

Abstract
Mutations that disrupt function of the human inwardly rectifying potassium channel KIR2.1 are associated with the craniofacial and digital defects of Andersen-Tawil Syndrome, but the contribution of Kir channels to development is undefined. Deletion of mouse Kir2.1 also causes cleft palate and digital defects. These defects are strikingly similar to phenotypes that result from disrupted TGFβ/BMP signaling. We use Drosophila melanogaster to show that a Kir2.1 homolog, Irk2, affects development by disrupting BMP signaling. Phenotypes of irk2 deficient lines, a mutant irk2 allele, irk2 siRNA and expression of a dominant-negative Irk2 subunit (Irk2DN) all demonstrate that Irk2 function is necessary for development of the adult wing. Compromised Irk2 function causes wing-patterning defects similar to those found when signaling through a Drosophila BMP homolog, Decapentaplegic (Dpp), is disrupted. To determine whether Irk2 plays a role in the Dpp pathway, we generated flies in which both Irk2 and Dpp functions are reduced. Irk2DN phenotypes are enhanced by decreased Dpp signaling. In wild-type flies, Dpp signaling can be detected in stripes along the anterior/posterior boundary of the larval imaginal wing disc. Reducing function of Irk2 with siRNA, an irk2 deletion, or expression of Irk2DN reduces the Dpp signal in the wing disc. As Irk channels contribute to Dpp signaling in flies, a similar role for Kir2.1 in BMP signaling may explain the morphological defects of Andersen-Tawil Syndrome and the Kir2.1 knockout mouse.
Keywords: Inwardly rectifying potassium channel, TGFβ, Dpp, BMP, Drosophila, Mouse
INTRODUCTION
Mutations in inwardly rectifying K+ channels are associated with patterning defects. For example, mutations that disrupt Kir2.1 are associated with the morphological defects of Andersen-Tawil Syndrome (ATS): cleft palate, micrognathia, hypertelorism, dental abnormalities, clinodactyly, syndactyly and shortened phalanges (Andersen et al., 1971; Tawil et al., 1994; Sansone et al., 1997; Canún et al., 1999; Yoon et al., 2006a; Yoon et al., 2006b). Furthermore, deletion of the mouse Kir2.1 gene (Kcnj2 – Mouse Genome Informatics) causes cleft palate and narrow maxilla (Zaritsky et al., 2000).
Inwardly rectifying K+ channels comprise four subunits (Choe, 2002; MacLean et al., 2002). Mutations that disrupt Kir2.1 cause periodic paralysis, heart arrhythmia and morphological defects in individuals with ATS. The most severe defects in such individuals are caused by dominant-negative Kir2.1 subunits that complex with other subunits and alter the selectivity filter, affecting K+ conductivity of the entire heteromeric channels (MacLean et al., 2002; Bichet et al., 2003; McLerie and Lopatin, 2003). The electrophysiological consequences of dysfunctional Kir2.1 are understandable, but the mechanism underlying the developmental abnormalities is unclear.
Despite the growing body of evidence for a role of K+ channels in development, the mechanism by which they influence pattern formation is not understood. Similar cleft palate and digit defects can be caused by loss of transforming growth factor β (TGFβ)/bone morphogenetic protein (BMP), Wnt-Wingless (Wg) or Notch signaling (Jiang et al., 1998; Tucker et al., 1998a; Tucker et al., 1998b; Dudas et al., 2004; Liu et al., 2005; Bandyopadhyay et al., 2006; Casey et al., 2006; Richardson et al., 2009; Xu et al., 2010; Menezes et al., 2010; Ferretti et al., 2011; He et al., 2011; Jin et al., 2011; Lin et al., 2011). We tested the hypothesis that inhibiting Kir2.1 channels interferes with TGFβ/BMP signaling.
The TGFβ/BMP superfamily has orthologous pathways in multicellular organisms (Padgett et al., 1987; Sampath et al.,, 1993; Derynck et al., 1985; Mason et al., 1985; Ohta et al., 1987). In Drosophila, Dpp is a BMP homolog that is required for embryonic development, growth and patterning of adult structures, including the wing (Gelbart, 1982; Letsou et al., 1995; O’Connor et al., 2006; Blair, 2007). Dpp binds type 1 and type 2 kinase receptors (Nellen et al., 1994; Letsou et al., 1995; Ruberte et al., 1995). Upon Dpp binding, type 2 receptors phosphorylate type 1 receptors (thickveins), which phosphorylate Mothers against Dpp (Mad) to propagate the signal intracellularly (Kim et al., 1997).
Drosophila is an excellent system for determining the mechanism underlying developmental defects, because conserved developmental signaling pathways are well-defined and non-redundant. Kir2.1 has three homologs in Drosophila. Irk2 is 50% identical and 69% similar to Kir2.1. Two other homologs, Irk1 and Irk3, may form heterotetromeric channels with Irk2 as with some Kir channels in mammals. Electrophysiological and expression studies demonstrate that these channels function similarly to mammalian Kir channels (Döring et al., 2002). In this study, we test the hypothesis that Irk2 function is necessary for developmental signaling. We use an Irk2 dominant-negative allele, Irk2-deficient alleles, an Irk2 p-element allele and RNAi to show that Irk channels are necessary for patterning and growth of the Drosophila wing. We conclude that disruption of Irk channels leads to reduction in Dpp signaling and wing defects. These studies explain the mechanism by which K+ channels regulate development and provide one possible explanation for the defects in individuals with ATS and in Kir2.1 knockout mice.
MATERIALS AND METHODS
Maintenance of Drosophila stocks
Stocks were maintained on cornmeal food at 25°C or 18°C in a Percival incubator model 122 vL (Percival Scientific).
Generation of the UAS-Irk2DN and UAS-Irk2WT fly strains
irk2A from Berlinw1118 fly cDNA was cloned into the EcoRI and XhoI sites of the pUAST vector. PCR was performed with cDNA template and primers (GGAATTCCATGCGTTTCAATTTCTCC and CCGCTCGAGCGGCTAGGA GGCCTGGTCAGA) to add EcoRI and XhoI sites. Sequencing ensured fidelity of the construct. UAS-Irk2 DN was constructed by cutting irk2A out of UAS-irk2 WT with EcoRI and XhoI, and ligating into pET. The GYG of pET-Irk2A template plasmid was mutated to AAA using a QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) with the following primers: ACGCAGCACACTATTGCCGCTGCCGTCCGAACCACCTCG and CGAGGTGGTTCGGACGGCAGCGGCAATAGTGTGCTGCGT. Irk2-DN was removed from the pET vector with EcoRI and XhoI restriction enzymes and ligated into pUAST. All constructs were sequenced to verify the GYG to AAA mutations. We injected UAS-Irk2 WT or UAS-Irk2 DN plasmid with transposase DNA into 1-hour-old Berlinw1118 embryos. Matured injected flies were crossed to Berlinw1118 and progeny with the transgene were selected by eye color. irk1-AAA and irk3-AAA were generated with the same strategy using primer pairs: ACCCAGACGACGATAGCCGCTGCCAATC/CGTCACATAGCGATTGGCAGCGGCTATC (Irk1-AAA) and ATCGAGTCCAAGATACGAGTCTACATCATC/GATGATGTAGACTCGTATCTTGGACTCGATGGA (Irk3-AAA).
