

Abstract
Ehlers-Danlos syndrome is a heterogeneous connective tissue condition characterized by varying degrees of skin hyperextensibility, joint hypermobility, and vascular fragility. Joint dislocations, musculoskeletal pain, atrophic scars, easy bleeding, vessel/viscera rupture, severe scoliosis, and obstetric complications may occur. These manifestations are secondary to abnormal collagen, with specific molecular defects in types I, III, and V collagen; they may also be related to tenascin-X, which has been identified in some patients. Ehlers-Danlos syndrome has been classified into 6 types, with variable degrees of joint instability, skin hyperextensibility, wound healing difficulty, and vascular fragility. Diagnosis begins with recognition of the signs and symptoms of global hypermobility and referring appropriate patients for genetic consultation. It is important to accurately identify patients with Ehlers-Danlos syndrome to initiate appropriate musculoskeletal treatment, optimize anesthetic and postoperative management, perform appropriate vascular screening, and help families address their concerns with other families and advocacy groups.
Keywords: Ehlers-Danlos, ligament laxity, instability
Ehlers-Danlos syndrome (EDS) is a connective tissue disorder characterized by varying degrees of skin hyperextensibility, joint hypermobility, and vascular fragility. The syndrome was given its name in 1949 after the descriptions from 2 physicians, Danish dermatologist Edvard Ehlers16 and French dermatologist Henri-Alexandre Danlos,13 were merged.30 Inheritance is autosomal dominant, autosomal recessive, or X-linked. EDS is likely underdiagnosed; therefore, it is difficult to estimate the incidence. The overall incidence may be approximately 1 in 5000 individuals worldwide.2
Differentiating patients with “loose joints” within the spectrum of normal versus patients with EDS is important because of the associated manifestations and complications that occur in patients with EDS. The purpose of this article is to describe the etiology and classification, discuss the criteria for diagnosis, review treatment principles, and summarize treatment outcomes. This information can be used to improve the musculoskeletal and overall care of athletes with EDS.
Etiology and Classification
In many cases, EDS is shown to be caused by mutations that result in qualitatively abnormal collagen, with manifestations in collagen-rich tissues such as the skin, ligaments, joints, and vessels.8 Types I, III, and V collagen may be affected, and specific mutations have been described for most types (Table 1).61
Table 1.
Classification and genetic causes of Ehlers-Danlos syndrome.5
| Type | Major Criteria | Minor Criteria | Inheritance | Genetic Defect |
|---|---|---|---|---|
| Classical | Skin hyperextensibility, widened atrophic scars, joint hypermobility | Smooth skin, molluscoid pseudotumors, subcutaneous spheroids, joint hypermobility, easy bruising, tissue fragility, positive family history | Autosomal dominant | Abnormal type V collagen (COL5A1 and COL5A2 genes) |
| Hypermobility | Skin involvement, generalized joint hypermobility | Recurrent joint dislocations, chronic limb pain, positive family history | Autosomal dominanta | Reduction in tenascin X (responsible for a small percentage of hypermobility cases) |
| Vascular | Thin skin; arterial, intestinal, or uterine fragility; extensive bruising; characteristic facial appearance | Acrogeria, small joint hypermobility, tendon or muscle rupture, clubfoot, early onset varicose veins, arteriovenous fistulae, pneumothorax, gingival recession, positive family history | Autosomal dominant | Structural defects of type III collagen (COL3A1 gene) |
| Kyphoscoliosis | Generalized joint laxity, severe muscle hypotonia, scoliosis, scleral fragility | Tissue fragility, easy bruising, arterial rupture, Marfanoid habitus, microcornea, osteopenia, positive family history | Autosomal recessive | Deficiency of lysyl hydroxylase (collagen modifying enzyme) |
| Arthrochalasia | Severe joint hypermobility with subluxations and congenital hip dislocations | Skin hyperextensibility, tissue fragility, easy bruising, muscle hypotonia, kyphoscoliosis, osteopenia | Autosomal dominant | Deficiency of chains in type I collagen (skipped exon 6 in COL1A1 or COL1A2 genes) |
| Dermatosparaxis | Skin fragility, redundant skin | Soft skin, easy bruising, premature rupture of fetal membranes, umbilical or inguinal hernias | Autosomal recessive | Deficiency of procollagen I (ADAMST2 gene) |
Most are autosomal dominant; tenascin X mutations are autosomal recessive.
The revised classification was developed in Villefranche in 1997 to refine the previous Berlin nosology for EDS (Table 1).5 This classification delineates 6 basic types of EDS: classical, hypermobile, vascular, kyphoscoliosis, arthrochalasia, and dermatosparaxis. The classical and hypermobile types account for more than 90% of cases,23 but it is not established which is most common.40 The vascular type is the third-most common and may affect 1 in 250 000 people.2 The kyphoscoliosis, arthrochalasia, and dermatosparaxis types are rare.23 Approximately 30 cases of the arthrochalasia type, 60 cases of the kyphoscoliosis type, and 12 cases of the dermatosparaxis types have been reported worldwide.2 Other types of EDS are even less common, occurring in one to a few families. Classification into a type is based on the presence of major and minor criteria. Major criteria, such as skin involvement and generalized joint hypermobility, are higher in diagnostic specificity because they are infrequent in other conditions and the general population. Minor criteria such as recurrent dislocations, chronic pain, and positive family history are less specific.5
Classical Type
The classical type is characterized by the presence of skin hyperextensibility with atrophic scars in individuals with joint hypermobility (Table 1).5 Inheritance is autosomal dominant. Mutations in the COL5A1 and COL5A2 genes responsible for type V collagen synthesis are present in 50% of cases.31
Hypermobility Type
The hypermobility type is also characterized by hypermobile joints and hyperextensile and/or smooth velvety skin (Table 1). However, skin hyperextensibility is less prominent and more variable than that in the classical type.5 More than 90% of patients are female.47
The hypermobility type is associated with the most debilitating musculoskeletal manifestations,47 and joint pain is reported by 100% of patients.42 Three clinical phases have been described.11 The hypermobility phase with marked ligamentous laxity begins during the first months of life.11 The pain phase starts during the second decade and is characterized by a relative decrease in hypermobility and the development of joint muscle and back pain.11 Chronic pain and joint instability progressively limit daily activities. The stiffness phase with progressive limitation of joint motion develops later.11
The mutation of the hypermobility type is not known in most cases. Mutations in the TNXB gene, resulting in the reduction of tenascin X, cause a small percentage of these cases.61 Tenascin X is a large extracellular matrix protein associated with collagen fibrils involved in extracellular matrix maintenance.60 A tenascin X deficiency results in fragmentation of elastic fibers, reduction of collagen, failure of fibroblasts to correctly deposit collagen, and loose packing of collagen fibrils.61 Most cases are autosomal dominant, but tenascin X mutations are autosomal recessive.
