

Abstract
Mouse primordial germ cells (PGCs) erase global DNA methylation (5mC) as part of the comprehensive epigenetic reprogramming that occurs during PGC development. 5mC plays an important role in maintaining stable gene silencing and repression of transposable elements (TE) but it is not clear how the extensive loss of DNA methylation impacts on gene expression and TE repression in developing PGCs. Using a novel epigenetic disruption and recovery screen and genetic analyses, we identified a core set of germline-specific genes that are dependent exclusively on promoter DNA methylation for initiation and maintenance of developmental silencing. These gene promoters appear to possess a specialised chromatin environment that does not acquire any of the repressive H3K27me3, H3K9me2, H3K9me3 or H4K20me3 histone modifications when silenced by DNA methylation. Intriguingly, this methylation-dependent subset is highly enriched in genes with roles in suppressing TE activity in germ cells. We show that the mechanism for developmental regulation of the germline genome-defence genes involves DNMT3B-dependent de novo DNA methylation. These genes are then activated by lineage-specific promoter demethylation during distinct global epigenetic reprogramming events in migratory (∼E8.5) and post-migratory (E10.5-11.5) PGCs. We propose that genes involved in genome defence are developmentally regulated primarily by promoter DNA methylation as a sensory mechanism that is coupled to the potential for TE activation during global 5mC erasure, thereby acting as a failsafe to ensure TE suppression and maintain genomic integrity in the germline.
Keywords: DNA methylation, Genome defence, Epigenetic reprogramming, Mouse
INTRODUCTION
DNA methylation is a key epigenetic system that is associated with stable transcriptional silencing, genomic imprinting and cellular differentiation (Bird, 2002; Reik, 2007). In mammals, DNA methylation occurs primarily at genomic CpG dinucleotides and is essential for embryonic development (Li et al., 1992; Okano et al., 1999). Methylation of CpGs is established by the de novo methyltransferases DNMT3A and DNMT3B and is subsequently maintained by the maintenance methyltransferase DNMT1. During mouse development, the bulk of global DNA methylation is established by embryonic day (E) 6.5 and is thought to contribute to stable lineage commitment (Borgel et al., 2010; Meissner et al., 2008; Mohn et al., 2008). However, after their specification at ∼E6.25-7.25, mouse primordial germ cells (PGCs) undergo extensive erasure of DNA methylation at ∼E8.5 when PGCs are migrating into the hind-gut endoderm (Seki et al., 2005), and at E10.5-11.5 shortly after the germ cells colonise the genital ridges (Hajkova et al., 2002; Popp et al., 2010). The global DNA demethylation in PGCs appears to be part of a comprehensive epigenetic reprogramming that also includes extensive changes in histone modifications (Seki et al., 2007; Hajkova et al., 2008). The erasure of DNA methylation is required for resetting genomic imprints, and might play a role in reprogramming the developmental potential of PGCs to a totipotent state (Hackett et al., 2012).
At the level of gene regulation, although promoter DNA methylation is associated with transcriptional repression, in many cases it is not clear whether methylated promoters represent a cause or a consequence of gene silencing (Walsh and Bestor, 1999; Bird, 2002). Indeed, at most methylated promoters, DNA methylation acts to reinforce repression that is initially established through chromatin-based silencing mechanisms (e.g. polycomb) (Feldman et al., 2006; Fouse et al., 2008). Moreover, DNA methylation at low CpG-density promoters is readily bypassed by activating signals, whereas CpG-dense promoters, for which DNA methylation is correlated with significant transcriptional silencing, are rarely methylated in vivo (Metivier et al., 2008; Meissner et al., 2008; Weber et al., 2007). Nonetheless, two recent genome-wide studies have identified genes that rely on DNA methylation for transcriptional silencing in either early embryos or embryonic fibroblasts, and these gene sets are enriched for germline-specific genes (Borgel et al., 2010; Velasco et al., 2010). This observation is consistent with DNA methylation regulating the expression of the germline-specific genes Dazl, Mvh (Ddx4 – Mouse Genome Informatics) and Sycp3 as loss of DNA methylation at these promoters is associated with their transcriptional activation in the developing germline (Maatouk et al., 2006). Thus, DNA methylation appears to play a specific role in contributing to the silencing of some germline-specific genes, although it is uncertain whether this mechanism initiates developmental gene silencing or only ‘locks in’ stable repression at these loci, perhaps in parallel with chromatin-based silencing. Interestingly, whether there is any developmentally advantageous reason for germline-specific genes to be preferentially regulated by promoter DNA methylation is also unclear.
In addition to a role at some gene promoters, DNA methylation has an important role in maintaining stable silencing of mobile transposable elements (TEs) and reducing their threat to genomic integrity (Reik, 2007). Around 40% of the mammalian genome is composed of TEs (Waterston et al., 2002), which can propagate through the genome via either a ‘cut and paste’ mechanism or an RNA intermediate (Goodier and Kazazian, 2008). To successfully propagate through generations, TEs must be active in germ cells or during early embryonic development, and are therefore strongly selected for germline expression. However, several mechanisms have evolved to limit the mutagenic potential of TEs in the germline (Ollinger et al., 2010; Zamudio and Bourc’his, 2010). Mice carrying mutations in Dnmt1, or in the germline-restricted accessory factor for de novo DNA methylation Dnmt3L, lose DNA methylation at TE sequences resulting in massive upregulation of LINE1 and intracisternal A-type particle (IAP) retroelements in the developing germline (Walsh et al., 1998; Bourc’his and Bestor, 2004). The ‘genome defence’ hypothesis argues that the primary developmental role of DNA methylation is to contribute to TE silencing rather than to regulate developmental gene expression (Yoder et al., 1997). The transient erasure of repressive epigenetic mechanisms, and in particular of DNA methylation, in PGCs therefore presents an unparalleled opportunity for TEs to become active in the developing germline and represents a potential risk to transgenerational genomic stability.
In a screen to identify genes that are regulated directly and primarily by promoter DNA methylation, we identified several key components of the TE defence machinery. We demonstrate that these genes are dependent on promoter CpG methylation to direct their developmental expression pattern, and show that expression of these genes is activated during two phases of DNA demethylation in PGCs. Intriguingly, these gene promoters appear to contain a specialised chromatin domain that does not acquire repressive H3K27me3, H3K9me2, H3K9me3 or H4K20me3 histone modifications when silenced by DNA methylation. This reliance on DNA methylation acts as a highly tuned sensor of global DNA demethylation in PGCs and thereby primes germ cells to suppress opportunistic TE activity. These findings provide a developmental rationale for germ cells using DNA methylation to regulate gene expression: this mechanism is used as a feedback loop to protect genomic integrity in the germline during potentially hazardous reprogramming events.
MATERIALS AND METHODS
Cell culture
DNA methylation deficient E9.5 Dnmt1n/n p53–/– primary embryonic fibroblasts, control p53–/– (Dnmt1n/+), and Dnmt3b–/– mouse embryonic fibroblasts (MEFs) and primary MEFs (C57BL/6) were cultured in Dulbecco’s modified Eagle’s medium supplemented with l-glutamine, non-essential amino acids, sodium pyruvate, β-mercaptoethanol and 15% foetal calf serum and passaged at sub-confluence (Lande-Diner et al., 2007). Dnmt3b–/– MEFs were derived at E12.5 and E13.5 from heterozygous crosses of mice carrying one floxed and one deleted copy of Dnmt3b and lacking any Cre-containing alleles (Dodge et al., 2005). Embryonic stem (ES) cells (E14/J1) were cultured as previously described (Ollinger et al., 2008). Embryoid body (EB) differentiation was induced through leukaemia inhibitory factor (LIF) withdrawal and cell aggregation in hanging drops for 2 days, followed by suspension culture for 5 days, and then culture as adherent colonies. Where indicated, retinoic acid (1 μM) was applied for 72 hours to induce differentiation.
Primordial germ cell isolation
PGCs were harvested from outbred mice carrying the Oct4-GFP transgene. The dissected hind-gut (E9.5) or urogenital ridges (E10.5 and E13.5) from embryos were trypsinised and sorted by fluorescence-activated cell sorting (FACS) for GFP using a FACSAriaII SORP (Becton Dickinson). Purity of PGCs was determined by OCT4 (POU5F1 – Mouse Genome Informatics) staining (supplementary material Fig. S8).