Drosophila strains
The Vienna Drosophila RNAi Center (VDRC) provided stocks that express short RNA hairpins complementary to irk channel genes under control of an inducible upstream activating sequence (UAS) promoter (Dietzl et al., 2007; Yapici et al., 2008). The GAL4 activator controls expression of genes behind UAS. Ubiquitous expression of GAL4 (and thus the irk siRNA or irkX-AAA subunits) was achieved with the daughterless or actin promoter. Wing-directed expression was achieved with MS1096-GAL4 or engrailed-GAL4. To accomplish irk siRNA, VDRC stocks irk2 108140, irk1 28430 and Irk3 101174 were mated to the flies with appropriate GAL4 driver. The UAS-P35 stock was a gift from Sally Kornbluth (Duke University, NC, USA). All balancer strains, GAL4 driver strains [MS1096 (w[1118], P{w[+mW.hs]=GawB}Bx[MS1096]), engrailed, daughterless, A9 and actin], dpphr92 and tkv7 were obtained from the Bloomington Drosophila Stock Center.
Generation of Irk2 Df strains
An irk2-deficient stock [Df(3R)Irk2exel 6194{w[+rC]=xp-U} exel 6194)] was from the Exelixis custom deficiency generation system (Parks et al., 2004). For clarity, we refer to this strain as irk2DfA. The irk2DfA deficiency covers 280 kb and removes 25 genes, including Irk2 and is homozygous lethal. We used the same scheme (Parks et al., 2004) to generate a second irk2 deficiency. FLP recombinase expression was induced in Drosophila larva carrying P(ry+t7.2:hsFLP)12, P(WH)f002619 and PBac(RB)e01487 at 37°C in daily 1 hour increments. FLP expression induced recombination between PiggyBac P(WH)f002619 and PBac(RB)e01487 generating flies that lack the genomic region between the two insertion sites, including irk2 (designated Irk2DfB). irk2DfA/irk2DfB flies are viable and lack irk2 entirely without removing more than five surrounding genes. The deletion was verified by PCR.
Irk2 in situ
Standard irk2 in situ was performed (Lécuyer et al., 2008). The irk2 probe sequence was: AGGCCTGGTCAGAATGATTGTGGGACAGTTGACGCATGGTAAAGGTGGTTTCTGGTGTGCGGAATCCCTCCTGGATCTTGTAGATCTCGTTCAATTCCCGAGCACTGCACAACGGAGTGTCCACCTGAGTGGTTTCGTTAAAGCGAGCGTAGTCGATTTCATAGGCCTGCAGATCCTTGTTGTACAACACCACTGGATCGAAACGATGTCCCCAAAGGATCTCA.
Quantitative RT-PCR
Total RNA was isolated from third instar larvae (SV Total RNA Isolation System, Promega). Total cDNA was reverse transcribed in vitro using Super Script III reverse transcriptase (Invitrogen, Grand Island, NY). Real-time PCR was accomplished with 100 ng of template cDNA, the Syber Green Assay kit (Applied Biosystems) and 200 nM primers in a Step One plus Real-Time PCR thermal cycling block (Applied Biosystems, Carlsbad, CA). Primer sequences are as follows: RP49, AAGAAGCGCACCAAGCACTTCATC and TCTGTTGTCGATACCCTTGGGCTT; irk1, GCCATCGTTTCGTGAATGTGGTGT and AGTGTCCACGTCGTAGGTGTTGTT; irk2, ATGGCCGGAATAGTGTTTGCCAA and GGCAAATTACGGCGTGTTTGGAGA; irk3, TTATTCTCTGGCCCGATGTGGTGT and GGCAGCATTGAAGAGTTTCGCTGT.
The data were obtained using Step one software V2.1 (Applied Biosystems, Carlsbad, CA). Fold difference was calculated with ΔΔ CT (Livak and Schmittgen, 2001).
Immunohistochemistry and TUNEL staining
Wing discs were isolated from third instar larvae. Discs were stained with anti-phopho-Smad1/5 Ser463/465 (Cell Signaling) (Cao et al., 2006) and 1:250 goat anti-rabbit IgG CY3 (Millipore). Anti-Spalt staining was accomplished with 1:250 anti-Spalt (Halachmi et al., 2007) from Adi Salzberg (Technion-Israel Institute of Technology, Haifa, Israel) and 1:200 goat-anti-rabbit IgG CY3. Achaete was stained with 1:5 mouse-anti-achaete [Developmental Studies Hybridoma Bank (DSHB), Iowa (Skeath and Carroll, 1991)], a 1:200 dilution of the secondary antibody, Alexa Fluor 488 anti-mouse IgG (Invitrogen). Wingless was stained with 1:20 mouse-anti-wingless [DSHB, Iowa (Brook and Cohen, 1996)]. The secondary antibody was 1:200 goat-anti-mouse IgG CY5 (Millipore). Apoptosis was detected using DeadEnd Fluorometric TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) system (Promega, Madison, WI). Wing discs were mounted with Vectashield mounting medium (Vector Laboratory, Burlingame, CA).
Imaging
Wing discs were imaged at 20× magnification with a Zeiss Axioscope A1 (Carl Zeiss Microscopy LLC, NY) in bright field with a Texas-red filter for anti-pMAD, anti-Spalt and anti-Wingless staining, and a GFP filter for TUNEL and Achaete staining. An AxioCam HRc digital camera with the Axioscope A1 computer program photographed images. For the intensity analysis of anti-pMad staining, control and experimental samples were processed and imaged in parallel with identical settings for exposure (33.2 ms). The fluorescent microscopy images were analyzed with Slidebook (Intelligent Imaging Innovations). Peak intensity was determined by subtracting the minimum fluorescence from the maximum fluorescence intensity in an anterior-to-posterior cross-section of the wing disc. Five independent cross-sections were used for each disc. Anti-Wingless and anti-Achaete stained wing discs were imaged with CY5 and GFP filters, respectively.
Kir2.1 mouse
Generation of the Kir2.1-knockout mouse has been described previously (Zaritsky et al., 2000). Bone and cartilage were dissected from newborn pups and placed into 95% ethanol. Ethanol was replaced with 0.03% Alcian Blue, 80% ethanol and 20% acetic acid. Tissue was washed with 95% ethanol before incubation in 2% KOH. Tissue was incubated in 0.03% Alizarin Red. Skeletons were cleared in 20% glycerol. All mouse protocols were approved by the UCSF IACUC.
Statistical analysis
Error bars represent the standard error of the mean (s.e.m.). Raw P values were determined using a two-tailed Student’s t-test. Each experiment consisted of at least three repeated trials. The number of flies tested is given for each figure in the legend.
RESULTS
Kir2.1 mouse phenotypes
Deletion of the gene that encodes Kir2.1 in mice results in patterning defects that are similar to BMP knockout phenotypes. For example, Zaritsky et al. reported that homozygous Kir2.1 knockout mice have cleft palates (Zaritsky et al., 2000). We further characterized skeletal deformations in homozygous Kir2.1 knockout animals (n=13) (Fig. 1). The anterior and posterior palatine processes and vomer bones are reduced in size. In addition to the defects that were previously published, we found digit defects in all homozygous Kir2.1 knockout animals (Fig. 1). It has previously been reported that heterozygous Kir2.1 animals appear normal. However, dissection of heterozygous Kir2.1 mice revealed reduction in the size of anterior and posterior palatine processes and apparent decreased ossification in 98% of heterozygous pups (n=40) (Fig. 1C,D). By contrast, no palate or digit defects were found in wild-type siblings (n=41). The developmental defects of Kir2.1 knockout mice are similar to the cleft palate defects of Tgfb2 and Tgfb3 knockout animals (Sanford et al., 1997; Taya et al., 1999). Disruption of TGFβ co-factors, regulators and receptors also leads to cleft palate in mice (Peters et al., 1998; Dudas et al., 2004; Bjork et al., 2010). Kir2.1 knockout digital defects are similar to the BMP2/4 limb conditional knockout (Bandyopadhyay et al., 2006). Other signaling pathways, such as Notch and Wnt, also contribute to these developmental processes (Jiang et al., 1998; Casey et al., 2006; Richardson et al., 2009; Xu et al., 2010). We tested the hypothesis that defective inwardly rectifying K+ channels interfere with TGFβ/BMP signaling using the genetic tools of Drosophila melanogaster. The role of Dpp, a Drosophila BMP ligand, is well characterized in the wing (Segal and Gelbart, 1985; Blair, 2007), where Drosophila homologs of Kir2.1 are expressed (supplementary material Fig. S1) (MacLean et al., 2002). Thus, Drosophila provides an ideal system for investigating whether inwardly rectifying K+ channel function is necessary for BMP signaling.