Vascular Type
The vascular type is characterized by fragile viscera and therefore has the most serious consequences, such as arterial, intestinal, or uterine rupture (Table 1). Other characteristics include thin and translucent skin, acrogeria (skin wrinkling in the hands and feet due to the loss of subcutaneous fat), small joint hypermobility, and a characteristic facial appearance (Table 1). A thin slender face with prominent bones, sunken cheeks, and lobeless ears is typical.5 Large joint hypermobility and skin hyperextensibility are not prominent features.
Arterial rupture is the most common cause of death.36 Spontaneous arterial rupture has a peak incidence in the third and fourth decades, and life expectancy averages 48 years.36 Midsized arteries of the thorax and abdomen are most commonly involved,39 but stroke from ruptured cerebral vessels may also occur. Hollow viscera of the gastrointestinal tract or uterus may rupture. Surgical repair is difficult because of tissue fragility.
Inheritance is autosomal dominant. The vascular type is the result of structural defects in the pro-alpha1(III) chain of collagen type III encoded by COL3A1.19,36 Type III collagen is abundant in major blood vessels46 and is also present in the gastrointestinal tract, uterus, and skin.
Kyphoscoliosis Type
The kyphoscoliosis type is characterized by severe hypotonia and scoliosis (Table 1). Inheritance is autosomal recessive. Scoliosis is secondary to muscle hypotonia and ligament laxity. Severe progression can occur, and operative treatment is often required.32 Hypotonia can be very pronounced, leading to gross motor development and loss of ambulation by the second and third decades.5 Blue sclera may be present. This type is secondary to mutations in the gene encoding the collagen-modifying enzyme lysyl hydroxylase.58 A diagnostic urine test is available.
Arthrochalasia Type
This type is characterized by severe hypermobility and joint dislocations (Table 1).5 Congenital bilateral hip dislocations are present in almost all patients, and joint hypermobility is severe. Other characteristics include short stature, large fontanelles, and typical facies with epicanthic folds, down-slanting palpebral fissures, puffy eyelids, blue sclera, and micrognathia.14 Inheritance is autosomal dominant. This type is secondary to specific mutations of COL1A1 and COL1A2.48 These mutations result in the deficient processing of the amino terminus of the precursor procollagen molecule to type I collagen.48
Dermatosparaxis Type
The dermatosparaxis type is characterized by fragile and sagging, redundant skin (Table 1).5 The skin fragility and bruising are substantial, but wound healing is not impaired. Inheritance is autosomal recessive. This type is caused by deficiency of procollagen I N-terminal peptidase due to mutations of the ADAMST2 gene.5
Diagnosis
Accurate diagnosis of EDS allows appropriate screening for vascular complications, enhances musculoskeletal management, and may reduce complications such as wound dehiscence and aortic or viscera rupture. However, many patients are undiagnosed or have a long delay in diagnosis. Clinicians must be able to recognize patients with global ligamentous laxity that require additional evaluation.
Diagnosis begins with a complete history and physical examination. Symptoms such as joint dislocations, subluxations, pain, easy bruising, easy bleeding, or poor wound healing may be elicited. Cardiovascular and family histories are important. The physical examination includes evaluation of range of motion, scoliosis, pes planus, and the skin. Blue sclera may be present. The skin is evaluated for a soft consistency, dystrophic scars,4 striae (Figure 1), brown discoloration secondary to hemosiderin deposition at areas of repetitive trauma and bruising,14 and subcutaneous spheroids.56 Skin hyperextensibility is tested in the volar surface of the forearm by pulling up until resistance is felt.4 Joint hypermobility is assessed using the Beighton criteria7 (Figures 2-6; Table 2). On this scale, a maximum of 9 points is possible, and a score of > 4 defines hypermobility. A cardiac examination and usually an echocardiogram are performed to assess valve and vessel abnormalities, including mitral valve prolapse and aortic dilatations. Autonomic dysfunction, such as postural orthostatic tachycardia, may be seen.
Figure 1.
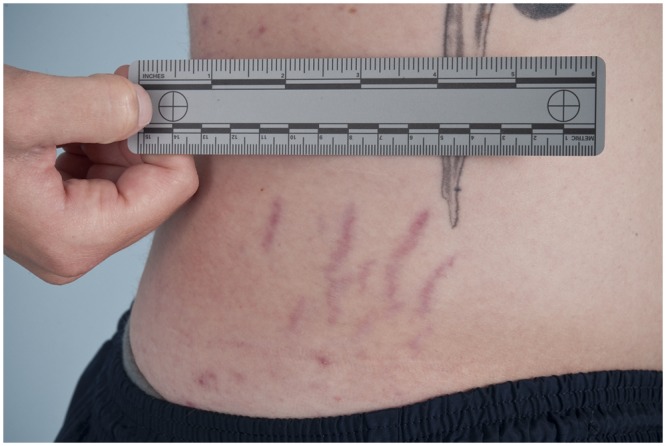
Abdominal striae in a male patient with Ehlers-Danlos syndrome.
Figure 2.