Aza-recovery assay
Low density cells were treated with 1 μM 5aza-dC (5aza-deoxycytidine) for 3 days, with fresh 5aza-dC-containing media added daily. Cells were subsequently washed and allowed to recover with normal media in situ for 5 days and then passaged every 3 days.
DNA constructs and luciferase assays
Indicated promoter regions were PCR amplified and directionally cloned into pGL3-basic (Promega) using the BglII and MluI sites. In vitro methylation of constructs was carried out with SssI (CpG) methylase (New England Biolabs). Luciferase reporter assays were performed using the Dual Luciferase Assay Kit (Promega), according to the manufacturer’s instructions.
Quantitative RT-PCR
Total RNA from cell lines or tissues was isolated with TRIzol reagent (Invitrogen). After Turbo DNase treatment (Ambion), first strand cDNA was transcribed with superscript III reverse transcriptase (Invitrogen). Quantitative real-time PCR was carried out with Brilliant II SYBR green qPCR mix (Stratagene) using an iCycler (Bio-Rad) and relative quantification was determined using the Pfaffl method (Pfaffl, 2001). RT-PCR was performed with Platinum Taq (Invitrogen) for an optimised number of cycles. Primers are shown in supplementary material Table S2.
Bisulphite sequencing
Genomic DNA was extracted from cells by phenol-chloroform after proteinase K/SDS digestion. Bisulphite conversion was carried using the EZ DNA Methylation Gold Kit (Zymo Research) following the manufacturer’s instructions. We used nested primer sets to amplify a specific region of interest with Platinum Taq (Invitrogen) (supplementary material Table S3). A single band was excised, gel extracted, and cloned into pGEM-T-easy (Promega) for sequencing.
Methylated DNA immunoprecipitation (MeDIP)
MeDIP was performed as previously reported, with modifications (Borgel et al., 2010). Briefly, 200 ng genomic DNA was sonicated to 500 bp (range 300 bp to 1 kb) with a Bioruptor (Diagenode) and denatured at 95°C for 5 minutes. DNA was immunoprecipitated overnight at 4°C with 0.5 μl 5mC-antibody (Eurogentec), washed, and purified through MinElute columns (Qiagen). Enrichment of specific genomic loci relative to input was determined by qPCR.
Single cell RT-PCR
Reverse transcribed single cell cDNA libraries were generated as previously described (Tang et al., 2009). Gene expression was determined by gene-specific qPCR from three PGC libraries at each developmental stage.
Chromatin immunoprecipitation (ChIP)
Native chromatin was prepared from cell nuclei in NBR buffer (85 mM NaCl, 5.5% sucrose, 10 mM Tris, 3 mM MgCl2, 1.5 mM CaCl2, 0.2 mM PMSF, 1 mM DTT). Nuclei were treated with micrococcal nuclease (MNase) (Worthington) to generate mono-, di and tri-nucleosomes and chromatin was released overnight at 4°C. Immunoprecipitation was carried out with antibodies specific to H3K27me3 (Millipore 07449), H3K9me2 (Active Motif 39239), H3K9me3 (Abcam ab8898), H4K20me3 (Active Motif 39181) or rabbit IgG (Santa Cruz), followed by antibody-optimised washes. Samples from native ChIP were purified through affinity columns (Qiagen) and quantified by qPCR using primers shown in supplementary material Table S3.
Microarray and bioinformatics analysis
Total RNA was biotinylated and amplified with a TotalPrep RNA Amplification Kit (Illumina). Pre- and post-labelled RNA was run on a RNA bionalyser chip (Agilent) to confirm RNA integrity and hybridised to Illumina mouse Ref-8 v2.0 BeadChips. Microarray gene expression profiles were generated in quadruplicate from Dnmt1n/n p53–/– and p53–/– MEFs, and from duplicate biological replicates of 5aza-dC treated and 5aza-dC recovery NIH3T3 cells. Background subtraction, normalisations and statistical analyses were carried out using BeadStudio data analysis software modules. Microarray expression changes were calculated as a ratio of normalised expression between samples, with a P<0.01 detection threshold and t-test used to determine significance. Microarray expression data is deposited at the GEO database at NCBI under accession number GSE38307. A stringent sixfold expression change threshold was applied to enrich for bona fide methylation-dependent genes. Gene ontology analysis was performed using the DAVID functional annotation package (http://david.abcc.ncifcrf.gov/). Raw DNA methylation sequencing data from pMEFs was mapped to CpG-dense (ICP and HCP) promoters, with R statistical analysis software (http://www.r-project.org) used to determine CpG methylation levels at target genes (Meissner et al., 2008).
RESULTS
Identification of novel methylation-dependent genes
Previous studies have identified numerous genes that are differentially expressed in response to DNA demethylation using, for example, Dnmt1n/n fibroblasts, Dnmt3b–/– fibroblasts or Dnmt3b–/– embryos (Jackson-Grusby et al., 2001; Velasco et al., 2010; Borgel et al., 2010). However, the complex interactions that exist between epigenetic silencing systems are likely to result in different gene sets being identified depending on the cell types or DNA methyltransferases studied. Moreover, many genes upregulated in genetic deletions probably represent indirect hits, as the majority of upregulated genes are already unmethylated (Jackson-Grusby et al., 2001). In order to identify a minimal set of genes whose expression is primarily and causally regulated by promoter DNA methylation in different cell types, we generated and integrated gene expression datasets from multiple experimental conditions in which genome-wide loss of DNA methylation is induced in different ways.
We initially developed an epigenetic disruption and recovery screen, based on the demethylation-inducing agent 5aza-dC (Fig. 1A). We predicted that genes regulated directly by DNA methylation would be de-repressed by transient exposure to 5aza-dC and would remain continually expressed (and hypomethylated) during an extended recovery period, on the basis that somatic cells lack significant de novo methyltransferase activity (Okano et al., 1999). This approach separates bona fide methylation-dependent genes from those genes that are activated indirectly or by the apoptotic effects of 5aza-dC, which are predicted to re-impose repression during cellular recovery from drug exposure (Palii et al., 2008). In addition, we reasoned that genes that are regulated primarily and causally by DNA methylation would undergo large (off → on) expression changes in this assay and thus imposed a stringent sixfold threshold to select for upregulated genes (supplementary material Fig. S1). Increasing the threshold stringency (i.e. taking the most upregulated genes) selects for somatically methylated CpG island genes, which are promising methylation-dependent candidates (supplementary material Fig. S1A). We profiled global gene expression in NIH3T3 fibroblasts after 5aza-dC exposure and identified 344 significantly upregulated genes (Aza-Up). Following a 14 day recovery, only 49 of these genes remained de-repressed (Rec-Up) (Fig. 1A). These data suggest that the majority of genes activated by 5aza-dC (>85%) are re-repressed upon 5aza-dC withdrawal, with most genes (70%) becoming fully re-silenced (supplementary material Fig. S1). Notably, when mapped to a global promoter methylation dataset (Meissner et al., 2008), Rec-Up genes were highly enriched in genes associated with methylated CpG-dense promoters in MEFs (P<0.01), a likely prerequisite for regulation by DNA methylation, whereas control and Aza-Up gene sets showed no such enrichment (Fig. 1B). The surprisingly small numbers of genes that remain de-repressed after recovery from 5aza-dC have probably undergone a stable and heritable epigenetic switch to activate their expression during the 5aza-dC treatment.
Fig. 1.
