental
Crystal data
C13H9ClN2O3
M r = 276.67
Monoclinic,
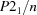
a = 9.6019 (7) Å
b = 13.0688 (10) Å
c = 9.6412 (7) Å
β = 103.853 (1)°
V = 1174.64 (15) Å3
Z = 4
Mo Kα radiation
μ = 0.33 mm−1
T = 130 K
0.49 × 0.20 × 0.18 mm
Data collection
Bruker SMART APEX diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 2004 ▶) T min = 0.855, T max = 0.943
10822 measured reflections
2808 independent reflections
2557 reflections with I > 2σ(I)
R int = 0.019
Refinement
R[F 2 > 2σ(F 2)] = 0.042
wR(F 2) = 0.116
S = 1.08
2808 reflections
172 parameters
H-atom parameters constrained
Δρmax = 0.76 e Å−3
Δρmin = −0.26 e Å−3
Data collection: SMART (Bruker, 2002 ▶); cell refinement: SAINT (Bruker, 2002 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and local programs.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812036082/bt6822sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812036082/bt6822Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812036082/bt6822Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C13—H13A⋯O1 | 0.95 | 2.26 | 2.859 (2) | 120 |
| N1—H1A⋯O3i | 0.88 | 2.29 | 3.1312 (17) | 159 |
Symmetry code: (i) 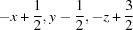 .
.
Acknowledgments
The authors acknowledge financial assistance for this project from the Higher Education Commission of Pakistan.
supplementary crystallographic information
Comment
Aromatic polyimides are distinguished as high performance polymers owing to excellent thermal, mechanical, and chemical properties (Yang et al., 1999, More et al., 2010). They are not only used as beneficial substitutes for metals or ceramics in presently used goods but also as new materials in novel technological applications (Litvinov et al., 2010). Nevertheless, infusibility and insolubility are some of the shortcomings due to the highly regular and rigid polymer backbones and the formation of intermolecular hydrogen bonding, causing deterioration in processability and applications (Sheng et al., 2009, Choi et al., 1992). In order to improve upon these drawbacks, recent research has aimed at improving their processability and solubility without an intense loss in the chemical, thermal, and mechanical properties. For this, improvement of solubility is targeted through diminishing the cohesive energy by lowering the interchain interactions. To achieve this, designing and synthesizing new diamines or dicarboxylic acids is proposed to produce a great variety of soluble and processable polyimides (Hsiao et al., 2004). Incorporating substituted pendant groups which reduce dense chain packing and interchain interactions increases the solubility of resulting polyimides (Liaw et al., 2005, Li et al., 2007). As part of our enduring interest in solubility of aromatic polyimides by structural modification, we are reporting a chloro substituted pendant group having inbuilt amide functionality, which enhances the solubility of polyimides without worsening the inherent properties of polyimides. The molecular structure of the title compound (Figure 1) is closely related to that of the bromo- (Saeed et al., 2011) and iodo-compound (Wardell et al., 2006). The two aromatic rings are almost coplanar with a dihedral angle of 4.63 (6)°, and the nitro group is also coplanar, the associated C4–C5–N2–O2 torsion angle is 6.7 (2)°. The molecular conformation is stabilized by a rather strong intramolecular C13–H···O1 bond. Crystal packing shows a strong intermolecular N1–H···O3(-x + 0.5, y - 0.5, -z + 1.5) hydrogen interaction with H···O3 2.29 Å and N–H···O 159.1° that links molecules into endless zigzag chains extended along the b axis (Figure 2).