Fig. 1.
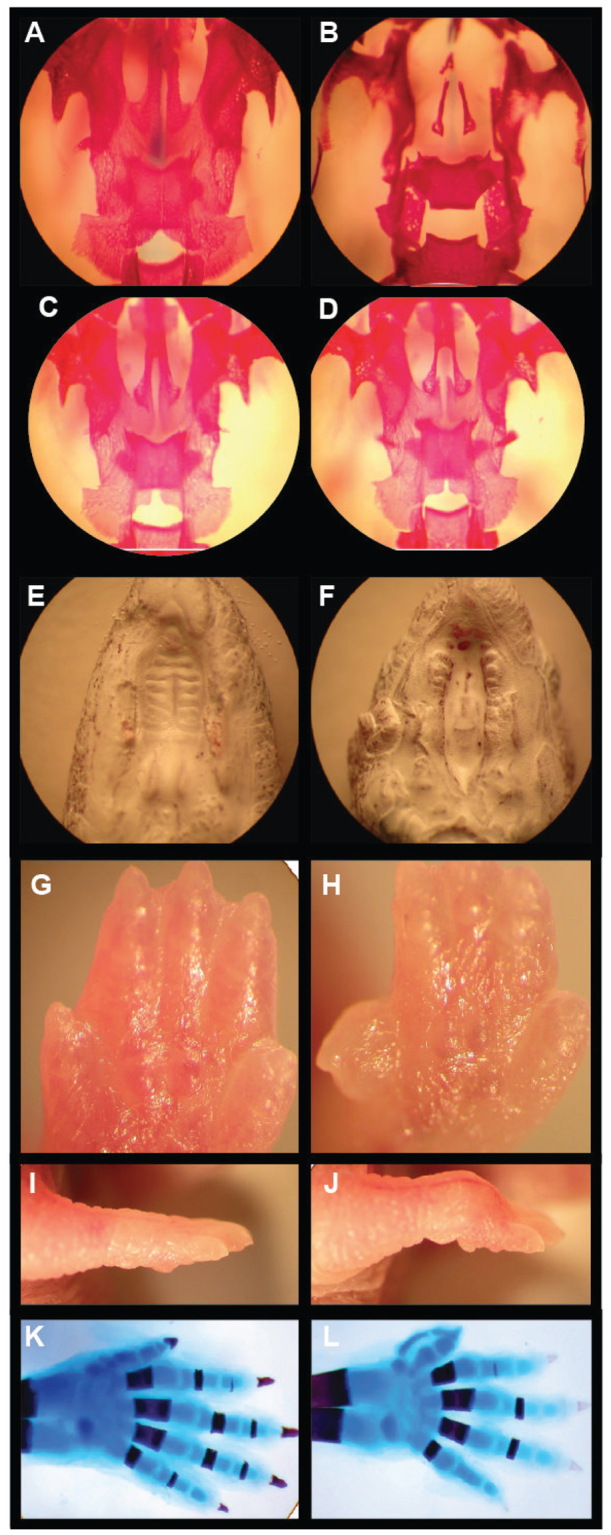
Knockout of mouse Kir2.1 causes cleft of the secondary palate and patterning defects of the skeletal digits. (A-D) Ventral view of Alizarin Red (bone) staining of palates of newborn wild-type (A), Kir2.1 knockout (B) and Kir2.1 heterozygous knockout (C,D) pups. (E,F) Ventral view of wild-type palate (E) and Kir2.1 knockout cleft of secondary palate (F). (G,H) Forelimb of wild-type (G) and Kir2.1 knockout (H). (I,J) Ventral view of a wild-type (I) and Kir2.1 knockout (J) forepaw. (K,L) Whole-mount forelimb skeletons from newborn animals stained with Alcian Blue and Alizarin Red: wild-type (K); preaxial digit duplication of the forelimb is shown in the Kir2.1 knockout (L). The dorsal view of the right limb is shown. Anterior is upwards.
Irk2 deficiency disrupts wing patterning
To define the role of Irk2 in development, we examined the phenotypes of flies that lack irk2 and the surrounding region from 19260K to 19350K of chromosome 3R (supplementary material Fig. S2). irk2-deficient flies (irk2DfA/irk2DfB) were compared with wild-type flies. When Irk2DfA/TM6 is mated to irk2DfB/TM6, more than the expected 33% of the progeny are Irk2DfA/Irk2DfB, indicating that Irk2 is not necessary for viability (n=380). The wings from irk2-deficient flies have wing venation defects: incomplete or branched posterior cross veins, incomplete L5 vein, bifurcations of L3 and L4 veins, and wing bristle transformations (Fig. 2; Table 1). Thirty-nine percent of the irk2-deficient flies have held-out wings from hinge defects (supplementary material Fig. S3). Rarely, irk2-deficient wings are notched or smaller than wild type (Fig. 2C). The penetrance and severity of wing phenotypes in Irk2-deficient flies were dependent both on sex and the temperature during development. Phenotypes were most severe in males raised at low temperatures. At 25°C, 38% of male irk2-deficient flies had wing defects (n=172). Seventeen percent of males that were heterozygous for either Irk2 deficiency had wing venation defects (n=189 and 121). Sixty-two percent of flies harboring a p-element insertion in irk2 (Irk2G8696) had wing venation defects or small wings and 48% had wing hinge defects when raised at 25°C. All male Irk2G8696 flies had wing bristle transformations (Fig. 2A,D; supplementary material Fig. S3; Table 1).
Fig. 2.