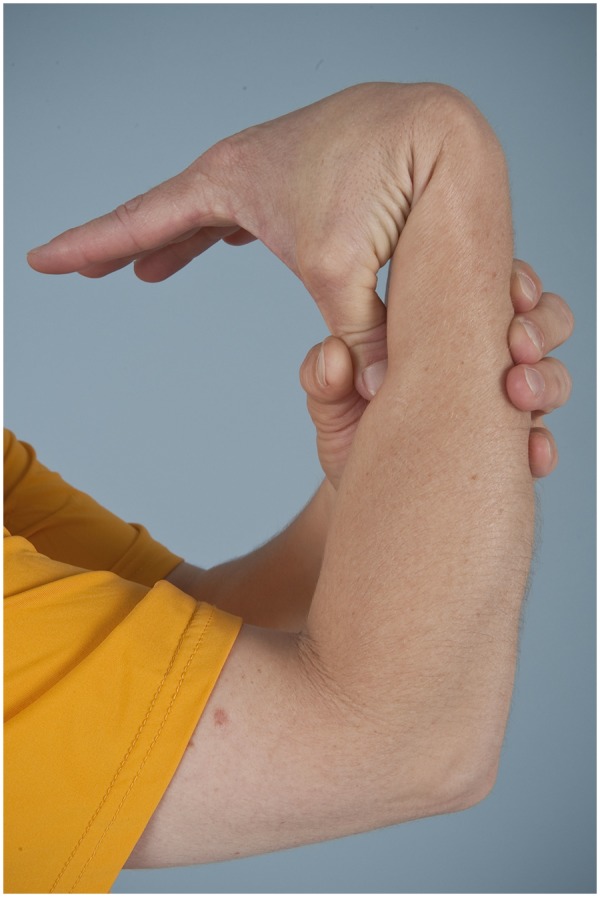
Apposition of the thumb to the flexor aspect of the forearm.
Figure 3.
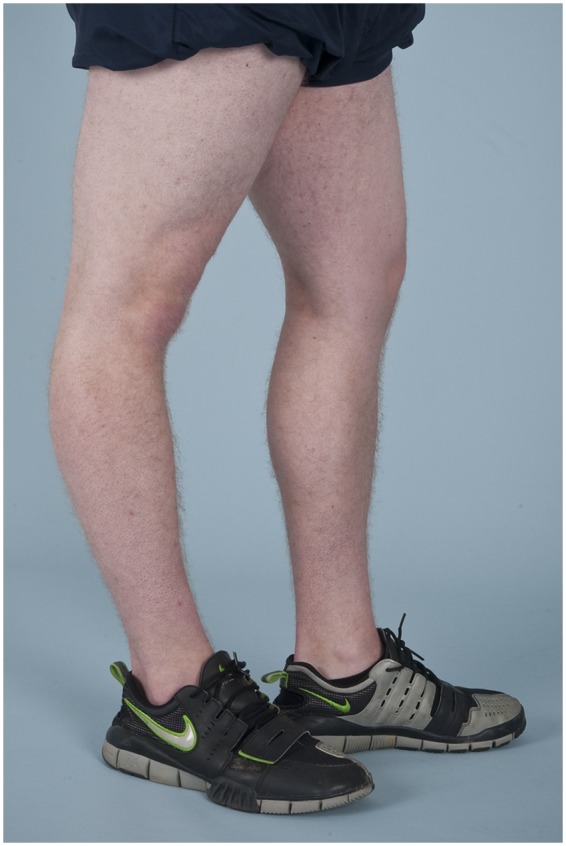
Knee hyperextension (genu recurvatum) in a male patient with Ehlers-Danlos syndrome.
Figure 4.

Female patient with Ehlers-Danlos syndrome placing palms flat on floor while maintaining knees in full extension.
Figure 5.

Dorsiflexion of the fifth digit beyond 90° in a male patient with Ehlers-Danlos syndrome.
Figure 6.
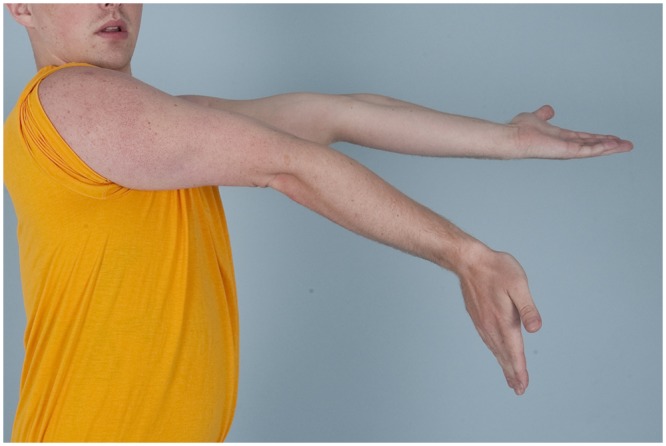
Elbow hyperextension in a male patient with Ehlers-Danlos syndrome.
Table 2.
Beighton criteria for generalized joint hypermobility.7
| Finding | Score (Points) |
|---|---|
| Passive apposition of the thumb to the flexor aspect of the forearm (1 point for each hand) | 2 |
| Passive dorsiflexion of the fifth digit beyond 90° (1 point for each hand) | 2 |
| Hyperextension of the elbow beyond 10° (1 point for each arm) | 2 |
| Hyperextension of the knees beyond 10° (1 point for each leg) | 2 |
| Forward flexion of the trunk with the knees extended, while the palms easily rest flat on the floor | 1 |
A genetics consultation should be strongly considered for hypermobile patients ≥ 4 years old with Beighton scores of ≥ 5, recurrent dislocations in the setting of a positive family history or minimal trauma, recurrent postoperative instability, or a history/family history of artery or visceral rupture. Although EDS is generally a clinical diagnosis based on clinical findings and family history, confirmatory genetic testing is available for the vascular, kyphoscoliosis, arthrochalasia, and dermatosparaxis types. Geneticists will typically use the revised classification to achieve a diagnosis.5 In general, the presence of 1 or more major criteria is necessary or highly indicative for diagnosis.5 For the vascular type, the presence of 2 major criteria is highly indicative of the diagnosis, and confirmatory genetic testing should be performed.5 For the kyphoscoliosis type, the presence of 3 major criteria in an infant is suggestive of the diagnosis, and genetic testing via a urinary analysis assay is warranted.5
Finally, an ophthalmology consultation may also be initiated if there are visual symptoms, severe involvement, concern for the kyphoscoliosis type, or a broader connective tissue diagnosis. Cardiology consultation may also be indicated for cardiac signs or symptoms.