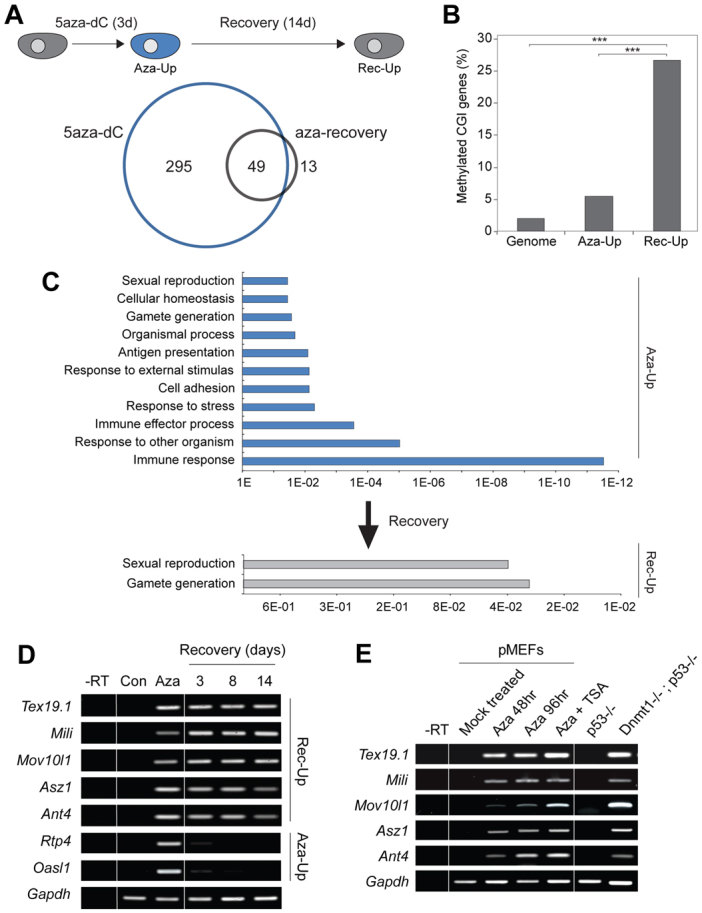
Identification of core methylation-dependent genes. (A) Schematic of the 5aza-dC-recovery assay. Cells are exposed to 5aza-dC for 3 days (3d) and allowed to recover for 14 days (14d). Below: Venn diagram showing overlap between genes upregulated after 5aza-dC (Aza-Up; blue) and after recovery (Rec-Up; grey). (B) Percentage of genes with somatically methylated CpG island (CGI) promoters in Aza-Up or after 14 days recovery (Rec-Up) (Meissner et al., 2008). ***P<0.001. (C) Gene ontology of Aza-Up and Rec-Up gene set, with P-values along the x-axis. Multiple categories of genes are activated by 5aza-dC exposure but only germline-associated categories remain enriched after recovery, when only methylation-dependent genes are predicted to remain. (D) RT-PCR validation of selected germline Rec-Up genes from microarray analysis that remain active during recovery and of control somatic Aza-Up genes that re-impose silencing after 5aza-dC exposure, probably owing to indirect activation. Gapdh is a loading control. (E) RT-PCR showing that genome-defence genes are activated by demethylation in multiple contexts including in Dnmt1n/n MEFs and by 5aza-dC (± the histone deacetylase inhibitor TSA) in primary MEFs. –RT, without reverse transcriptase.
Gene ontology analysis showed that the 344 genes upregulated by 5aza-dC treatment (Aza-Up) were enriched for eleven different categories, including immune response, immune effector processes, response to stress, gamete generation and sexual reproduction (supplementary material Table S4). By contrast, only two gene ontology classes were enriched in the Rec-Up gene set, sexual reproduction and gamete generation, suggesting that the Rec-Up gene set of methylation-dependent candidates corresponds to a distinct group of genes with common functional annotation in germline development (Fig. 1C). We confirmed our findings from the microarray data (Rec-Up dataset) by RT-PCR showing that the Tex19.1, Mili (Piwil2 – Mouse Genome Informatics), Mov10l1 and Asz1 (also known as Gasz) germline-specific genes remain de-repressed after 5aza-dC withdrawal, as does Ant4 (Slc25a31 – Mouse Genome Informatics), which has previously been shown to be methylation dependent (Rodic et al., 2005) (Fig. 1D). By contrast, representative genes (Rtp4 and Oasl1) of the Aza-Up dataset were initially activated by 5aza-dC and rapidly silenced after 5aza-dC withdrawal, implying these are not methylation-dependent targets. Importantly, we found that following 5aza-dC-induced DNA demethylation, the promoters of the Tex19.1 and Mili Rec-Up genes become hypomethylated, and are not re-methylated during recovery (supplementary material Fig. S2). We have employed the 5aza-dC recovery assay on a number of somatic cell lines, primary MEFs and CMT-93 mouse rectal carcinoma cells, and continuous Tex19.1 de-repression is representative of active germline-specific genes in each case (J.A.H., D.S.D., R.R.M., unpublished observations).
Genome-defence genes are regulated directly by CpG methylation
To refine further our Rec-Up gene set to a core subset that is crucially reliant on DNA methylation in multiple contexts, we used mouse embryonic fibroblasts (MEFs) derived from hypomorphic Dnmt1n/n; p53–/– embryos, which lack significant DNA methylation (Lande-Diner et al., 2007). We profiled global gene expression from Dnmt1n/n cells and intersected upregulated genes with Rec-Up genes. This identified 26 genes that are strongly activated by DNA demethylation in different experimental and cellular contexts (supplementary material Table S1). These core genes are highly enriched (13/26) for germline-specific expression. Strikingly, five of the six germline-associated genes that have functionally characterised roles are involved in genome defence against transposable elements (TE) in the germline, either via piwi-interacting RNA (piRNA) biogenesis or alternative mechanisms (Ollinger et al., 2008; Aravin et al., 2007; Frost et al., 2010; Ma et al., 2009; Aravin et al., 2008). Although previous genome-wide studies have described an association between DNA methylation and germline-associated genes (Borgel et al., 2010; Velasco et al., 2010), the genes identified in these studies do not appear to be co-expressed at any one specific stage of germ cell development, and do not appear to have a common functional role during gametogenesis. Given the remarkable enrichment for known germline genome-defence genes (Tex19.1, Mili, Mov10l1 and Asz1) in our Rec-Up data set, we hypothesised that the shared epigenetic mechanism (DNA methylation) of regulating their expression might be related to a common molecular function in the developing germline.
We confirmed that Tex19.1, Mili, Mov10l1 and Asz1 are all ectopically expressed in Dnmt1n/n MEFs (Fig. 1E). Transcript mapping demonstrated that transcription in Dnmt1n/n MEFs initiated from canonical transcriptional start sites (TSSs), ruling out ectopic expression from cryptic promoters (supplementary material Fig. S3). To investigate whether DNA methylation is the initiating mechanism for silencing Tex19.1, Mili and Mov10l1 rather than a stable ‘lock’, we utilised early passage embryonic stem (ES) cells lacking the de novo methyltransferases DNMT3A and DNMT3B. Notably, Tex19.1 and Mov10l1 failed to become fully repressed, and Mili only exhibited partial repression during late embryoid body (EB) differentiation of Dnmt[3a, 3b]–/– ES cells, whereas all were rapidly silenced during differentiation of parental J1 ES cells (Fig. 2A). By contrast, Oct4 was silenced in both wild-type and double mutant EBs. Even after 15 days differentiation, Dnmt[3a, 3b]–/– EBs are intrinsically unable to silence Tex19.1, Mili and Mov10l1 expression, suggesting that de novo promoter methylation is the critical mark mediating the onset of repression during differentiation. We were unable to assess whether DNA methylation initiated silencing of Asz1 in the EB differentiation assay as we could not detect Asz1 expression in wild-type ES cells, although we could detect strong expression of Asz1 in Dnmt1n/n ES cells (supplementary material Fig. S3).
Fig. 2.