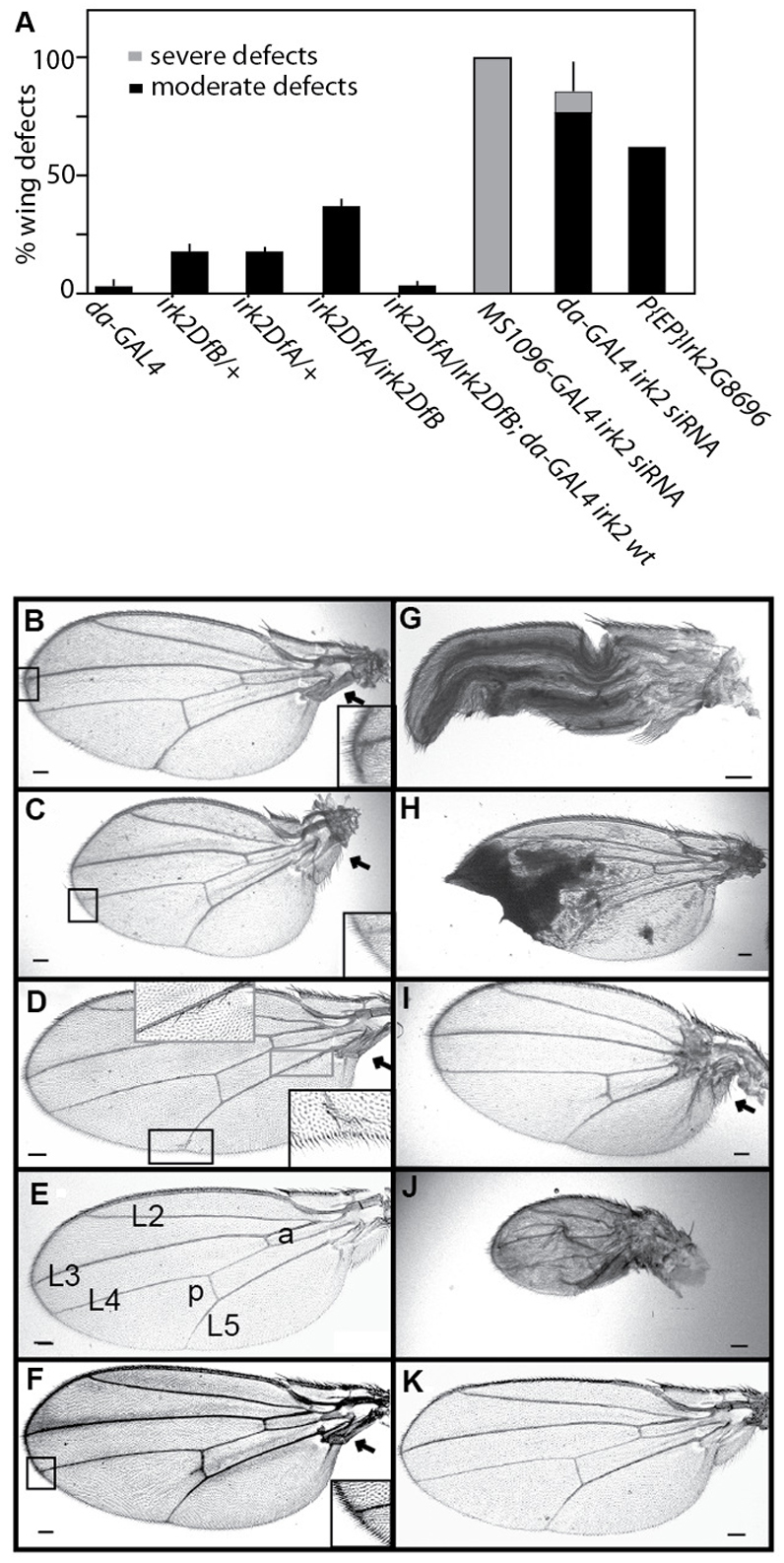
Reduced Irk2 function wing phenotypes. (A) Quantification of wing defects when irk2 function is decreased (n≥100 flies). Error bars indicate s.e.m. (B) Bifurcation of L2 and L3 wing veins from Irk2DfA/Irk2DfB female. (C) Reduced wing size, angled hinge and bifurcation from L4 of Irk2DfA/Irk2DfB female. (D) Bristle transformations and incomplete L5 from male irk2G8696. (E) Wild-type male wing L2-5 longitudinal veins; anterior (a) and posterior (p) crossveins are labeled. (F) irk2DfA/irk2DfB male fly wing. (G) Wing expansion defects from daughterless-GAL4 Irk2 siRNA female. (H) Ubiquitous expression of irk2 siRNA causes patches of wing tissue necrosis. (I) Small wing and hinge defects from MS1096-GAL4 Irk2 siRNA female. (J) Small wing, hinge and venation defects from MS1096-GAL4 Irk2 siRNA male. (K) Rescued male irk2DfA/irk2DfB; engrailed-GAL4 UAS-irk2WT fly wing. Arrows indicate wing hinge defects. Scale bars: 100 μm.
Table 1.
Wing venation defects
To verify that venation defects were the result of irk2 deficiency, irk2WT was expressed ectopically in whole Irk2-deficient flies. Wing venation defects other than L5 defects are completely rescued by the ectopic expression of Irk2WT. Only 22% of wings of irk2DfA/irk2DfB; UAS-irk2WT flies had L5 venation defects compared with 92% of irk2-deficient flies that had wing venation defects when raised under the same conditions (Table 1; Fig. 2F). To test for the tissue specificity of the requirement for Irk2, irk2WT was expressed using engrailed-GAL4 in the posterior compartment of irk2-deficient wing discs. Wing L5 venation defects were decreased to 37% in flies that express exogenous irk2WT in an irk2-deficient background. All other irk2Df/Df wing defects were rescued by engrailed-GAL4; UAS-Irk2WT (Fig. 2F,K), indicating that loss of irk2 function is responsible for wing venation phenotypes and the requirement is partially fulfilled if irk2 is expressed in the posterior compartment of the wing. As a comparison, only 4% of the male flies harboring the engrailed-GAL4 driver without the irk2 transgene had wing venation defects (Table 1). We verified that endogenous irk2 was deleted and that exogenous Irk2 was provided in rescued flies by amplifying irk2 DNA from single flies with primers specific to either genomic Irk2 or the UAS-irk2 transgene (Fig. 2). These data show that irk2 is required non-cell-autonomously in the wing disc to specify wing venation patterning and that the loss of irk2 is responsible for venation phenotypes that are observed in irk2-deficient flies.
Irk2 siRNA expression disrupts wing patterning
To characterize the tissue-specific requirement for Irk2, we expressed small-interfering irk2 RNA (siRNA) under the control of either ubiquitous (daughterless) or wing-directed (MS1096) GAL4 drivers. When irk2 siRNA was expressed ubiquitously using daughterless-GAL4, we found defects in 84% of the wings: 73% of the wings had venation defects, 9% failed to expand correctly (Fig. 2G) and 4% of the wings had tissue necrosis (Fig. 2H). Wing-directed expression of irk2 siRNA causes reduction in wing size, wing venation defects and hinge defects that result in the held-out wings phenotype for all surviving flies (Fig. 2; supplementary material Fig. S3). We designated venation defects and reduction in wing size as moderate defects; any additional defects are designated as severe. Male flies that express Irk2 siRNA have more severe wing phenotypes than female flies of the same genotype. Ubiquitous expression of irk2 siRNA can lead to wing expansion defects, whereas wing-directed irk2 siRNA does not. The difference in phenotypes suggests that expansion of the wing requires Irk function outside the wing whereas patterning requires Irk channel function in the wing disc itself.
Phenotypes that result from ubiquitous or wing-directed expression of the irk2 siRNA are more severe and more penetrant than phenotypes associated with irk2 deficiency. We reasoned that expression of irk2 siRNA could affect the transcription of other irk subunits (irk1 and irk3). To determine whether irk2 siRNA changed the expression of irk1 and irk3, we used quantitative RT-PCR to quantify irk1, irk2 and irk3 mRNA from flies that ubiquitously express Irk2 siRNA and compared mRNA levels with those from flies with the daughterless-GAL4 driver without the Irk2 siRNA transgene. irk2 mRNA is reduced 85.7% in flies that express irk2 siRNA demonstrating that the irk2 siRNA effectively reduces expression of irk2. Surprisingly, irk1 was increased 14-fold and irk3 mRNA levels are increased 4.6-fold in flies that express the irk2 siRNA (supplementary material Fig. 4A). To determine whether a similar increase in irk1 and irk3 expression occurred in irk2-deficient flies, we compared irk1, irk2 and irk3 mRNA levels in wild-type and irk2-deficient flies. As expected, no Irk2 mRNA was detected in irk2-deficient flies. irk3 mRNA levels increased 3.4-fold, but irk1 transcript levels were not significantly different in irk2-deficient flies compared with wild-type flies (supplementary material Fig. S4B). Phenotypes of the irk2-deficient wings are less severe than those of irk2 siRNA. This leads to the conclusion that Irk subunits are not entirely functionally equivalent. To block the function of the entire Irk channel, we generated a dominant-negative Irk2 subunit.