Differential Diagnosis
The differential diagnosis for patients with hypermobility is extensive. Causes of hypermobility include benign joint hypermobility syndrome, Loeys-Dietz syndrome,50 and Marfan syndrome.50 Other conditions associated with ligamentous laxity that demonstrate other, more prominent manifestations include osteogenesis imperfecta, skeletal dysplasias, developmental syndromes, pseudoxanthoma elasticum,50 and cutis laxa syndrome.50
Benign Joint Hypermobility Syndrome
Generalized joint hypermobility is common, particularly in children. Patients with joint hypermobility and other findings may meet criteria for a diagnosis of benign joint hypermobility syndrome (BJHS). BJHS describes the combination of joint hypermobility associated with symptoms such as chronic musculoskeletal pain, joint instability, soft tissue injuries, osteopenia, fatigue, and anxiety. Diagnosis of BJHS is made by applying the revised Brighton criteria (Table 3). According to these criteria, a diagnosis of BJHS is made in the presence of 2 major criteria, 1 major and 2 minor criteria, 4 minor criteria, or 2 minor criteria in the presence of an unequivocally affected first-degree relative.22
Table 3.
| Major Criteria | Minor Criteria |
|---|---|
| Beighton score of ≥ 4/9 | Beighton score of 1,2, or 3 |
| Arthralgia for > 3 months in ≥ 4 joints | Arthralgia of ≥ 3 months in 1 to 3 joints or back pain of ≥ 3 months; spondylosis, spondylolysis, spondylolisthesis |
| Dislocation/subluxation in > 1 joint or in 1 joint on > 1 occasion | |
| Soft tissue rheumatism in ≥ 3 lesions (eg, epicondylitis, tenosynovitis, bursitis) | |
| Marfanoid habitus (tall, slim, span:height ratio of > 1.03, upper:lower segment ratio of < 0.89, arachnodactyly) | |
| Abnormal skin: striae, hyperextensibility, thin skin, papyraceous scarring | |
| Eye signs: drooping eyelids or myopia or antimongoloid slant | |
| Varicose veins, hernias, or uterine/rectal prolapse |
Differentiating between patients with hypermobility-type EDS, benign joint hypermobility syndrome, and generalized joint hypermobility (in children) can be difficult. Although generalized joint hypermobility in children is likely polygenetic in origin with environmental influences,50 BJHS and hypermobility-type EDS may actually be the same entity,21 as assumed by the BJHS criteria.22 Tofts et al50 suggested using the diagnosis joint hypermobility syndrome for any patient with symptomatic joint hypermobility in addition to any contributing connective tissue disorder. This approach allows appropriate attention to be placed on both the symptoms from hypermobility and those related to other manifestations.
Manifestations and Treatment
The diagnosis of EDS allows clinicians to take into consideration the underlying tissue abnormalities and modify the treatment regimens of the individual manifestations accordingly. These manifestations include joint hypermobility, propensity to increased bleeding, vascular pathology, and obstetric complications.
Joint Hypermobility
Hypermobility is secondary to an increase in connective tissue length and elasticity, which results in increased joint range of motion and distractibility.29 Complications of hypermobility include symptomatic subluxation, dislocation, and tendon ruptures. Nonoperative therapy is the primary treatment modality. The goal of rehabilitation is to enhance musculotendinous strength, neuromuscular coordination, and joint proprioception to maximize function, minimize symptoms, and improve joint stability.26,44 Therapy should be initiated with a protected range and gradually increased to avoid instability and pain.26 Prophylactic braces and pads may be helpful.
Shoulder instability in patients with EDS is common.25,29,45 Unlike patients with traumatic instability, patients with hyperlaxity and instability are more likely to experience recurrent subluxation than dislocation.29 The evaluation of shoulder instability has been well described.45 However, because shoulder symptoms in hypermobile patients are not always the result of instability, rotator cuff impingement and other conditions need to be considered.29 Nonoperative treatment should be maximized before surgery is indicated.25 When surgery is required, open inferior capsular shift is the gold standard, but the results of arthroscopic procedures may now achieve similar results.29
Patellar instability also may occur in patients with EDS. A survey of EDS patients found patellar instability in 57% of patients with the hypermobile type.1 Treatment of patellar instability is similar to shoulder instability; nonoperative measures should be maximized first. Reconstructive procedures must take into account the pathologic tissue laxity. Other knee symptoms may also occur, with one study showing an incidence of chronic knee pain in 85% of patients with EDS.55
Bleeding
Symptoms of easy bruising and bleeding tendencies range from bleeding after teeth brushing to excessive bleeding after minor trauma or surgery. These manifestations are secondary to poor connective tissue support of the blood vessels and direct involvement of the vessel wall, rather than to abnormal platelet function or coagulation defects. Laboratory coagulation evaluations (including prothrombin time, partial thromboplastin time, clotting factors, platelet function, and bleeding time) are usually within normal limits,14 except in some patients with the vascular type.3 The Hess test result is often positive.14 This test is performed by applying a blood pressure cuff to the upper arm and inflating it to a level between the diastolic and systolic values for 5 minutes. The development of more than 10 petechiae is abnormal and indicative of capillary fragility.27 Therefore, intramuscular injections are discouraged in patients with EDS because of the risk of ecchymosis.
Skin
Skin involvement is most severe in the classical type.38 Tissue fragility results in the splitting of the skin after relatively minor trauma. Lacerations and incisions heal slowly and may result in widened or atrophic scars. Prevention is the best approach for minimizing skin complications. A safe home environment (with protective pads over pressure points or on sharp furniture edges) should be created, particularly for younger patients.