Experimental
All the chemicals were of analytical grade and no further purification was carried out before their usage. 1.275 g (0.01 mole) of 4-chloroaniline, 25 ml dichloromethane and 1.39 ml of triethylamine were charged in 100 ml, three-necked, round-bottomed flask fitted with a condenser, a nitrogen inlet tube, a thermometer and a magnetic stirrer. The mixture was stirred at 273-278K for 30 minutes. A solution of 1.85 g (0.01mole) of 4-nitrobenzoyl chloride in 25 ml dichloromethane was added dropwise and stirring was continued for further 45 minutes under same conditions. The temperature was then raised to room temperature along with stirring for further 30 minutes. Product was precipitated by pouring the flask content into water. The product was filtered, washed with 5% NaOH solution, further washing with hot water was carried out and solid product was dried overnight under vacuum at 343K. The product was recrystallized from an ethanol-tetrahydrofuran(1:1)
Refinement
Hydrogen atoms were clearly derived from difference Fourier maps and then refined at idealized positions riding on the carbon or nitrogen atoms with isotropic displacement parameters Uiso(H) = 1.2U(C/Neq) and N—H 0.88 / C—H 0.95 Å.
Figures
Fig. 1.

Molecular structure of the title compound. Displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

Crystal packing viewed along [100] with hydrogen bonding pattern indicated as dashed lines. H-atoms not involved are omitted.
Crystal data
| C13H9ClN2O3 | F(000) = 568 |
| Mr = 276.67 | Dx = 1.564 Mg m−3 |
| Monoclinic, P21/n | Melting point: 141 K |
| Hall symbol: -P 2yn | Mo Kα radiation, λ = 0.71073 Å |
| a = 9.6019 (7) Å | Cell parameters from 5166 reflections |
| b = 13.0688 (10) Å | θ = 2.7–28.3° |
| c = 9.6412 (7) Å | µ = 0.33 mm−1 |
| β = 103.853 (1)° | T = 130 K |
| V = 1174.64 (15) Å3 | Prism, yellow |
| Z = 4 | 0.49 × 0.20 × 0.18 mm |
Data collection
| Bruker SMART APEX diffractometer | 2808 independent reflections |
| Radiation source: sealed tube | 2557 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.019 |
| φ and ω scans | θmax = 27.9°, θmin = 2.7° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 2004) | h = −11→12 |
| Tmin = 0.855, Tmax = 0.943 | k = −16→17 |
| 10822 measured reflections | l = −12→12 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.042 | Hydrogen site location: difference Fourier map |
| wR(F2) = 0.116 | H-atom parameters constrained |
| S = 1.08 | w = 1/[σ2(Fo2) + (0.0652P)2 + 0.6505P] where P = (Fo2 + 2Fc2)/3 |
| 2808 reflections | (Δ/σ)max < 0.001 |
| 172 parameters | Δρmax = 0.76 e Å−3 |