Expression of dominant-negative Irk2 causes severe wing defects
Inwardly rectifying K+ channel subunits can form heterotetrameric channels in mammals. Incorporation of a single subunit with a mutated selectivity filter (GYG to AAA) into the complex completely blocks ion flow through the channels in mammals (Bichet et al., 2003). We used the UAS/GAL4 system to express dominant-negative Irk transgenes to block the Irk channel in a tissue- and time-specific manner. A transgenic animal expressing this Irk2 (Irk2DN) gene is expected to demonstrate the effects of the loss of the entire channel. By contrast, in an irk2 null, Irk1 and Irk3 could theoretically form a functional channel without Irk2, thereby at least partially compensating for reduced Irk2 function. We generated three transgenic Irk2DN lines: Irk2DN5.1, Irk2DN5.2 and Irk2DN13. Ubiquitous expression of Irk2DN or irk2WT was accomplished by mating transgenic UAS-irk2WT/balancer and UAS-Irk2DN/balancer flies with actin-GAL4 or daughterless-GAL4 transgenic flies. An approximately equal number of transgenic and balancer adult progeny result from the UAS-irk2WT cross. No Irk2DN5.1- or Irk2DN5.2-expressing flies survive from the UAS-Irk2DN cross whereas over 90 balancer sibling adults survive. Irk2DN13-expressing flies survive to adulthood when raised at 25°C, but only 20% of Irk2DN13 survive when flies are raised at 18°C (Fig. 3A). Larvae that express Irk2DN are marked with actin-GFP and can be compared in survival to their balancer siblings after hatching. Only 17% of the larva expected to ubiquitously express Irk2DN hatch and none of these survive 48 hours after hatching (Fig. 3B). Endogenous Irk2 functions in the developing wing, so we directed expression of Irk2DN to block Irk channels in the wing disc.
Fig. 3.
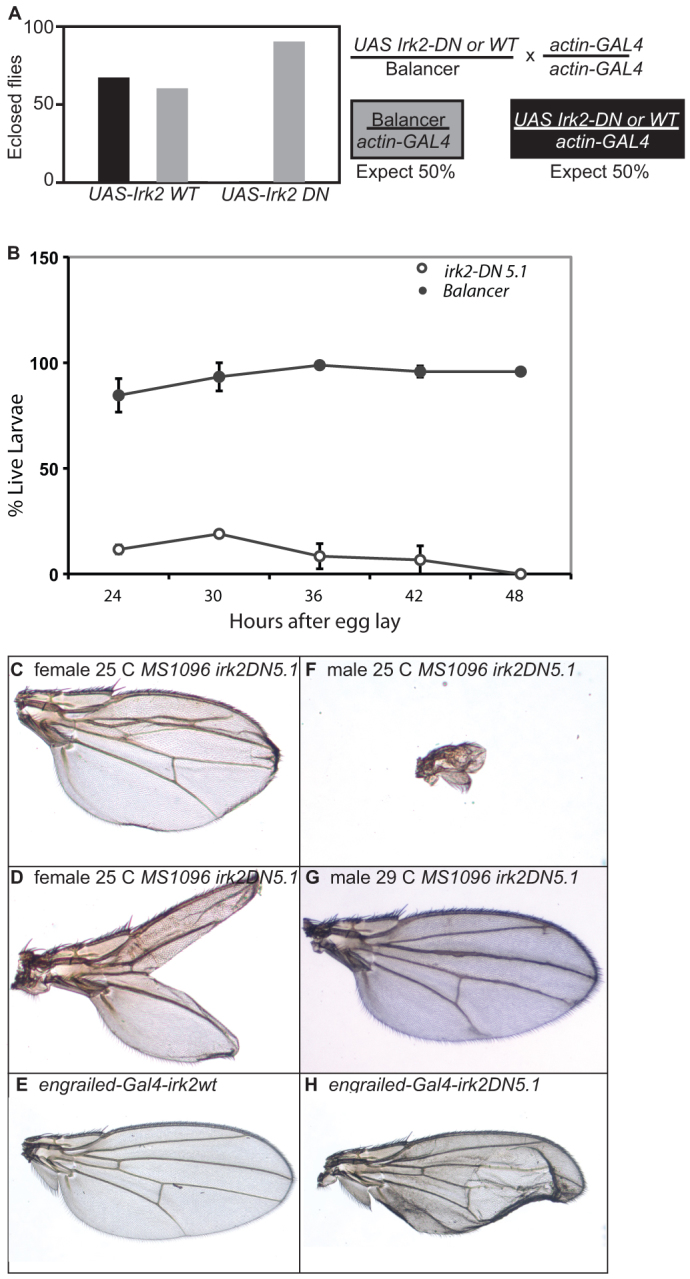
Phenotypes of flies expressing Irk2DN. (A,B) Graphs of adult (A) and larval (B) survival of actin-GAL4 UAS-Irk2WT, actin-GAL4 UAS-Irk2DN and siblings without transgene. Error bars indicate s.e.m. (C) L2-3 fusion in wing from MS1096-GAL4 Irk2DN5.1 female at 25°C. (D) Bifurcated wing from MS1096-GAL4 Irk2DN5.1 at 25°C. (E) engrailed-GAL4 UAS-irk2WT. (F) Wing of MS1096-GAL4 Irk2DN5.1 male at 25°C. (G) MS1096-GAL4 UAS Irk2DN5.1 at 29°C. (H) engrailed-GAL4 UAS Irk2DN5.1 at 25°C.
We expressed Irk2DN and irk2WT in wing discs using MS1096-GAL4 and engrailed-GAL4 (Gullaud et al., 2003) to block Irk channels during development. Wing defects include blisters, fusion of longitudinal veins, thickened veins and complete or partial ablation of the wing blade (Fig. 3). Defects were more severe and penetrant in males raised at lower temperatures. To control for sex differences, only female defects are reported in quantification of defects. We report wing defects from two different transgenic lines: Irk2DN5.1 and Irk2DN5.2. Wing-directed expression of Irk2DN causes moderate and severe wing defects. Moderate wing defects include wings that are small, collapsed or blistered. Moderate defects also include venation defects such as branching of the posterior cross-vein, L2-3 collapse, L4 bifurcation, incomplete L5 and wing bristle transformations. Severe defects include partial or complete loss of the wing blade.
All of the flies that express Irk2DN in transgenic line 5.1 have wing defects with 6% of the wings classified with moderate defects and 94% with severe defects. Expression of Irk2DN in transgenic line 5.2 resulted in 42% moderate wing defects and 4% severe defects (Fig. 3I). Less than 25% of MS1096-GAL4 UAS-irk2WT flies have minor venation defects, similar to MS1096-GAL4 without a UAS transgene. Expression of Irk2DN5.2 using the engrailed-GAL4 driver causes wing venation and blistering defects, whereas expression of Irk2WT with the same driver does not cause defects (Fig. 3E,H). As Irk2DN expression causes defects that are more severe than loss of Irk2, we conclude that Irk2 forms a heteromeric channel that is necessary for patterning of the adult wing in Drosophila.
We reasoned that heteromeric channels could be made up of a combination of Irk1, Irk2 and/or Irk3 subunits. Thus, Irk1 and Irk3 could form a partially functional channel when lacking the Irk2 subunit. If this was the case, we would expect that reduced irk1 and irk3 function should cause similar phenotypes to irk2Df, siRNA or Irk2DN. Ubiquitous expression of irk1 siRNA results in 88% lethality and ubiquitous expression of irk3 siRNA results in 98% lethality (n=145) (Fig. 4A). As irk2-deficient flies survive to adulthood, it is likely that Irk1 and Irk3 play a more substantial role in embryonic development than does Irk2. It is likely that ubiquitous expression of Irk2DN causes embryonic lethality by blocking the function of a heteromeric channel that includes Irk1 and/or Irk3.