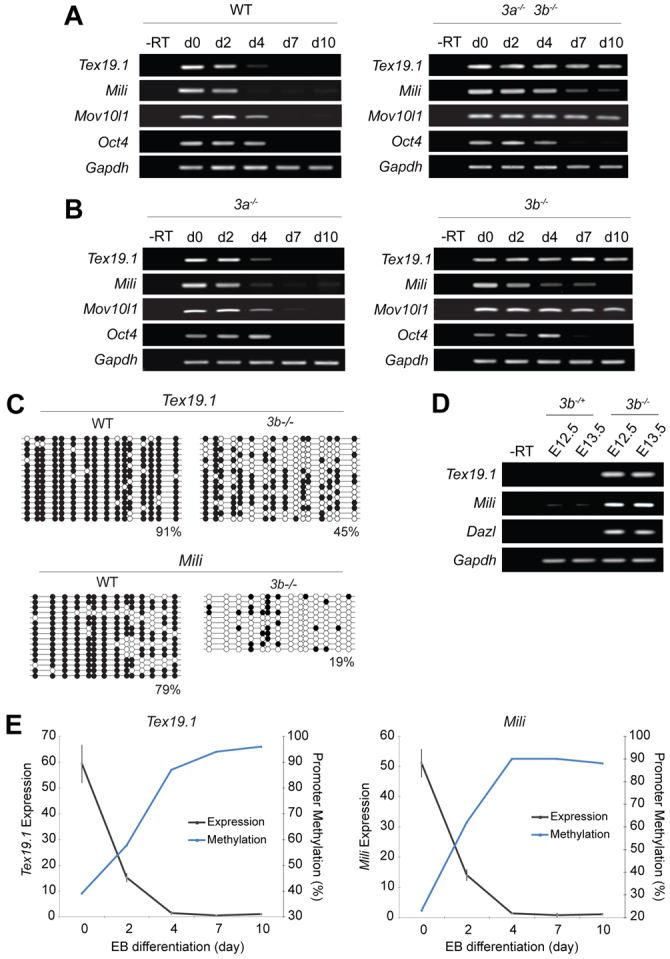
Dynamics of DNA methylation at genome-defence genes. (A) RT-PCR showing expression upon EB differentiation of wild-type (WT) or Dnmt3a/3b–/– ES cells. Note that Oct4 does not require Dnmt3a or Dnmt3b for silencing. (B) RT-PCR in Dnmt3a- and Dnmt3b-mutant ES cells. Transcriptional silencing fails in the absence of DNMT3B. (C,D) Tex19.1 and Mili are highly demethylated and ectopically expressed in Dnmt3b–/– embryos at E12.5. Filled circles indicate methylated CpG and empty indicate unmethylated CpG. Percentages indicate average DNA methylation. (E) The Tex19.1 and Mili promoters are progressively de novo methylated (right-hand y-axis, blue line) coincident with the onset of transcriptional silencing (left-hand y-axis, grey line) during EB differentiation of ES cells. d, day; –RT, without reverse transcriptase.
To determine the relative contribution of each de novo methyltransferase in initiating gene silencing, we induced EB formation in ES cells lacking either Dnmt3a or Dnmt3b. This showed that Tex19.1, Mili and Mov10l1 specifically require DNMT3B, but not DNMT3A, for full silencing and de novo methylation during ES cell differentiation (Fig. 2B,C; supplementary material Fig. S4). However, Mili was partially silenced in Dnmt3b–/– ES cells, which might be indicative of additional silencing pathways that are operative in differentiating ES cells. Furthermore, we derived fibroblasts from Dnmt3b–/– embryos and found that Tex19.1 and Mili were hypomethylated and fully de-repressed, implying that these loci do not initiate appropriate silencing in somatic tissues in the absence of DNMT3B in vivo (Fig. 2C,D). Notably, global profiling revealed that our null Dnmt3b allele results in de-repression of a different subset of germline-specific genes (and to a greater extent) compared with a hypomorphic compound-heterozygote Dnmt3b allele previously reported (Velasco et al., 2010). We found that the Asz1 and Dazl genome-defence genes are also de-repressed in Dnmt3b–/– embryonic fibroblasts (supplementary material Fig. S4). Mov10l1 is also upregulated in Dnmt3b–/– MEFs, although it was below the sixfold expression threshold (supplementary material Table S5). Thus, silencing of many germline genome-defence genes during somatic differentiation appears to be initiated by the de novo DNA methyltransferase DNMT3B.
It has been reported that silencing of some genes, including Oct4, occurs in a stepwise manner, with trans-acting repressors and histone modifications establishing a hetrochromatinised silenced state and promoter methylation only occurring later, as a secondary repressive lock against reactivation (Feldman et al., 2006). To investigate whether DNA methylation was the primary or a secondary regulatory mechanism at Tex19.1 and Mili, we tracked the dynamics of promoter methylation and gene silencing during ES cell differentiation. We observed that the acquisition of promoter DNA methylation occurs in parallel with the onset of transcriptional silencing at Tex19.1 and Mili (Fig. 2E). Treatment of day 7 EBs with 5aza-dC was sufficient to reactivate Tex19.1, Mili, Mov10l1 and other silenced genome-defence genes but not Oct4 (supplementary material Fig. S5). Taken together, this suggests that tissue-specific promoter DNA methylation is a primary mechanism for regulating the expression pattern of a number of genes that function in germline genome defence.
Genome-defence genes are depleted of chromatin modifications
To investigate whether chromatin-based silencing mechanisms also operate at genome-defence genes, we performed native chromatin-immunoprecipitation (ChIP) against the best characterised repressive histone modifications in NIH3T3 cells, in which these genes are silent (Fig. 1A-D). Compared with a positive control, HoxC10, the repressive H3K27me3 mark, is absent from the promoters of Tex19.1, Mili and Mov10l1 with levels comparable to the Gapdh negative control (Fig. 3A). Moreover, retinoic acid (RA) differentiation of Eed–/– ES cells, which lack H3K27me3, did not impair silencing of Tex19.1, implying that H3K27me3 is functionally dispensable for repression at this locus (Fig. 3E) (Montgomery et al., 2005). Further analysis of the heterochromatin-associated marks H3K9me3 and H4K20me3 revealed that Tex19.1, Mili and Mov10l1 are also depleted of these modifications, and are marked comparably to unmodified Gapdh (Fig. 3B,C) (Mikkelsen et al., 2007). We also found that H3K9me2, which has been reported to be functionally upstream of DNA methylation at some loci, was depleted from tested gene promoters (Fig. 3D) (Tachibana et al., 2008). Importantly, ChIP for histone H3 gave comparable or greater enrichment of Tex19.1, Mili and Mov10l1 than positive controls, indicating that the reduction of unmodified histones we observe at these loci is not symptomatic of nucleosome-free regions (supplementary material Fig. S6). These genes therefore appear to possess specialised chromatin at their promoters in non-expressing somatic cells that is selectively depleted of many repressive histone modifications. Thus, methylation of their CpG-dense promoters probably represents the predominant mechanism for regulating the developmental and tissue-specific expression of these genes. Consistent with this, promoter reporters for Tex19.1 and Mili, but not Oct4, drive strong expression in somatic cell lines derived from either mesodermal or neural lineages, implying that their germline-restricted expression in vivo is not a consequence of tissue-specific availability of transcription factors or repressors (supplementary material Fig. S7). In vitro methylation of Tex19.1 and Mili reporters strongly repressed expression up to 150-fold in this assay.
Fig. 3.