Viscera Involvement
Manifestations of EDS are seen in multiple viscera, including the cardiovascular, respiratory, ocular, and gastrointestinal systems. Cardiovascular manifestations range from no involvement to varicose veins, aortic regurgitation, mitral valve prolapse, conduction disturbances, arterial aneurysms, and arterial rupture.27 An echocardiogram should be considered in any patient with EDS.14 Wenstrup et al57 found aortic root dilatation in 33% of those with classical-type EDS and in 26% of those with hypermobile-type EDS. Given the increase risk of aortic dilatation, regular imaging may be warranted, but imaging guidelines are not well established. Echocardiogram may have a limited ability to visualize the aorta, so other studies may also be considered. Visceral injury and rupture is a severe complication for patients with vascular-type EDS. This complication must be considered in affected patients with abdominal pain. Other gastrointestinal complications include diverticulosis, megacolon, hernias (hiatus, umbilical hernia, diaphragmatic, inguinal), gastroesophageal reflux disease, and esophagitis.27
Respiratory system manifestations include restrictive lung disease secondary to kyphoscoliosis, oral and laryngeal tissue fragility, and spontaneous pneumothorax.34 Ocular complications include strabismus, glaucoma, retinal detachment, retinal hemorrhages, and globe rupture.
Pain
Pain is common, particularly in classical- and hypermobile-type EDS.52 Stanitski et al47 reported joint pain in 88% of patients and limited walking in 61%, whereas Voermans et al52 reported pain in 90% and functional impairment in 87%. The pain may be secondary to hypermobility, dislocations, or previous surgery. In some patients, pain may be nociceptive (because of ongoing stimulation of nociceptors and related to primary joint damage).20 However, in other patients, pain is neuropathic (secondary to primary lesion or dysfunction in the nervous system).9 Complex regional pain syndrome has been reported in patients with classical- or hypermobility-type EDS.49
Pain management should be a prominent aspect of treatment. Nociceptive pain may respond better to anti-inflammatory treatment, and neuropathic pain may respond better to antidepressants, antiepileptics, and opioids.9 Conventional therapy modalities such as phonophoresis41 and electric stimulation12 may also be helpful. Assist devices may be required.47
Scoliosis
Scoliosis is another manifestation of EDS and may be present in patients with any type of EDS. However, deformity in patients with the kyphoscoliosis type may be particularly severe and develop after rapid progression. Jasiewicz et al28 reviewed 11 patients with EDS who underwent surgical treatment of kyphoscoliosis. The mean onset of deformity was 1.6 years (range, 1-2.5 years), and the mean age at the time of surgery was 13.8 years. The mean preoperative thoracic and lumbar Cobb angles were 109° (range, 83°-142°) and 75° (range, 0°-108°), respectively. Six patients underwent posterior fusion alone, and 5 underwent combined anterior and posterior fusion. Surgery was associated with an increased risk (relative to idiopathic scoliosis) of postoperative sagittal imbalance: 4 of 11 patients required revision surgery because of imbalance or instrumentation failure.28
In an evaluation of patients with hypermobile and classical EDS, 52% of 58 patients had scoliosis.47 Curves were minimal to moderate, and no patient had a curve > 50°. Substantial back or neck pain was present in 82% of patients with scoliosis and in 71% of patients without spinal deformity.
Vascular complications may be increased (relative to the general population) during anterior spine surgery.59 Yang et al59 reported vascular complications after anterior spine surgery in 3 patients with classical- and vascular-type EDS. Sources of bleeding included a torn external iliac artery, avulsed segmental arteries, and a compromised aorta. All vessels required repair with sutures and synthetic grafts. To minimize complications, McMaster32 recommended using hypotensive anesthesia, avoiding blunt dissection, using electrocautery liberally, carefully dissecting segmental arteries, and limiting discectomies.
Obstetrics
EDS may be associated with an increased risk of complications during pregnancy and delivery.54 The most severe complications are seen in patients with the vascular type, and pregnancy may be life-threatening to the mother because of aorta or uterus rupture. The pregnancy-related maternal mortality for each pregnancy in such patients is 20%.35 Overall, pregnancy in patients with classical- and hypermobility-type EDS is generally well tolerated.54
Neuromuscular Involvement
Mild to moderate neuromuscular involvement is common in patients with EDS.53 Examples of neuromuscular involvement include muscle weakness, reduction of vibration sense, impaired proprioceptive sensory feedback, abnormal needle electromyography, and mild myopathic features on muscle biopsy.53 Deficiencies in postural control have also been reported,17 and postural training with emphasis on core lumbar stabilization may be helpful in management.
A correlation between EDS and Chiari I malformations has been described.33 Patients who exhibit frequent headaches, dizziness, blurred vision, or other symptoms of Chiari malformations should be evaluated for this association.
Other Considerations
In addition to addressing the specific manifestations of EDS, other considerations need to be taken into account, such as ensuring multidisciplinary care, athletic participation, and surgical issues.
General Treatment
EDS can be debilitating to patients and challenging to manage. Multidisciplinary treatment is required, including mental health support for declining psychological well-being. Patients often appreciate providers who treat them as partners in treatment, listen to information they discover, and provide connections to support groups.18 Support groups include the Ehlers-Danlos National Foundation and local groups. Internet resources include the National Library of Medicine, the Ehlers-Danlos Syndrome Network, and the Ehlers-Danlos National Foundation.
Athletic Participation
Sports participation requires careful consideration because bruising, bleeding, dislocations, and joint injury may result. Cardiac, thoracic, and abdominal vascular imaging should be considered before clearing the athlete for most competitive sports. In addition, avoiding collision sports is recommended for all patients with pronounced bruising, vascular-type EDS, or moderate to severe forms of other types.44 Pool and low-impact activities are preferred to high-impact activities that increase joint stress. Many patients choose sports that benefit from flexibility, such as cheerleading, gymnastics, ballet, and swimming. Protection with pads and braces may prevent lacerations. Overhead and collision athletes may benefit from a maintenance rotator cuff and scapular stabilization program. To enhance joint stability, patients with hypermobility type may benefit from low resistance exercise for improving muscle strength.