Fig. 4.

Irk1 and Irk3 siRNA phenotypes. (A) Ubiquitous expression of Irk1 and Irk3 siRNA causes lethality. (B) Wing-directed Irk1 and Irk3 siRNA causes wing venation defects and a reduction in wing size. (C) Irk1-AAA or Irk3-AAA ubiquitous expression does not decrease survival. (D) Quantification of Irk1 mRNA in wild-type, actin-GAL4-UAS Irk1WT and actin-GAL4-UAS Irk1AAA. (E) Quantification of Irk2 mRNA in wild-type, actin-GAL4-UAS Irk2WT and actin-GAL4-UAS Irk2AAA. (F) Quantification of Irk3 mRNA in wild-type, actin-GAL4-UAS Irk3WT and actin-GAL4-UAS Irk3AAA. Data are mean±s.e.m.
To examine the contribution of Irk1 and Irk3 to the development of the wing, we expressed irk1 and irk3 siRNA in the wing with MS1096-GAL4. Wing-directed expression of Irk1 and Irk3 siRNA in the wing results in venation defects in 100% of wings from animals of both genotypes (n=117 and n=92, respectively) (Fig. 4B). Expression of Irk3 siRNA caused a reduction in wing size and hinge defects in addition to venation defects in 40% of the wings. Wing defects that result from reduced irk1 and irk3 support the conclusion that Irk1, Irk2 and Irk3 form a heteromeric channel that is necessary for wing development.
To determine if the conserved GYG of Irk1 and Irk3 play the same structural role as for Irk2, we generated mutant alleles of both, changing the conserved GYG to AAA. Ubiquitous expression of irk1-AAA, irk1WT, irk3-AAA or irk3WT did not decrease survival or cause wing defects (n=127 Irk1-AAA, 129 Irk1WT, 132 Irk3-AAA and 124 Irk3WT) nor did wing-directed expression of mutant irk1-AAA or irk3-AAA cause wing defects (n=107 and 113) (Fig. 4C). We ensured that the Irk2DN and Irk2WT constructs were expressed using QRT-PCR (n=3 trials) (Fig. 4D-F). We conclude that Irk1, Irk2 and Irk3 are important to channel function, but play different structural roles in the heteromeric Irk channel.
Reduced Dpp signaling enhances Irk2DN phenotypes
Disruption of irk2 function with siRNA, irk2 deficiency or expression of Irk2DN causes defects in the wing that are similar to defects caused by compromised Dpp signaling (Gelbart, 1982; Spencer et al., 1982; Irish and Gelbart, 1987; Letsou et al., 1995; Cook et al., 2004; del Alamo Rodríguez et al., 2004). A possible explanation for Irk2 developmental defects is that Irk channels are necessary for proper Dpp signaling. We undertook genetic interaction studies by disrupting the function of Dpp (BMP ligand) or Thickveins (Tkv, BMP type 1 receptor) in an Irk2DN background.
To determine how defective Dpp signaling affects the Irk2DN wing phenotype, either the Irk2DN or the irk2WT transgene was expressed in wings of dpphr92/+ or tkv7/+ flies. Owing to differences in phenotype severity, we report defects from females only. Defective Dpp signaling enhances Irk2DN phenotypes (Fig. 5). When Irk2DN5.2 is expressed in a tkv7/+ mutant background, total wing defects increased from 46% to 72%. When Irk-2DN5.2 is expressed in a dpphr92/+ background, 84% of flies have wing defects and 29% are severely defective, whereas without compromised Dpp signaling, only 4% are classified as severe in the Irk2DN5.2 line (Fig. 5). In the more severe Irk2DN5.1 transgenic line, 94% of flies have severe wing defects and when Irk2DN5.1 is expressed in a tkv7/+ background, 99% of the flies have severe wing defects. Similarly, 97% of flies have severe wing defects in Irk2DN5.1; dpphr92/+ flies. Expression of the wild-type Irk2 transgene caused minor wing venation defects in 20% of the flies, but these defects did not increase in penetrance or severity when the transgene was expressed in the dpphr92/+ or tkv7/+ background (16% and 15%, respectively). Thus, Irk2DN phenotypes are enhanced by reducing function of Dpp or its type 1 receptor, Tkv.
Fig. 5.
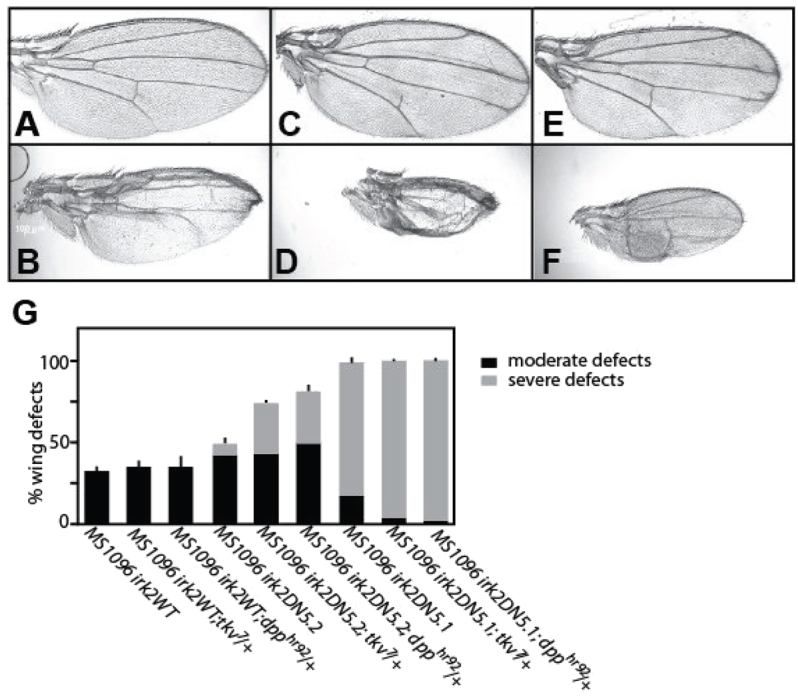
Reduced Dpp or Thickveins function enhances Irk2DN phenotypes. (A,B) Irk2DN5.2 female incomplete L5. (C) Thickened, bifurcated veins of dpphr92/+ female wing. (D) Irk2DN5.2/+; dpphr92/+ female wing is small and missing veins. (E) Thickened, bifurcated L2, L3 and L4 veins, and incomplete anterior crossvein of tkv7/+ female wing. (F) Irk2DN5.2/+; tkv7/+ female wing is small and missing veins. (G) Quantification of wing defects of MS1096-GAL4 UAS-irk2WT, UAS-Irk2DN5.1, UAS-Irk2DN 5.2 and with dpphr92/+ or tkv7/+. n>100 for all genotypes. Data are mean±s.e.m.