| 0 restraints | Δρmin = −0.26 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.86234 (4) | 0.07922 (3) | 0.98180 (4) | 0.02702 (14) | |
| O1 | 0.81457 (12) | 0.60046 (9) | 0.92017 (14) | 0.0290 (3) | |
| O2 | 0.38514 (13) | 1.02953 (9) | 0.81384 (14) | 0.0317 (3) | |
| O3 | 0.20735 (12) | 0.93858 (9) | 0.69700 (13) | 0.0268 (3) | |
| N1 | 0.61843 (14) | 0.49653 (10) | 0.87249 (14) | 0.0212 (3) | |
| H1A | 0.5241 | 0.4972 | 0.8458 | 0.025* | |
| N2 | 0.33274 (14) | 0.94804 (11) | 0.76579 (15) | 0.0216 (3) | |
| C1 | 0.68462 (17) | 0.58903 (12) | 0.88325 (17) | 0.0208 (3) | |
| C2 | 0.58628 (16) | 0.68089 (12) | 0.84829 (16) | 0.0193 (3) | |
| C3 | 0.64838 (16) | 0.77663 (12) | 0.88474 (17) | 0.0210 (3) | |
| H3A | 0.7479 | 0.7812 | 0.9287 | 0.025* | |
| C4 | 0.56698 (17) | 0.86526 (12) | 0.85776 (17) | 0.0215 (3) | |
| H4A | 0.6090 | 0.9305 | 0.8833 | 0.026* | |
| C5 | 0.42207 (16) | 0.85577 (11) | 0.79222 (16) | 0.0193 (3) | |
| C6 | 0.35741 (16) | 0.76187 (13) | 0.75241 (17) | 0.0217 (3) | |
| H6A | 0.2584 | 0.7578 | 0.7064 | 0.026* | |
| C7 | 0.44026 (17) | 0.67427 (12) | 0.78121 (17) | 0.0224 (3) | |
| H7A | 0.3977 | 0.6092 | 0.7553 | 0.027* | |
| C8 | 0.68397 (16) | 0.39913 (12) | 0.89933 (16) | 0.0198 (3) | |
| C9 | 0.59677 (17) | 0.31396 (12) | 0.85279 (17) | 0.0220 (3) | |
| H9A | 0.4996 | 0.3238 | 0.8032 | 0.026* | |
| C10 | 0.65021 (17) | 0.21567 (13) | 0.87802 (17) | 0.0225 (3) | |
| H10A | 0.5907 | 0.1580 | 0.8466 | 0.027* | |
| C11 | 0.79284 (17) | 0.20303 (12) | 0.95039 (17) | 0.0203 (3) | |
| C12 | 0.88114 (16) | 0.28597 (13) | 0.99628 (17) | 0.0213 (3) | |
| H12A | 0.9784 | 0.2756 | 1.0452 | 0.026* | |
| C13 | 0.82716 (17) | 0.38472 (13) | 0.97057 (17) | 0.0215 (3) | |
| H13A | 0.8875 | 0.4420 | 1.0014 | 0.026* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0277 (2) | 0.0195 (2) | 0.0341 (2) | 0.00400 (14) | 0.00786 (16) | 0.00411 (15) |
| O1 | 0.0156 (5) | 0.0224 (6) | 0.0469 (7) | −0.0015 (4) | 0.0033 (5) | 0.0064 (5) |
| O2 | 0.0278 (6) | 0.0180 (6) | 0.0468 (8) | −0.0001 (5) | 0.0041 (5) | −0.0024 (5) |
| O3 | 0.0182 (5) | 0.0259 (6) | 0.0344 (6) | 0.0029 (4) | 0.0029 (5) | −0.0003 (5) |
| N1 | 0.0142 (6) | 0.0192 (6) | 0.0288 (7) | 0.0001 (5) | 0.0023 (5) | 0.0008 (5) |
| N2 | 0.0194 (6) | 0.0216 (7) | 0.0248 (6) | 0.0008 (5) | 0.0069 (5) | 0.0004 (5) |
| C1 | 0.0177 (7) | 0.0216 (8) | 0.0232 (7) | −0.0007 (6) | 0.0051 (6) | 0.0026 (6) |
| C2 | 0.0177 (7) | 0.0203 (7) | 0.0202 (7) | −0.0005 (5) | 0.0053 (5) | 0.0019 (5) |
| C3 | 0.0153 (6) | 0.0227 (8) | 0.0238 (7) | −0.0022 (6) | 0.0024 (6) | 0.0009 (6) |