Considerations During Surgery
There are several important perioperative considerations in patients with EDS. Cardiac evaluation with possible echocardiogram should be completed before surgery,15 and prophylactic antibiotics for mitral valve prolapse should be administered as needed. A blood type and cross-match should be ordered, and an evaluation for clotting abnormalities should be considered.30 Concerns regarding the safety of epidural anesthesia for patients with EDS have been raised, given the increased risks of bleeding15 and potential decreased efficacy (particularly in hypermobile patients).24 However, epidural blocks have been shown to be safe and efficacious for obstetric patients with EDS.10
In the operating room, careful positioning is required to prevent injury, subluxation, or dislocation.30 Needle sticks should be minimized, and consideration should be given to placing central monitoring devices under ultrasound guidance.30 Instrumentation of the nose, mouth, or esophagus is carefully performed to avoid bleeding and airway compromise. Hyperopic intubation should be considered.30 Low airway pressures are maintained to avoid iatrogenic pneumothorax, and spontaneous ventilation is preferred.30 Hypertension is avoided to prevent vessel damage.30
Intraoperatively, meticulous hemostasis is necessary to decrease vascular complications. Special care is taken during retraction, ligation, and vessel repair. Tourniquets may be used, but time and pressure should be kept to a minimum to avoid vessel rupture.51 Postoperatively, wounds should be followed closely for hematoma development, and anticoagulation should be considered with caution.
Particular attention must be given to the skin. Skin retractors should be minimized,55 and surgical closure may be difficult because sutures may tear out of the wound edges.6 Multilayered closure is helpful with wound closure strips, and nylon sutures are preferred over staples.55 Sutures may need to be left in place twice as long as normal.6 Brief immobilization may also prevent additional tearing.37 Wound dehiscence in the vascular type may be particularly severe. Wound-healing complications are common and may contribute to an elevated infection rate, which may be 6 times higher than normal.55
Postoperatively, multimodal pain management may be helpful. Rehabilitation is individualized. Range of motion is less aggressive than standard protocols to avoid stretching of repaired or reconstructed tissue, and stiffness does not typically develop.
Surgery may be associated with increased complications in patients with EDS relative to the general population.43,55 In a survey of 44 patients with EDS who underwent 214 procedures on the shoulder, elbow, knee, and ankle, pain-relieving procedures were successful in 50%, shoulder stabilization procedures were successful in 47%, and knee stabilization procedures were successful in 54%.55 In addition, 11% developed wound-healing problems and 6% had postoperative infections.55 Rose et al43 reported the results of 12 total knee replacements (8 for instability and 4 for arthritis) in patients with EDS; a 70% satisfaction rate (vs 90% in patients without EDS) was reported because of continued instability or pain.43
Case Reports
Two cases are provided to demonstrate the importance of establishing a diagnosis of EDS to improve treatment outcomes.
Case 1
A 41-year-old female softball player on active military duty sustained bilateral anterior cruciate ligament injuries as a result of noncontact twisting events during a rugby game. She underwent bilateral reconstructions with anterior tibial tendon allografts staged 5 months apart without acute complications. Postoperatively, she noted increasing severity and frequency of instability in both knees without a history of interval trauma and was referred for a second opinion. On closer evaluation, her medical history was significant for migraines, Raynaud phenomenon, polyarthralgia with effusions, and numerous drug allergies. Her knee examination was consistent with bilateral anterior cruciate ligament instability. In addition, examination also revealed hypermobile joints, a Beighton score of 9 out of 9 (Figures 2-4), widened scars, and soft skin. A genetics consultation was obtained, and she was diagnosed with classical-type EDS. The patient underwent staged revision bilateral anterior cruciate ligament reconstructions with bone-tendon-bone autografts, followed by a conservative rehabilitation program. Bone-tendon-bone autografts were selected because of the advantages of bone healing to maximize stability. Although allograft may intuitively seem to be a better choice for patients with EDS, the allograft may be from older donors, and the patient’s healing response may be abnormal because of the underlying disease. In addition, the ligamentization process that occurs after graft implantation may offset any potential benefit of allograft use. The patient’s instability resolved, but she had persistent knee pain and occasional swelling postoperatively.
Case 2
A 22-year-old right-hand-dominant male overhead athlete on active military duty presented with chronic right shoulder pain and a sensation of instability for 2 years despite attempted treatment with physical therapy. There was no history of a discrete injury or overuse. He reported increasing pain in all weight-bearing joints, particularly the ankles and knees. His history was significant for migraine headaches, depression, and palpitations with atypical chest pain. Family history was positive for “loose joints” in his sister. On physical examination, he had abdominal striae (Figure 1), a Beighton score of 9 out of 9 (Figures 5 and 6), multidirectional instability of both shoulders, and a positive O’Brien sign. Radiographs were unremarkable. Magnetic resonance imaging showed a posterior inferior labral tear. A genetics consultation was obtained, and he was diagnosed with EDS. Surgery was indicated for persistent symptomatic multidirectional instability. He underwent an arthroscopic labral reconstruction and capsular placation, followed by a conservative rehabilitation program. The patient’s shoulder symptoms improved markedly.
Conclusion
In summary, EDS is a heterogeneous connective tissue condition consisting primarily of skin hyperextensibility, ligamentous laxity, and vascular fragility. The genetic cause of several types has been identified, with mutations affecting collagen types I, II, and III, as well as tenascin X. Providers should remember primary features of this syndrome and make appropriate referrals to obtain a diagnosis. Because manifestations and complications can be severe, early diagnosis can improve management and minimize complications. Furthermore, providers can play an important role in counseling patients regarding exercise and sports activity to minimize episodes of joint instability.