Dpp signaling is disrupted by aberrant Irk2 function
Enhancement of Irk2DN phenotypes by reduced Dpp or Tkv function suggests that Dpp signaling is compromised in flies with defective Irk channel function. To explore this possibility, we looked at Dpp signaling in the larval wing imaginal disc, a precursor to the adult wing. Dpp-bound Tkv phosphorylates the C-terminal of Mothers against Dpp (Mad) in two stripes that form the border between the anterior and posterior compartments of the wing disc (Teleman and Cohen, 2000). To determine whether Dpp signaling is interrupted by disruption of irk2 function, we used an antibody to measure phosphorylation of the C-terminal site of Mad (p-Mad). Two stripes of p-Mad are intact in Irk2 deficient and Irk2 siRNA-expressing wing discs, but the stripe intensity is lessened compared with wild type (Fig. 6). TUNEL staining shows that reducing function of irk2 does not cause apoptosis, and therefore is not the cause of decreased p-Mad (Fig. 6). Spalt (Sal), a transcriptional target of Dpp, was reduced in irk2-DfA/irk2-DfB wing discs and was not detectable in Irk2DN-expressing wing discs, supporting the conclusion that Dpp signaling is disrupted by blocking Irk channels (Fig. 7). By contrast, patterns of two genes that are not regulated by Dpp, wingless and achaete, are intact in irk2-deficient wing discs (Fig. 7).
Fig. 6.
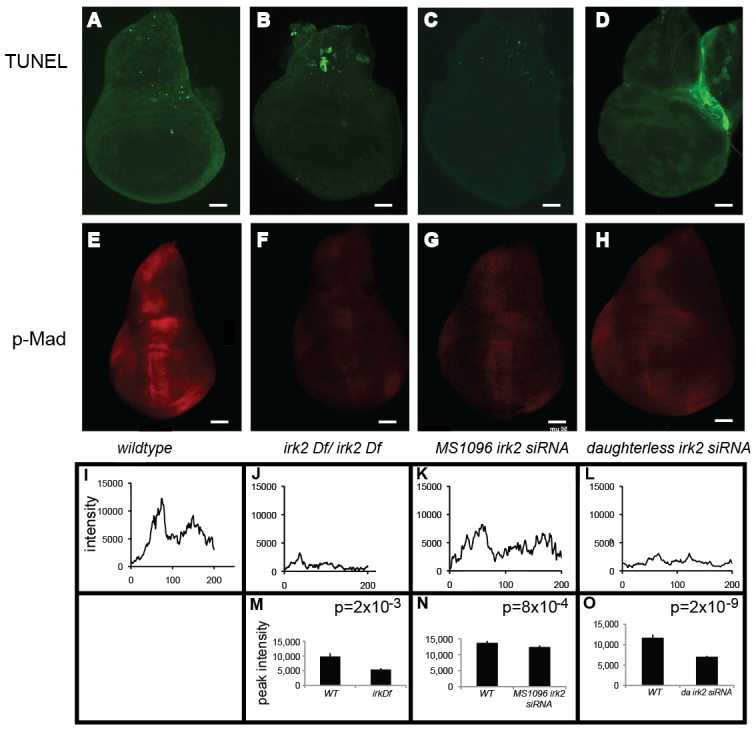
Reduced Mad phosphorylation in Irk2DfA/Irk2DfB and Irk2 siRNA wing discs. (A-D) TUNEL-stained wing discs. Anterior is rightwards. (E-H) Anti-p-Mad stained wing discs. (I-L) Relative fluorescence intensity across a posterior to anterior cross-section of the anti-p-Mad-stained wing disc shown in E-H. (M-O) Graphs of average peak intensity of control and irk2DfA/irk2DfB (M), control and MS1096-GAL4 irk2 siRNA (N), and control and daughterless-GAL4 Irk2 siRNA (O). Control and experimental discs were stained and imaged in parallel. Graphs represent average peak intensities for n>7 anti-p-Mad-stained discs. Peak intensity is determined by subtracting minimum from maximum fluorescence intensity in a posterior to anterior cross-section of the anti-p-Mad stained wing disc. Data are mean±s.e.m. Scale bars: 50 μm.
Fig. 7.
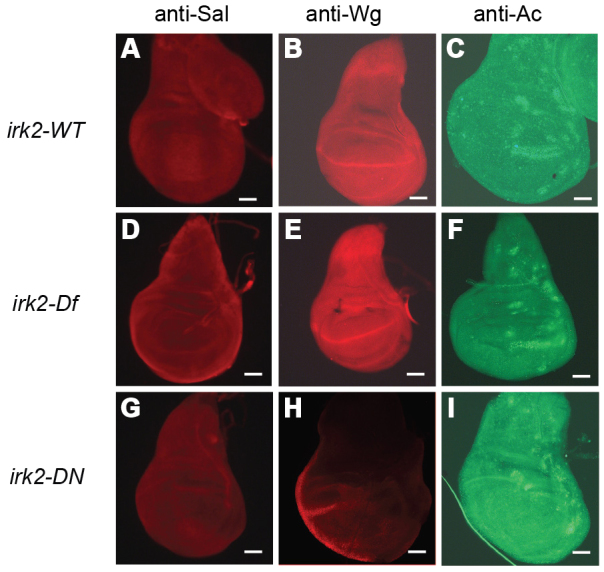
Immunohistochemistry demonstrates that Dpp signaling is reduced in Irk2DN and Irk2DfA/Irk2DfB. (A-I) Wild-type wing discs (A-C), irk2DfA/irk2DfB (D,E) and Irk2DN5.1 (G-I) stained with anti-Spalt (A,D,G), anti-Wingless (B,E,H) and anti-Achaete (C,F,I). Scale bars: 50 μm.
Expression of Irk2DN causes the most severe wing phenotypes and is predicted to block the Irk channel completely. We found that when MS1096-Gal4 drives expression of Irk2DN, phosphorylation of Mad is completely blocked in the anterior/posterior boundary of wing pouch (Fig. 8A-C). p-Mad staining was intact when flies expressed the irk2WT transgene. Irk2DN expressed by other wing-directed Gal4 drivers (Marquez et al., 2001), en-Gal4;Irk2DN5.1, A9-Gal4;Irk2-DN13 and A9-Gal4;Irk2-DN5.1, also profoundly decreases p-Mad staining and can decrease the size of the wing disc (supplementary material Fig. S5). Less that 5% of en-Gal4; Irk2-DN5.1 and A9;Gal4-DN5.1 flies survive past early larval stage.
Fig. 8.
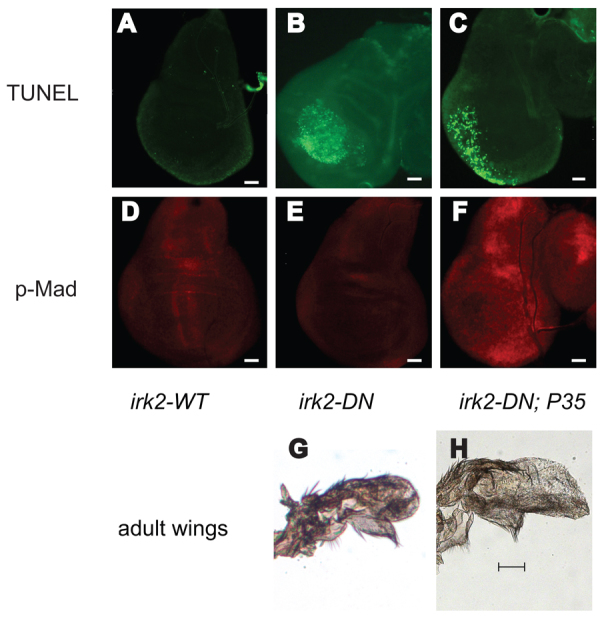
Irk2DN expression causes apoptosis and eliminates p-Mad staining in the third instar larval imaginal wing disc. (A-C) TUNEL stained wing disc. (D-F) Anti-p-Mad stained wing disc. (A,D) MS1096-GAL4-Irk2WT. (B,E) MS1096-GAL4-Irk2DN. (C,F) MS1096-GAL4-UAS-Irk2DN; P35. n>10 discs. (G) MS1096-GAL4-UAS-Irk2DN; P35 male wing. (H) Control MS1096-GAL4-UAS-Irk2DN male wing. Scale bars: 50 μm.