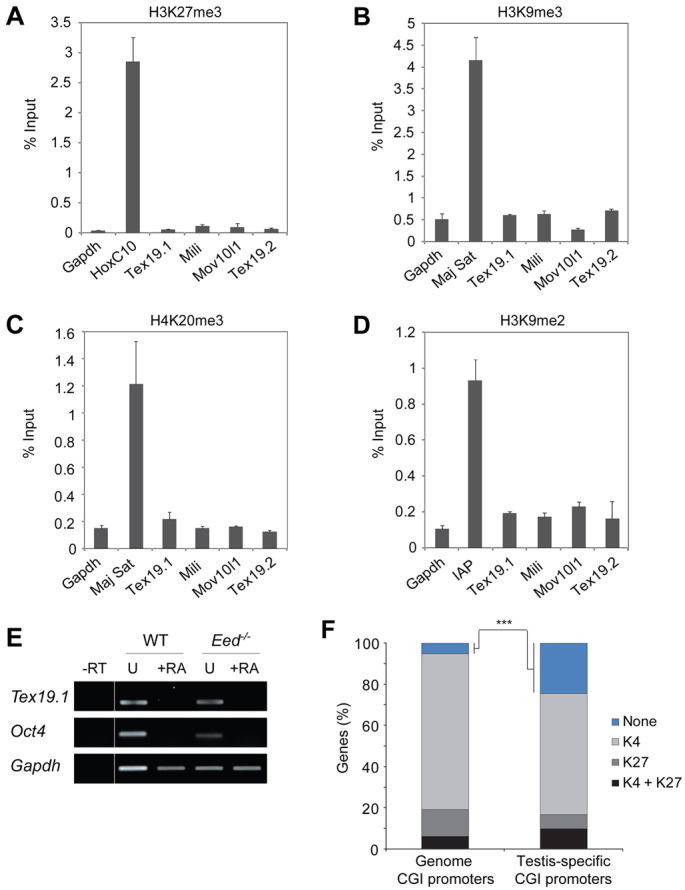
Genome-defence genes are associated with specialised promoter chromatin. (A-D) Native ChIP demonstrating that the repressive chromatin modifications H3K27me3, H3K9me3, H4K20me3 and H3K9me3 are depleted from the promoter regions of methylated and silenced genome-defence genes. Gapdh marks the background levels expected of no significant modification. Hoxc10, major satellite (Maj Sat) and IAP are positive controls for respective modifications. (E) RT-PCR demonstrating that Tex19.1 is silenced during ESC differentiation by retinoic acid (+RA) in the absence of polycomb (Eed–/–). –RT, without reverse transcriptase; U, untreated. (F) Germline-specific genes in general are highly enriched for promoters that lack histone modifications in MEFs. Blue, no modification; light grey, H3K4me3; dark grey, H3K27me3; black, bivalent modification (H3K4me3 plus H3K27me3). ***P<0.001.
The general depletion of repressive histone modifications at Tex19.1, Mili and Mov10l1 prompted us to investigate whether unmodified promoter histones was a common feature of silenced germline-specific genes. We mapped a dataset of testis-specific CpG-dense promoter genes (defined as expressed more than tenfold in testis than all somatic tissues in the BioGPS database) to their histone modification state for active (H3K4me3), repressive (H3K27me3) and bivalent (H3K4me3 and H3K27me3) marks in MEFs (Mikkelsen et al., 2007). We find that testis-specific genes are highly enriched in promoters that lack these histone modifications (Fisher’s exact test, P=0.00019) (Fig. 3F). This finding might partly explain the general association between germline genes and regulation by DNA methylation reported here and in other studies (Maatouk et al., 2006; Borgel et al., 2010; Velasco et al., 2010).
Promoter CpG methylation dynamics during PGC development
Although immunostaining with anti-5mC antibodies suggests that PGCs might undergo extensive loss of DNA methylation at ∼E8.5 (Seki et al., 2005), the genomic loci that lose DNA methylation during this process have not yet been identified. Although partial demethylation of some single-copy loci is evident in E9.5 PGCs (Guibert et al., 2012), repetitive elements, imprinted loci and selected single-copy gene promoters are all still fully methylated in E10.5 PGCs (Hajkova et al., 2002; Maatouk et al., 2006; Lee et al., 2002). By contrast, the genomic targets corresponding to the ∼E11.0 demethylation event include gene promoters, repetitive elements and imprinted genes (Hajkova et al., 2002; Maatouk et al., 2006). To investigate the dynamics of DNA methylation at germline genome-defence genes during PGC development in vivo, we tracked the methylation status and expression of these genes in PGCs isolated from E10.5 and E13.5 embryonic genital ridges using a transgenic Oct4-GFP marker (supplementary material Fig. S8).
We used methylated DNA immunoprecipitation (meDIP) to evaluate promoter methylation in PGCs and in control somatic cells and epiblast stem cells (EpiSCs). Unexpectedly, we found that the tested genes fell into one of two categories: either they are methylated in PGCs at E10.5 and demethylated by E13.5 as reported for Dazl (Maatouk et al., 2006), or they are already demethylated in PGCs at E10.5 prior to post-migratory epigenetic reprogramming (Fig. 4A; supplementary material Fig. S9). The former group (including Mov10l1, Asz1 and Tex19.2) probably represent novel methylation-dependent genes that utilise global demethylation to direct expression in post-migratory PGCs (supplementary material Fig. S9). However, intriguingly, the latter group of methylation-dependent genes (Tex19.1, Mili) were already hypomethylated in PGCs at E10.5, prior to global methylation erasure (Fig. 4A). These ‘early demethylation’ genes are also strongly expressed in PGCs at E10.5, whereas loci undergoing demethylation after E10.5, such as Dazl, were repressed and exhibited strong transcriptional upregulation by E13.5 (Fig. 4B). Importantly, all tested genes are methylated in EpiSCs, which are derived from the pre-gastrulation epiblast prior to PGC specification.
Fig. 4.

DNA demethylation in PGCs during two reprogramming events (A) Methylated DNA immunoprecipitation (meDIP) showing the relative levels of promoter methylation at Tex19.1, Mili and Dazl in soma, epiblast stem cells (EpiSC), and E10.5, E13.5 female (F) and E13.5 male (M) PGCs. Notably, Tex19.1 and Mili are demethylated in E10.5 PGCs whereas Dazl remains highly methylated. Error bars represent s.e.m. (B) qRT-PCR expression analysis from pooled PGCs. Expression levels of Tex19.1, Mili and Dazl in PGCs correlate with promoter methylation levels (A). Normalised relative to 18S RNA. Error bars represent s.e.m. (C) Bisulphite sequencing showing promoter DNA methylation at Tex19.1, Mili and Dazl during primordial germ cell (PGC) development and matched somatic cells from genital ridges (lower panel). (D) DNA methylation profile of Tex19.1 in the embryonic portion of E6.5 (epiblast), E7.5 and E8.5 embryos. In C and D, filled circles indicate methylated CpG and empty indicate unmethylated CpG.
We confirmed the differential demethylation dynamics between Tex19.1/Mili and Dazl promoters by bisulphite sequencing these loci in purified PGC populations. Consistent with our meDIP data, the Dazl promoter is methylated in PGCs at E10.5 but Tex19.1 and Mili promoters are hypomethylated (Fig. 4C). Furthermore, in agreement with the meDIP data, the Tex19.2 promoter behaves differently to Tex19.1 and follows similar DNA methylation kinetics to Dazl (supplementary material Fig. S9). The Tex19.1 and Mili promoters therefore become hypomethylated earlier than Dazl during PGC development, and are already hypomethylated in E10.5 PGCs. Tex19.1 is also hypomethylated and expressed in migrating PGCs isolated from the hind-gut region of younger embryos at E9.5 (supplementary material Fig. S10). Thus, Tex19.1 and Mili are good candidate targets for the DNA demethylation event that is reported to occur in E8.5 PGCs.
To test whether loci that are hypomethylated at E10.5 escape de novo methylation before PGC specification (at ∼E6.5), and are therefore never methylated in the germline, we initially assessed the methylation dynamics of Tex19.1 during post-implantation development. Although isolation and bisulphite sequencing of pure PGC DNA from E7.5 embryos is technically prohibitive, bisulphite sequencing of epiblast and embryonic tissue between E6.5 and E8.5 showed that the Tex19.1 promoter is fully methylated at E7.5 and E8.5 (when Tex19.1 becomes silenced), and is in an intermediate methylated state at E6.5, indicating that de novo methylation commences at this locus prior to PGC specification (Fig. 4D). These data are consistent with a recent report demonstrating that Mili, Mov10l1, Asz1, Dazl and Miwi (Piwil1 – Mouse Genome Informatics) are also all highly methylated in E6.5 epiblast in vivo and therefore undergo lineage-specific demethylation in the PGCs (Borgel et al., 2010). Furthermore, although TEX19.1 protein and mRNA is expressed in the pluripotent epiblast cells in E6.5 embryos, Tex19.1 is downregulated during gastrulation, can only be weakly detected in E7.5 embryos, and is not detectable in the hind-gut region of E8.5 embryos where PGCs are present (supplementary material Fig. S10). This suggests that Tex19.1 expression is downregulated during PGC specification. Thus, the genome-defence genes appear to undergo de novo methylation in the post-implantation epiblast, followed by lineage-specific loss of promoter DNA methylation either in migratory PGCs (Tex19.1, Mili) or post-migratory PGCs (Tex19.2, Mov10l1, Asz1, Dazl).