References
- 1. Ainsworth SR, Aulicino PL. A survey of patients with Ehlers-Danlos syndrome. Clin Orthop Relat Res. 1993;286:250-256 [PubMed] [Google Scholar]
- 2. Anonymous Ehlers-Danlos syndrome. Genetics Home Reference. http://ghr.nlm.nih.gov/condition/ehlers-danlos-syndrome Accessed April 1, 2012
- 3. Anstey A, Wilkinson JD, Pope FM. Ehlers-Danlos syndrome with recurrent bruising. J R Soc Med. 1990;83:800-801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beighton P. The Ehlers-Danlos syndromes. In: Beighton P, ed. McKusick’s Heritable Disorders of Connective Tissue. St. Louis, MO: Mosby; 1993:189-251 [Google Scholar]
- 5. Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Am J Med Genet. 1998;77:31-37 [DOI] [PubMed] [Google Scholar]
- 6. Beighton P, Horan FT. Surgical aspects of the Ehlers-Danlos syndrome: a survey of 100 cases. Br J Surg. 1969;56:255-259 [DOI] [PubMed] [Google Scholar]
- 7. Beighton P, Solomon L, Soskolne CL. Articular mobility in an African population. Ann Rheum Dis. 1973;32:413-418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Byers PH. Ehlers-Danlos syndrome: recent advances and current understanding of the clinical and genetic heterogeneity. J Invest Dermatol. 1994;103:47S-52S [DOI] [PubMed] [Google Scholar]
- 9. Camerota F, Celletti C, Castori M, Grammatico P, Padua L. Neuropathic pain is a common feature in Ehlers-Danlos syndrome [letter]. J Pain Symptom Manage. 2010 [DOI] [PubMed] [Google Scholar]
- 10. Campbell N, Rosaeg OP. Anesthetic management of a parturient with Ehlers Danlos syndrome type IV. Can J Anaesth. 2002;49:493-496 [DOI] [PubMed] [Google Scholar]
- 11. Castori M, Camerota F, Celletti C, et al. Natural history and manifestations of the hypermobility type Ehlers-Danlos syndrome: a pilot study on 21 patients. Am J Med Genet A. 2010;152:556-564 [DOI] [PubMed] [Google Scholar]
- 12. Costello CT, Jeske AH. Iontophoresis: applications in transdermal medication delivery. Phys Ther. 1995;75:554-563 [DOI] [PubMed] [Google Scholar]
- 13. Danlos PM. Un cas de cutis laxa avec tumeurs par contusion chronique des coudes et des et des genoux (xanthome juvenile pseudo-diabetique de MM. Hallopeau et Mace de Lepinay). Bull Soc Fr Dermatol Syphiligr. 1908;19:70-72 [Google Scholar]
- 14. De Paepe A, Malfait F. Bleeding and bruising in patients with Ehlers-Danlos syndrome and other collagen vascular disorders. Br J Haematol. 2004;127:491-500 [DOI] [PubMed] [Google Scholar]
- 15. Dolan P, Sisko F, Riley E. Anesthetic considerations for Ehlers-Danlos syndrome. Anesthesiology. 1980;52:266-269 [DOI] [PubMed] [Google Scholar]
- 16. Ehlers E. Cutis Laxa, Neigung zu Hoemorrhagien in der Haut, Lockerung mehrerer Artikulationen. Dermatol Zr. 1901;8:173 [Google Scholar]
- 17. Galli M, Rigoldi C, Celletti C, et al. Postural analysis in time and frequency domains in patients with Ehlers-Danlos syndrome. Res Dev Disabil. 2011;32:322-325 [DOI] [PubMed] [Google Scholar]
- 18. Gawthrop F, Mould R, Sperritt A, Neale F. Ehlers-Danlos syndrome. BMJ. 2007;335:448-450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Germain DP. Clinical and genetic features of vascular Ehlers-Danlos syndrome. Ann Vasc Surg. 2002;16:391-397 [DOI] [PubMed] [Google Scholar]
- 20. Grahame R. Heritable disorders of connective tissue. Baillieres Best Pract Res Clin Rheumatol. 2000;14:345-361 [DOI] [PubMed] [Google Scholar]
- 21. Grahame R. Joint hypermobility and genetic collagen disorders: are they related? Arch Dis Child. 1999;80:188-191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27:1777-1779 [PubMed] [Google Scholar]
- 23. Hagberg C, Berglund B, Korpe L, Andersson-Norinder J. Ehlers-Danlos syndrome (EDS) focusing on oral symptoms: a questionnaire study. Orthod Craniofac Res. 2004;7:178-185 [DOI] [PubMed] [Google Scholar]
- 24. Hakim AJ, Grahame R, Norris P, Hopper C. Local anaesthetic failure in joint hypermobility syndrome [letter]. J R Soc Med. 2005;98:84-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hawkins RJ, Koppert G, Johnston G. Recurrent posterior instability (subluxation) of the shoulder. J Bone Joint Surg Am. 1984;66:169-174 [PubMed] [Google Scholar]
- 26. Hinton RY. Case study: rehabilitation of multiple joint instability associated with Ehlers-Danlos syndrome. J Orthop Sports Phys Ther. 1986;8:193-198 [DOI] [PubMed] [Google Scholar]
- 27. Hollister DW. Heritable disorders of connective tissue: Ehlers-Danlos syndrome. Pediatr Clin North Am. 1978;25:575-591 [DOI] [PubMed] [Google Scholar]
- 28. Jasiewicz B, Potaczek T, Tesiorowski M, Lokas K. Spine deformities in patients with Ehlers-Danlos syndrome, type IV: late results of surgical treatment. Scoliosis. 2010;5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson SM, Robinson CM. Shoulder instability in patients with joint hyperlaxity. J Bone Joint Surg Am. 2010;92:1545-1557 [DOI] [PubMed] [Google Scholar]
- 30. Johnston BA, Occhipinti KE, Baluch A, Kaye AD. Ehlers-Danlos syndrome: complications and solutions concerning anesthetic management. Middle East J Anesthesiol. 2006;18:1171-1184 [PubMed] [Google Scholar]
- 31. Malfait F, Wenstrup RJ, De Paepe A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet Med. 2010;12:597-605 [DOI] [PubMed] [Google Scholar]