| C4 | 0.0199 (7) | 0.0195 (7) | 0.0247 (7) | −0.0033 (6) | 0.0046 (6) | −0.0005 (6) |
| C5 | 0.0183 (7) | 0.0195 (7) | 0.0208 (7) | 0.0021 (6) | 0.0063 (5) | 0.0006 (5) |
| C6 | 0.0146 (6) | 0.0243 (8) | 0.0248 (8) | −0.0014 (6) | 0.0023 (5) | −0.0006 (6) |
| C7 | 0.0188 (7) | 0.0187 (7) | 0.0286 (8) | −0.0029 (6) | 0.0033 (6) | −0.0014 (6) |
| C8 | 0.0191 (7) | 0.0194 (7) | 0.0218 (7) | 0.0014 (6) | 0.0067 (6) | 0.0012 (6) |
| C9 | 0.0158 (7) | 0.0249 (8) | 0.0242 (7) | 0.0002 (6) | 0.0026 (6) | −0.0010 (6) |
| C10 | 0.0195 (7) | 0.0218 (8) | 0.0267 (8) | −0.0037 (6) | 0.0063 (6) | −0.0026 (6) |
| C11 | 0.0203 (7) | 0.0183 (7) | 0.0237 (7) | 0.0027 (6) | 0.0083 (6) | 0.0027 (6) |
| C12 | 0.0159 (7) | 0.0239 (8) | 0.0239 (7) | 0.0012 (6) | 0.0042 (6) | 0.0024 (6) |
| C13 | 0.0181 (7) | 0.0215 (7) | 0.0247 (7) | −0.0009 (6) | 0.0048 (6) | 0.0001 (6) |
Geometric parameters (Å, º)
| Cl1—C11 | 1.7488 (16) | C5—C6 | 1.387 (2) |
| O1—C1 | 1.222 (2) | C6—C7 | 1.384 (2) |
| O2—N2 | 1.2207 (19) | C6—H6A | 0.9500 |
| O3—N2 | 1.2339 (17) | C7—H7A | 0.9500 |
| N1—C1 | 1.358 (2) | C8—C13 | 1.395 (2) |
| N1—C8 | 1.4161 (19) | C8—C9 | 1.400 (2) |
| N1—H1A | 0.8800 | C9—C10 | 1.383 (2) |
| N2—C5 | 1.466 (2) | C9—H9A | 0.9500 |
| C1—C2 | 1.515 (2) | C10—C11 | 1.390 (2) |
| C2—C3 | 1.394 (2) | C10—H10A | 0.9500 |
| C2—C7 | 1.399 (2) | C11—C12 | 1.382 (2) |
| C3—C4 | 1.387 (2) | C12—C13 | 1.391 (2) |
| C3—H3A | 0.9500 | C12—H12A | 0.9500 |
| C4—C5 | 1.389 (2) | C13—H13A | 0.9500 |
| C4—H4A | 0.9500 | ||
| C1—N1—C8 | 127.36 (13) | C5—C6—H6A | 120.7 |
| C1—N1—H1A | 116.3 | C6—C7—C2 | 120.39 (14) |
| C8—N1—H1A | 116.3 | C6—C7—H7A | 119.8 |
| O2—N2—O3 | 123.50 (14) | C2—C7—H7A | 119.8 |
| O2—N2—C5 | 118.73 (13) | C13—C8—C9 | 119.58 (14) |
| O3—N2—C5 | 117.76 (13) | C13—C8—N1 | 123.64 (14) |
| O1—C1—N1 | 123.89 (14) | C9—C8—N1 | 116.77 (13) |
| O1—C1—C2 | 120.44 (14) | C10—C9—C8 | 120.91 (14) |
| N1—C1—C2 | 115.67 (13) | C10—C9—H9A | 119.5 |
| C3—C2—C7 | 119.53 (14) | C8—C9—H9A | 119.5 |
| C3—C2—C1 | 116.68 (13) | C9—C10—C11 | 118.56 (14) |
| C7—C2—C1 | 123.79 (14) | C9—C10—H10A | 120.7 |
| C4—C3—C2 | 120.96 (14) | C11—C10—H10A | 120.7 |
| C4—C3—H3A | 119.5 | C12—C11—C10 | 121.51 (14) |
| C2—C3—H3A | 119.5 | C12—C11—Cl1 | 119.40 (12) |
| C3—C4—C5 | 117.96 (14) | C10—C11—Cl1 | 119.09 (12) |
| C3—C4—H4A | 121.0 | C11—C12—C13 | 119.80 (14) |
| C5—C4—H4A | 121.0 | C11—C12—H12A | 120.1 |
| C6—C5—C4 | 122.55 (14) | C13—C12—H12A | 120.1 |
| C6—C5—N2 | 118.36 (13) | C12—C13—C8 | 119.63 (14) |
| C4—C5—N2 | 119.09 (14) | C12—C13—H13A | 120.2 |