Gene activation during distinct reprogramming phases in PGCs
The global loss of DNA methylation that occurs in ∼E11.0 PGCs has been shown to be responsible for inducing expression of key germline genes, such as Dazl and Mvh (Ddx4 – Mouse Genome Informatics), at this stage of PGC development. Indeed, expression of Tex19.2, which shows similar promoter demethylation kinetics to Dazl, is also activated in post-migratory PGCs (supplementary material Fig. S9). However, it is not clear whether the DNA demethylation in ∼E8.5 PGCs also acts as a trigger to induce developmental changes in PGC gene expression. We therefore used single-cell cDNA expression profiling to analyse Tex19.1 and Mili expression in E7.5-11.5 PGCs to test whether expression of these methylation-dependent genes coincided with the demethylation event at E8.5.
Interestingly, Tex19.1 and Mili are not expressed in PGCs at E7.5, consistent with these loci being methylated at this stage. However, we observed strong transcriptional activation of both loci starting from E8.5, immediately after the reported demethylation in PGCs at ∼E8.5 (Fig. 5A). In combination with the kinetics of Tex19.1 and Mili promoter methylation, these data strongly support a model whereby Tex19.1 and Mili undergo de novo methylation and silencing at ∼E6.5 and are then specifically demethylated in PGCs from ∼E8.0 resulting in their transcriptional activation. Thus, the global DNA demethylation event that occurs in PGCs at ∼E8.5 does appear to be associated with developmental regulation of PGC gene expression. The Tex19.1 and Mili promoters therefore represent novel loci regulated by the proposed lineage-specific DNA demethylation event in ∼E8.5 PGCs. The activation of TE-response genes in the germline at both ∼E8.5 and ∼E11.0 suggests that erasure of DNA methylation is used to regulate developmental gene expression in migratory and post-migratory PGCs and might represent a mechanism that helps defend the genome from active TEs during the epigenetic reprogramming events that occur during gametogenesis (Fig. 5B).
Fig. 5.

Transcriptional activation during reprogramming in PGCs. (A) Single-cell qRT-PCR analysis of PGCs between E8.5 and E11.5, and E8.5 somatic cells. Shown are three single-cell libraries from each time point with expression values normalised to Gapdh. Both Mili and Tex19.1 are transcriptionally activated between E8.5 and E9.5 prior to global to 5mC erasure at ∼E11.0. Error bars represent s.e.m. (B) Model of feedback loop between global 5mC erasure and genome-defence gene activation. At E6.25, PGCs are specified from epiblast cells that have already acquired global de novo methylation. At E8.5, initiation of DNA demethylation in PGCs activates some ‘early’ genome-defence genes (Tex19.1 and Mili), possibly to prime germ cells. At E11.5, genome-wide erasure of 5mC leads to activation of other genome-defence and germline-specific genes, which can suppress any unmasked transposable element (TE) activity during reprogramming, either post-transcriptionally or through the piRNA pathway. The coupling of genome-defence genes to potential TE activation through a common regulatory mechanism, DNA methylation, ensures that genomic integrity is maintained during 5mC erasure in the germline.
DISCUSSION
Identification of core methylation-dependent genes
Our initial aim was to unambiguously identify genes that are directly and causally regulated by promoter DNA methylation. We developed an ‘epigenetic disruption and recovery’ assay which selectively enriches for genes that functionally rely on promoter DNA methylation for silencing, and significantly reduces the indirect hits associated with previous genetic analyses and direct drug exposure (Jackson-Grusby et al., 2001; Palii et al., 2008). This recovery strategy might be useful in identifying genes that are epigenetically silenced by DNA methylation in other contexts, e.g. cancer and tissue specificity. We intersected recovery targets with genes also de-repressed in Dnmt1n/n MEFs and identified a limited number (26) of core methylation-dependent genes, which are highly enriched for germline expression. Using a combination of complementary genetic approaches we demonstrate that Tex19.1, Mili, Asz1 and Mov10l1 are causally regulated by promoter DNA methylation. The mechanism for silencing these genes during post-implantation development appears to involve the de novo DNA methyltransferase DNMT3B. Importantly, acquisition of DNA methylation at these promoters is both necessary for, and temporally coincides with, the initiation of transcriptional silencing at these loci. Indeed, silenced germline genes are preferentially associated with specialised promoter chromatin that is highly depleted of many repressive histone modifications, thus placing promoter CpG methylation as the predominant regulatory mechanism at these loci. It has been unclear whether promoter DNA methylation is only a secondary epigenetic mechanism for ‘locking in’ a prior silenced state to provide a stable epigenetic memory (Feldman et al., 2006; Fouse et al., 2008; Borgel et al., 2010; Mohn et al., 2008), or whether it also functions as a direct and causal regulatory mechanism per se. Our data supports a model whereby promoter DNA methylation is a primary mechanism for both initiating and maintaining silencing at a small number of germline-specific loci and is potentially the only epigenetic barrier to their ectopic expression.
Epigenetic reprogramming as a cue for gene expression in PGCs
Intriguingly, the methyl-dependent genes we identified appear to be activated by apparent global demethylation events during epigenetic reprogramming in PGCs at either ∼E8.5 or E10.5-11.5. The genome-wide reprogramming event that occurs in E10.5-11.5 PGCs has previously been associated with expression of key germline-specific genes, such as Mvh and Dazl (Maatouk et al., 2006). Here, we show that the epigenetic reprogramming that occurs in PGCs at ∼E8.5 might also be associated with developmental regulation of a different set of germline-associated genes, including Tex19.1 and Mili. The analysis of DNA methylation in post-implantation epiblast and single-cell expression data make Tex19.1 and Mili very good candidate loci for the 5mC reprogramming that is reported to occur in ∼E8.5 PGCs. However, loss of DNA methylation at these loci alone is not sufficient to explain the reported genome-wide reduction in 5mC-immunostaining in these cells, and it is not clear what other loci and genomic features might be losing DNA methylation at this stage of development. Indeed, one possibility is that DNA demethylation is a single progressive event lasting several days in PGCs, with specific genomic regions undergoing 5mC erasure in a temporally defined order, with Tex19.1 and Mili at the vanguard and demethylation of Dazl towards the end of the process.
What determines the different temporal activation profiles is not clear but might be related to specific gene functions during germ cell development, cis-acting elements or the chromatin state in PGCs. It is also not clear how loss of DNA methylation is brought about at the Tex19.1 and Mili promoters in ∼E8.5 PGCs. However, at this stage most PGCs are G2-arrested, suggesting that they might be targeted by active DNA demethylation mechanisms (Kurimoto et al., 2008; Hackett et al., 2012). The differential temporal activation of the closely related Tex19.1 and Tex19.2 paralogues in the developing PGCs is particularly striking given the proximity of these divergently transcribed genes on the chromosome (Kuntz et al., 2008), and analysis of the promoter and intergenic DNA sequence of this gene pair might provide some insight into how erasure of DNA methylation is targeted at different times in PGC development.
Developmental regulation of genome-defence genes
As genome-wide erasure of DNA methylation presents an opportunity for activation of TEs, it is likely that the coupled activation of Tex19.1, Mili, Mov10l1 and Asz1 functions as part of a fail-safe mechanism to ensure suppression of TE activity in the germline. Regulation of these genes by promoter methylation would therefore be inherently linked to their role as guardians of the genome by sensing the possibility of heightened TE activity during epigenetic reprogramming phases, and directing a coordinated response. It is possible that quantitative changes in DNA methylation at these loci in the developing germline might influence their level of expression at different stages in germ cell development. Moreover, the absence of significant regulation by chromatin-based mechanisms at these genes makes them particularly sensitive to global demethylation events, which may prime but do not directly activate other classes of genes. Consistent with this, germline-specific genes are often ectopically expressed in somatic cancers, in which dramatic alterations in DNA methylation distribution can occur (Simpson et al., 2005). The increased TE activity associated with hypomethylated cancer cells might contribute to the genomic instability that is a hallmark of cancers (Romanish et al., 2010). Whether ectopic germline gene activity in a cancer context is a driver of malignancy, or a neutral or even protective consequence of demethylation, remains to be fully determined (Janic et al., 2010). Moreover, although many of the germline-associated methylation-dependent genes appear to function in genome defence, it is not clear whether the non-germline-associated methylation-dependent genes that we have identified (supplementary material Table S1) are also functionally related, or whether co-regulation of these genes by DNA methylation is important for cellular responses to DNA hypomethylation in other contexts.