- 32. McMaster MJ. Spinal deformity in Ehlers-Danlos syndrome: five patients treated by spinal fusion. J Bone Joint Surg Br. 1994;76:773-777 [PubMed] [Google Scholar]
- 33. Milhorat TH, Bolognese PA, Nishikawa M, McDonnell NB, Francomano CA. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue. J Neurosurg Spine. 2007;7:601-609 [DOI] [PubMed] [Google Scholar]
- 34. Neldner KH. Inherited elastic tissue malformations. In: Arndt KA, LeBoit PE, Robinson JK, Wintroub BU, eds. Cutaneous Medicine and Surgery. Philadelphia, PA: Saunders; 1996:1770-1779 [Google Scholar]
- 35. Peaceman AM, Cruikshank DP. Ehlers-Danlos syndrome and pregnancy: association of type IV disease with maternal death. Obstet Gynecol. 1987;69:428-431 [PubMed] [Google Scholar]
- 36. Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673-680 [DOI] [PubMed] [Google Scholar]
- 37. Pinnell SR. The skin in Ehlers-Danlos syndrome [letter]. J Am Acad Dermatol. 1987;16:399-400 [DOI] [PubMed] [Google Scholar]
- 38. Pope FM. Ehlers-Danlos syndrome. Baillieres Clin Rheumatol. 1991;5:321-349 [DOI] [PubMed] [Google Scholar]
- 39. Prahlow JA, Wagner SA. Death due to Ehlers-Danlos syndrome type IV. Am J Forensic Med Pathol. 2005;26:78-82 [DOI] [PubMed] [Google Scholar]
- 40. Proske S, Hartschuh W, Enk A, Hausser I. [Ehlers-Danlos syndrome: 20 years experience with diagnosis and classification at the University skin clinic of Heidelberg]. J Dtsch Dermatol Ges. 2006;4:308-318 [DOI] [PubMed] [Google Scholar]
- 41. Reid DC, Cummings GE. Efficiency of ultrasound coupling agents. Physiotherapy. 1977;63:255-257 [PubMed] [Google Scholar]
- 42. Rombaut L, Malfait F, Cools A, De Paepe A, Calders P. Musculoskeletal complaints, physical activity and health-related quality of life among patients with the Ehlers-Danlos syndrome hypermobility type. Disabil Rehabil. 2010;32:1339-1345 [DOI] [PubMed] [Google Scholar]
- 43. Rose PS, Johnson CA, Hungerford DS, McFarland EG. Total knee arthroplasty in Ehlers-Danlos syndrome. J Arthroplasty. 2004;19:190-196 [DOI] [PubMed] [Google Scholar]
- 44. Schroeder EL, Lavallee ME. Ehlers-Danlos syndrome in athletes. Curr Sports Med Rep. 2006;5:327-334 [DOI] [PubMed] [Google Scholar]
- 45. Silliman JF, Hawkins RJ. Classification and physical diagnosis of instability of the shoulder. Clin Orthop Relat Res. 1993;291:7-19 [PubMed] [Google Scholar]
- 46. Solan K, Davies P. Anaesthetic and intensive care management of a patient with Ehlers-Danlos type IV syndrome after laparotomy. Anaesthesia. 2004;59:1224-1227 [DOI] [PubMed] [Google Scholar]
- 47. Stanitski DF, Nadjarian R, Stanitski CL, Bawle E, Tsipouras P. Orthopaedic manifestations of Ehlers-Danlos syndrome. Clin Orthop Relat Res. 2000;376:213-221 [DOI] [PubMed] [Google Scholar]
- 48. Steinmann B, Royce PM, Superti-Furga A. The Ehlers-Danlos syndrome. In: Royce PM, Steinmann B, eds. Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. New York: Wiley-Liss; 1993:351-407 [Google Scholar]
- 49. Stoler JM, Oaklander AL. Patients with Ehlers Danlos syndrome and CRPS: a possible association? Pain. 2006;123:204-209 [DOI] [PubMed] [Google Scholar]
- 50. Tofts LJ, Elliott EJ, Munns C, Pacey V, Sillence DO. The differential diagnosis of children with joint hypermobility: a review of the literature. Pediatr Rheumatol Online J. 2009;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tompkins MH, Bellacosa RA. Podiatric surgical considerations in the Ehlers-Danlos patient. J Foot Ankle Surg. 1997;36:381-387 [DOI] [PubMed] [Google Scholar]
- 52. Voermans NC, Knoop H, Bleijenberg G, van Engelen BG. Pain in Ehlers-Danlos syndrome is common, severe, and associated with functional impairment. J Pain Symptom Manage. 2010;40:370-378 [DOI] [PubMed] [Google Scholar]
- 53. Voermans NC, van Alfen N, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697 [DOI] [PubMed] [Google Scholar]
- 54. Volkov N, Nisenblat V, Ohel G, Gonen R. Ehlers-Danlos syndrome: insights on obstetric aspects. Obstet Gynecol Surv. 2007;62:51-57 [DOI] [PubMed] [Google Scholar]
- 55. Weinberg J, Doering C, McFarland EG. Joint surgery in Ehlers-Danlos patients: results of a survey. Am J Orthop (Belle Mead NJ). 1999;28:406-409 [PubMed] [Google Scholar]
- 56. Wenstrup RJ, de Paepe A. Ehlers-Danlos syndrome. http://www.ncbi.nlm.nih.gov/books/NBK1244/ Updated May 11, 2010 Accessed August 17, 2011
- 57. Wenstrup RJ, Meyer RA, Lyle JS, et al. Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genet Med. 2002;4:112-117 [DOI] [PubMed] [Google Scholar]
- 58. Wenstrup RJ, Murad S, Pinnell SR. Ehlers-Danlos syndrome type VI: clinical manifestations of collagen lysyl hydroxylase deficiency. J Pediatr. 1989;115:405-409 [DOI] [PubMed] [Google Scholar]
- 59. Yang JS, Sponseller PD, Yazici M, Johnston CE. Vascular complications from anterior spine surgery in three patients with Ehlers-Danlos syndrome. Spine (Phila Pa 1976). 2009;34:E153-E157 [DOI] [PubMed] [Google Scholar]
- 60. Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome [letter]. Am J Hum Genet. 2003;73:214-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zweers MC, Dean WB, van Kuppevelt TH, Bristow J, Schalkwijk J. Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with tenascin-X mutations. Clin Genet. 2005;67:330-334 [DOI] [PubMed] [Google Scholar]