| C7—C6—C5 | 118.60 (14) | C8—C13—H13A | 120.2 |
| C7—C6—H6A | 120.7 | ||
| C8—N1—C1—O1 | −0.7 (3) | N2—C5—C6—C7 | −177.98 (14) |
| C8—N1—C1—C2 | −179.73 (14) | C5—C6—C7—C2 | −0.4 (2) |
| O1—C1—C2—C3 | −11.2 (2) | C3—C2—C7—C6 | −0.7 (2) |
| N1—C1—C2—C3 | 167.93 (14) | C1—C2—C7—C6 | −179.81 (15) |
| O1—C1—C2—C7 | 167.99 (16) | C1—N1—C8—C13 | 14.2 (3) |
| N1—C1—C2—C7 | −12.9 (2) | C1—N1—C8—C9 | −167.02 (15) |
| C7—C2—C3—C4 | 1.2 (2) | C13—C8—C9—C10 | 0.8 (2) |
| C1—C2—C3—C4 | −179.64 (14) | N1—C8—C9—C10 | −178.06 (14) |
| C2—C3—C4—C5 | −0.6 (2) | C8—C9—C10—C11 | −0.2 (2) |
| C3—C4—C5—C6 | −0.5 (2) | C9—C10—C11—C12 | −0.3 (2) |
| C3—C4—C5—N2 | 178.45 (14) | C9—C10—C11—Cl1 | −179.48 (12) |
| O2—N2—C5—C6 | 172.37 (15) | C10—C11—C12—C13 | 0.2 (2) |
| O3—N2—C5—C6 | −6.8 (2) | Cl1—C11—C12—C13 | 179.39 (12) |
| O2—N2—C5—C4 | −6.7 (2) | C11—C12—C13—C8 | 0.4 (2) |
| O3—N2—C5—C4 | 174.13 (14) | C9—C8—C13—C12 | −0.9 (2) |
| C4—C5—C6—C7 | 1.0 (2) | N1—C8—C13—C12 | 177.89 (14) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C13—H13A···O1 | 0.95 | 2.26 | 2.859 (2) | 120 |
| N1—H1A···O3i | 0.88 | 2.29 | 3.1312 (17) | 159 |
Symmetry code: (i) −x+1/2, y−1/2, −z+3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: BT6822).
References
- Bruker (2002). SMART and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Choi, K.-Y., Yi, M. H. & Choi, S.-K. (1992). J. Polym. Sci. Part A Polym. Chem. 30, 1583–1588.
- Hsiao, S.-H. & Lin, K.-H. (2004). Polymer, 45, 7877–7885.
- Li, W., Li, S., Zhang, Q. & Zhang, S. (2007). Macromolecules, 40, 8205–8211.
- Liaw, D. J., Chang, F. C., Leung, M., Chou, M. Y. & Muellen, K. (2005). Macromolecules, 38, 4024–4029.
- Litvinov, V. M., Persyn, O., Miri, V. & Lefebvre, J. M. (2010). Macromolecules, 43, 7668–7679.
- More, A. S., Pasale, S. K. & Wadgaonkar, P. P. (2010). Eur. Polym. J. 46, 557–567.
- Saeed, S., Jasinski, J. P. & Butcher, R. J. (2011). Acta Cryst. E67, o279. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2004). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheng, S.-R., Pei, X.-L., Huang, Z.-Z., Liu, X.-L. & Song, C.-S. (2009). Eur. Polym. J. 45, 230–236.
- Wardell, J. L., Low, J. N., Skakle, J. M. S. & Glidewell, C. (2006). Acta Cryst. B62, 931–943. [DOI] [PubMed]
- Yang, G., Jikei, M. & Kakimoto, M.-A. (1999). Macromolecules, 32, 2215–2220.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812036082/bt6822sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812036082/bt6822Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812036082/bt6822Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report