Other genes involved in germline genome defence, such as Mvh and Mael, have previously been reported to be methylation dependent (Maatouk et al., 2006; Siomi et al., 2011) and some of the uncharacterised methylation-dependent germline genes we have identified might have as yet undiscovered roles in genome defence. Interestingly, although many of the identified genome-defence genes are implicated in the piRNA pathway for TE suppression, global de novo methylation via the piRNA pathway is not reported to commence until E16.5 (Kuramochi-Miyagawa et al., 2008). The activation of methylation-dependent piRNA pathway genes, such as Mili, as early as E9.5 might represent anticipation of future requirements for this pathway (Aravin et al., 2008). Alternatively, as some aspects of the piwi/piRNA pathway function to silence TE activity post-transcriptionally, it is also possible that this pathway is acting earlier than E16.5 (Reuter et al., 2011; Unhavaithaya et al., 2009; Xu et al., 2008). Interestingly, this period might have a key role in allowing TE transcription that, in turn, could target de novo methylation back to TE DNA, possibly via small RNAs (Seisenberger et al., 2010; Aravin et al., 2008). This mechanism would ensure that a powerful epigenetic response is mounted against active TEs and that they are fully silenced throughout the remainder of gametogenesis, at least in male germ cells. However, the relaxation of retrotransposon transcriptional repression induced during PGC epigenetic reprogramming might result in a greater reliance on post-transcriptional retrotransposon silencing mechanisms during these stages of PGC development. Indeed LINE-1 mRNA levels appear to be higher in PGCs than in neighbouring soma, supporting this possibility (Hayashi et al., 2008). Some of the genome-defence genes that are expressed in the developing PGCs might have piRNA-independent roles in regulating TE activity. For example, Tex19.1 appears to regulate TE activity through a post-transcriptional mechanism distinct from the piwi/piRNA pathway (Ollinger et al., 2008). The coordinated activation of multiple genome-defence mechanisms during PGC development is likely to provide a concerted defence against the hundreds of different TE elements present in the mouse genome (Waterston et al., 2002). Further experiments will be required to dissect the roles and specific TE targets of different genome-defence genes in developing PGCs.
In summary, we have identified a novel set of germline-associated genome-defence genes that rely directly on promoter DNA methylation to initiate and maintain their developmental regulation, independently of chromatin-based mechanisms. The tight coupling of DNA demethylation to the activation of these genome-defence genes in PGCs provides a powerful feedback loop that can help prevent transposable element activity and genome instability during global epigenetic reprogramming.
Supplementary Material
Acknowledgments
We thank W. Bickmore, W. Dean and N. Hastie for comments during manuscript preparation. We thank A. Bird, J. Selfridge, M. Okano and H. Cedar for cell lines; H. Scholer for Oct4 constructs; J. Nichols for Oct4-GFP reporter mice; and J. Zylicz for artwork. We dedicate this paper to the memory of our former and much missed colleague Danny Wolf.
Footnotes
Funding
Work in the R.R.M. laboratory (J.A.H., D.S.D., J.P.R., D.R., C.E.N.) and I.R.A. laboratory (J.R.) is supported by the Medical Research Council and Breakthrough Breast Cancer. Deposited in PMC for release after 6 months.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
J.A.H. performed microarray and expression analyses, meDIP in PGCs and bisulfite-sequencing. J.A.H. and J.P.R. carried out ChIP studies. I.R.A., J.R. and J.A.H. conducted mouse ESC and embryonic work. J.A.H. and C.E.N. carried out bioinformatics analyses. D.S.D., M.B., W.R. and M.A.S. provided essential experimental support, key mouse strains and advice on the manuscript. R.R.M. developed the original concept. J.A.H., I.R.A. and R.R.M. designed the experiments and wrote the manuscript.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.081661/-/DC1
References
- Aravin A. A., Hannon G. J., Brennecke J. (2007). The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 318, 761–764 [DOI] [PubMed] [Google Scholar]
- Aravin A. A., Sachidanandam R., Bourc’his D., Schaefer C., Pezic D., Toth K. F., Bestor T., Hannon G. J. (2008). A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 31, 785–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 [DOI] [PubMed] [Google Scholar]
- Borgel J., Guibert S., Li Y., Chiba H., Schübeler D., Sasaki H., Forné T., Weber M. (2010). Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 42, 1093–1100 [DOI] [PubMed] [Google Scholar]
- Bourc’his D., Bestor T. H. (2004). Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431, 96–99 [DOI] [PubMed] [Google Scholar]
- Chen J., Melton C., Suh N., Oh J. S., Horner K., Xie F., Sette C., Blelloch R., Conti M. (2011). Genome-wide analysis of translation reveals a critical role for deleted in azoospermia-like (Dazl) at the oocyte-to-zygote transition. Genes Dev. 25, 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge J. E., Okano M., Dick F., Tsujimoto N., Chen T., Wang S., Ueda Y., Dyson N., Li E. (2005). Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 280, 17986–17991 [DOI] [PubMed] [Google Scholar]
- Feldman N., Gerson A., Fang J., Li E., Zhang Y., Shinkai Y., Cedar H., Bergman Y. (2006). G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat. Cell Biol. 8, 188–194 [DOI] [PubMed] [Google Scholar]
- Fouse S. D., Shen Y., Pellegrini M., Cole S., Meissner A., Van Neste L., Jaenisch R., Fan G. (2008). Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG Complex, and Histone H3 K4/K27 trimethylation. Cell Stem Cell 2, 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost R. J., Hamra F. K., Richardson J. A., Qi X., Bassel-Duby R., Olson E. N. (2010). MOV10L1 is necessary for protection of spermatocytes against retrotransposons by Piwi-interacting RNAs. Proc. Natl. Acad. Sci. USA 107, 11847–11852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier J. L., Kazazian H. H., Jr (2008). Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135, 23–35 [DOI] [PubMed] [Google Scholar]
- Guibert S., Forne T., Weber M. (2012). Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 22, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett J. A., Zylicz J. J., Surani M. A. (2012). Parallel mechanisms of epigenetic reprogramming in the germline. Trends Genet. 28, 164–174 [DOI] [PubMed] [Google Scholar]
- Hajkova P., Erhardt S., Lane N., Haaf T., El-Maarri O., Reik W., Walter J., Surani M. A. (2002). Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 117, 15–23 [DOI] [PubMed] [Google Scholar]
- Hajkova P., Ancelin K., Waldmann T., Lacoste N., Lange U. C., Cesari F., Lee C., Almouzni G., Schneider R., Surani M. A. (2008). Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452, 877–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K., Chuva de Sousa Lopes S. M., Kaneda M., Tang F., Hajkova P., Lao K., O’Carroll D., Das P. P., Tarakhovsky A., Miska E. A., et al. (2008). MicroRNA biogenesis is required for mouse primordial germ cell development and spermatogenesis. PLoS ONE 3, e1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Grusby L., Beard C., Possemato R., Tudor M., Fambrough D., Csankovszki G., Dausman J., Lee P., Wilson C., Lander E., et al. (2001). Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 27, 31–39 [DOI] [PubMed] [Google Scholar]
- Janic A., Mendizabal L., Llamazares S., Rossell D., Gonzalez C. (2010). Ectopic expression of germline genes drives malignant brain tumor growth in Drosophila. Science 330, 1824–1827 [DOI] [PubMed] [Google Scholar]
- Kuntz S., Kieffer E., Bianchetti L., Lamoureux N., Fuhrmann G., Viville S. (2008). Tex19, a mammalian-specific protein with a restricted expression in pluripotent stem cells and germ line. Stem Cells 26, 734–744 [DOI] [PubMed] [Google Scholar]
- Kuramochi-Miyagawa S., Watanabe T., Gotoh K., Totoki Y., Toyoda A., Ikawa M., Asada N., Kojima K., Yamaguchi Y., Ijiri T. W., et al. (2008). DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 22, 908–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto K., Yabuta Y., Ohinata Y., Shigeta M., Yamanaka K., Saitou M. (2008). Complex genome-wide transcription dynamics orchestrated by Blimp1 for the specification of the germ cell lineage in mice. Genes Dev. 22, 1617–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande-Diner L., Zhang J., Ben-Porath I., Amariglio N., Keshet I., Hecht M., Azuara V., Fisher A. G., Rechavi G., Cedar H. (2007). Role of DNA methylation in stable gene repression. J. Biol. Chem. 282, 12194–12200 [DOI] [PubMed] [Google Scholar]
- Lee J., Inoue K., Ono R., Ogonuki N., Kohda T., Kaneko-Ishino T., Ogura A., Ishino F. (2002). Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development 129, 1807–1817 [DOI] [PubMed] [Google Scholar]
- Li E., Bestor T. H., Jaenisch R. (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926 [DOI] [PubMed] [Google Scholar]
- Ma L., Buchold G. M., Greenbaum M. P., Roy A., Burns K. H., Zhu H., Han D. Y., Harris R. A., Coarfa C., Gunaratne P. H., et al. (2009). GASZ is essential for male meiosis and suppression of retrotransposon expression in the male germline. PLoS Genet. 5, e1000635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maatouk D. M., Kellam L. D., Mann M. R., Lei H., Li E., Bartolomei M. S., Resnick J. L. (2006). DNA methylation is a primary mechanism for silencing postmigratory primordial germ cell genes in both germ cell and somatic cell lineages. Development 133, 3411–3418 [DOI] [PubMed] [Google Scholar]
- Meissner A., Mikkelsen T. S., Gu H., Wernig M., Hanna J., Sivachenko A., Zhang X., Bernstein B. E., Nusbaum C., Jaffe D. B., et al. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metivier R., Gallais R., Tiffoche C., Le Peron C., Jurkowska R. Z., Carmouche R. P., Ibberson D., Barath P., Demay F., Reid G., et al. (2008). Cyclical DNA methylation of a transcriptionally active promoter. Nature 452, 45–50 [DOI] [PubMed] [Google Scholar]
- Mikkelsen T. S., Ku M., Jaffe D. B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T. K., Koche R. P., et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F., Weber M., Rebhan M., Roloff T. C., Richter J., Stadler M. B., Bibel M., Schubeler D. (2008). Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 30, 755–766 [DOI] [PubMed] [Google Scholar]
- Montgomery N. D., Yee D., Chen A., Kalantry S., Chamberlain S. J., Otte A. P., Magnuson T. (2005). The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr. Biol. 15, 942–947 [DOI] [PubMed] [Google Scholar]
- Okano M., Bell D. W., Haber D. A., Li E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 [DOI] [PubMed] [Google Scholar]
- Ollinger R., Childs A. J., Burgess H. M., Speed R. M., Lundegaard P. R., Reynolds N., Gray N. K., Cooke H. J., Adams I. R. (2008). Deletion of the pluripotency-associated Tex19.1 gene causes activation of endogenous retroviruses and defective spermatogenesis in mice. PLoS Genet. 4, e1000199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollinger R., Reichmann J., Adams I. R. (2010). Meiosis and retrotransposon silencing during germ cell development in mice. Differentiation 79, 147–158 [DOI] [PubMed] [Google Scholar]
- Palii S. S., Van Emburgh B. O., Sankpal U. T., Brown K. D., Robertson K. D. (2008). DNA methylation inhibitor 5-Aza-2’-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol. 28, 752–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp C., Dean W., Feng S., Cokus S. J., Andrews S., Pellegrini M., Jacobsen S. E., Reik W. (2010). Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463, 1101–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W. (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432 [DOI] [PubMed] [Google Scholar]
- Reuter M., Berninger P., Chuma S., Shah H., Hosokawa M., Funaya C., Antony C., Sachidanandam R., Pillai R. S. (2011). Miwi catalysis is required for piRNA amplification-independent LINE1 transposon silencing. Nature 480, 264–267 [DOI] [PubMed] [Google Scholar]
- Reynolds N., Collier B., Maratou K., Bingham V., Speed R. M., Taggart M., Semple C. A., Gray N. K., Cooke H. J. (2005). Dazl binds in vivo to specific transcripts and can regulate the pre-meiotic translation of Mvh in germ cells. Hum. Mol. Genet. 14, 3899–3909 [DOI] [PubMed] [Google Scholar]
- Rodic N., Oka M., Hamazaki T., Murawski M. R., Jorgensen M., Maatouk D. M., Resnick J. L., Li E., Terada N. (2005). DNA methylation is required for silencing of ant4, an adenine nucleotide translocase selectively expressed in mouse embryonic stem cells and germ cells. Stem Cells 23, 1314–1323 [DOI] [PubMed] [Google Scholar]
- Romanish M. T., Cohen C. J., Mager D. L. (2010). Potential mechanisms of endogenous retroviral-mediated genomic instability in human cancer. Semin. Cancer Biol. 20, 246–253 [DOI] [PubMed] [Google Scholar]
- Seisenberger S., Popp C., Reik W. (2010). Retrotransposons and germ cells: reproduction, death, and diversity. F1000 Biol. Rep. 2, 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki Y., Hayashi K., Itoh K., Mizugaki M., Saitou M., Matsui Y. (2005). Extensive and orderly reprogramming of genome-wide chromatin modifications associated with specification and early development of germ cells in mice. Dev. Biol. 278, 440–458 [DOI] [PubMed] [Google Scholar]
- Seki Y., Yamaji M., Yabuta Y., Sano M., Shigeta M., Matsui Y., Saga Y., Tachibana M., Shinkai Y., Saitou M. (2007). Cellular dynamics associated with the genome-wide epigenetic reprogramming in migrating primordial germ cells in mice. Development 134, 2627–2638 [DOI] [PubMed] [Google Scholar]
- Simpson A. J., Caballero O. L., Jungbluth A., Chen Y. T., Old L. J. (2005). Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 5, 615–625 [DOI] [PubMed] [Google Scholar]
- Siomi M. C., Sato K., Pezic D., Aravin A. A. (2011). PIWI-interacting small RNAs: the vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 12, 246–258 [DOI] [PubMed] [Google Scholar]
- Tachibana M., Matsumura Y., Fukuda M., Kimura H., Shinkai Y. (2008). G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 27, 2681–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B. B., Siddiqui A., et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 [DOI] [PubMed] [Google Scholar]
- Unhavaithaya Y., Hao Y., Beyret E., Yin H., Kuramochi-Miyagawa S., Nakano T., Lin H. (2009). MILI, a PIWI-interacting RNA-binding protein, is required for germ line stem cell self-renewal and appears to positively regulate translation. J. Biol. Chem. 284, 6507–6519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco G., Hube F., Rollin J., Neuillet D., Philippe C., Bouzinba-Segard H., Galvani A., Viegas-Pequignot E., Francastel C. (2010). Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc. Natl. Acad. Sci. USA 107, 9281–9286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. P., Bestor T. H. (1999). Cytosine methylation and mammalian development. Genes Dev. 13, 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. P., Chaillet J. R., Bestor T. H. (1998). Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20, 116–117 [DOI] [PubMed] [Google Scholar]
- Waterston R. H., Lindblad-Toh K., Birney E., Rogers J., Abril J. F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., An P., et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 [DOI] [PubMed] [Google Scholar]
- Weber M., Hellmann I., Stadler M. B., Ramos L., Paabo S., Rebhan M., Schubeler D. (2007). Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457–466 [DOI] [PubMed] [Google Scholar]
- Xu M., You Y., Hunsicker P., Hori T., Small C., Griswold M. D., Hecht N. B. (2008). Mice deficient for a small cluster of Piwi-interacting RNAs implicate Piwi-interacting RNAs in transposon control. Biol. Reprod. 79, 51–57 [DOI] [PubMed] [Google Scholar]
- Yoder J. A., Walsh C. P., Bestor T. H. (1997). Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13, 335–340 [DOI] [PubMed] [Google Scholar]
- Zamudio N., Bourc’his D. (2010). Transposable elements in the mammalian germline: a comfortable niche or a deadly trap? Heredity (Edinb.) 105, 92–104 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.