MS1096-Gal4;UASIrk2DN5.1 wing discs are fragile and thinned in the wing pouch region that becomes the wing blade and hinge. TUNEL staining shows that apoptosis occurs in the wing pouch of MS1096-Gal4;UASIrk2DN wing discs. Very little apoptosis is detected in the wing discs of flies expressing irk2WT (Fig. 8). In Irk2DN-expressing wing discs, wingless and achaete patterns are normal surrounding the region that dies via apoptosis (Fig. 7).
Disruption of the K+ current with Irk2DN causes complete loss of the two stripes of p-Mad that form the proximal-distal axis of the wing disc and also causes apoptosis of many of the cells that are under the influence of the Dpp signal. At least two possibilities explain these phenotypes. First, as Dpp protects against apoptosis in the wing (Letsou et al., 1995; Adachi-Yamada et al., 1999; Adachi-Yamada and O’Connor, 2002; Moreno et al., 2002; O’Connor et al., 2006; Ziv et al., 2009), blocking the K+ current could interfere with Dpp signaling, leading to apoptosis and wing defects. Alternatively, blocking Irk could directly cause apoptosis, leading to reduced Dpp signaling. To test which of these possibilities explains Irk2DN wing defects, we blocked apoptosis in cells of the wing discs that express Irk2DN and asked whether Dpp signaling occurs and whether the wing defects are rescued. Exogenous expression of P35 blocks caspase activity, preventing apoptosis (Hay et al., 1994). TUNEL staining confirmed that apoptosis does not occur in the wing pouch cells that express both Irk2DN and P35 (Fig. 8). Wing pouch cells give rise to the wing blade. The severe wing phenotypes of MS1096-GAL4-Irk2DN;P35 are indistinguishable from those of wings that express only Irk2DN (Fig. 8). As blocking apoptosis in the wing pouch does not rescue the wing phenotype, we conclude that apoptosis is fully responsible for wing blade defects. Apoptosis occurs in periphery of the wing disc, but not in the center where both P35 and Irk2DN are expressed. Therefore, Irk2DN does not cause apoptosis cell-autonomously, but could cause apoptosis of peripheral cells by compromising a Dpp gradient (Adachi-Yamada et al., 1999). Together, these data suggest that Dpp signaling is disrupted by Irk2DN, which leads to apoptosis and Irk2DN wing defects.
DISCUSSION
The morphological defects associated with ATS suggest that Kir2.1 function is necessary for human development. We and others have shown that these defects are recapitulated in a mouse knockout of Kir2.1 (Zaritsky et al., 2000). Here, we show that reducing function of the Irk2 ion channel causes developmental defects in Drosophila, probably by disrupting Dpp signaling. Four points support this conclusion. First, mouse and fly Kir2.1/Irk2 phenotypes are similar to TGFβ/BMP mutant phenotypes. Second, reduced Dpp signaling enhances Irk2DN phenotypes. Third, phosphorylation of Mad and spalt expression are decreased in mutant Irk2 wing discs. Last, Irk2DN induces apoptosis of cells within the region of Dpp influence, but outside the region of Irk2DN;P35 expression.
Our data show that the Drosophila Irk channel is heteromeric. Loss of any Irk subunit alone causes minor defects compared with severe defects caused by an Irk2DN subunit predicted to block the channel. This suggests that, in the absence of one Irk subunit, the others partially compensate.
Irk2 phenotypes are more severe and penetrant when flies develop at colder temperatures, regardless of the mechanism by which Irk2 function is reduced: transgenic Irk2DN expression, irk2 deletion, p-element insertion or irk2 siRNA expression. We observed this temperature effect for three independent Irk2DN transgenic lines under the control of many different GAL4 drivers. Although the GAL4 system generally increases transgene expression at higher temperatures, Irk2DN phenotypes are most severe at 18°C, less severe at 25°C and least severe at 29°C. This suggests that the process by which Irk channels contribute to signaling is dependent on temperature.
Irk2 phenotypes are reminiscent of mutant dpp, tkv and punt wing phenotypes (Letsou et al., 1995; Zecca et al., 1995; de Celis, 1997; Adachi-Yamada et al., 1999). The L2-3 and L4-5 collapse and loss of the majority of the wing blade are similar to mutant dpp or its target, optomotor-blind (omb) (Cook et al., 2004; del Alamo Rodríguez et al., 2004; Tabata and Kornberg, 1994). irk2-deficient flies, mutant irk2 and irk2 siRNA flies have similar hinge phenotypes to those caused by mutant dpp alleles (Gelbart, 1982; Spencer et al., 1982; Irish and Gelbart, 1987). Furthermore, Irk2DN phenotypes are enhanced by reducing function of Dpp or its receptor. Together, these data suggest that irk2 wing phenotypes can be explained by disruption of Dpp signaling.
Reducing function of irk2 by deletion, siRNA or Irk2DN reduces p-Mad in the wing disc. We confirmed that loss of irk2 reduces Dpp signaling by measuring decreased levels of Spalt, a p-Mad transcriptional target. Activation of the Wg pathway negatively regulates Dpp signaling by reducing perdurance of p-Mad, presenting the possibility Wg is affected by Irk2 (Eivers et al., 2009a; Eivers et al., 2009b; Eivers et al., 2011). Expression of both wingless and acheaete are normal in irk2-deficient wing discs and outside the region where cells have died via apoptosis in Irk2DN-expressing wing discs. However, we have not ruled out the possibility that Wingless or other developmental pathways are also affected by Irk channel function.
We find that blocking apoptosis in the cells that express Irk2DN does not rescue the associated wing phenotypes. A wing can develop with normal size and patterning after x-irradiation-induced apoptosis of half of the wing disc cells during development (Hay et al., 1994; Huh et al., 2004; Martín et al., 2004; Pérez-Garijo et al., 2004; Ryoo et al., 2004). Dpp signaling is responsible for compensation for the lost apoptotic cells and preservation of patterning in damaged wing discs (Pérez-Garijo et al., 2005). The failure of Irk2DN;P35-expressing discs to compensate for apoptotic cells supports the hypothesis that Dpp signaling is disrupted in tissues that express Irk2DN.
How could Irk2 affect Dpp signaling? It could be that maintenance of membrane potential is important for the production, distribution or propagation of the Dpp signal. Alternatively, there may be communication between Irk channels and the Dpp signaling cascade upstream of Mad activation. Distribution and propagation of Dpp and other BMP/TGFβ signals are aided by the heparan sulfate proteoglycans (HSPGs) (Jackson et al., 1997; Grisaru et al., 2001; Paine-Saunders et al., 2002; Fujise et al., 2003). Changes in sodium concentrations inhibit sulfation of heparan sulfate and reduce the sensitivity of cells to FGF. It is interesting to speculate that a change in local K+ concentrations could interfere with HSPGs, receptor localization, stabilization of the receptor complex, phosphorylation events or production or distribution of the Dpp ligand (Fig. 9). It is also possible that Mad requires K+ for recruitment to the membrane as actin requires Ca2+ for that purpose (Lu et al., 2011). We have not directly established whether Irk2 is required cell-autonomously, but we favor the model that Irk2 function is required for Dpp signaling events outside the cell rather than for intracellular phosphorylation for two reasons. First, en-GAL4 expresses UAS-irk2WT in the posterior compartment of the wing disc, but rescues irk2 deficient phenotypes in the whole wing. Second, when Irk2DN and P35 are expressed together, apoptosis occurs well outside of the region of MS1096 expression, consistent with morphogen causation.
Fig. 9.
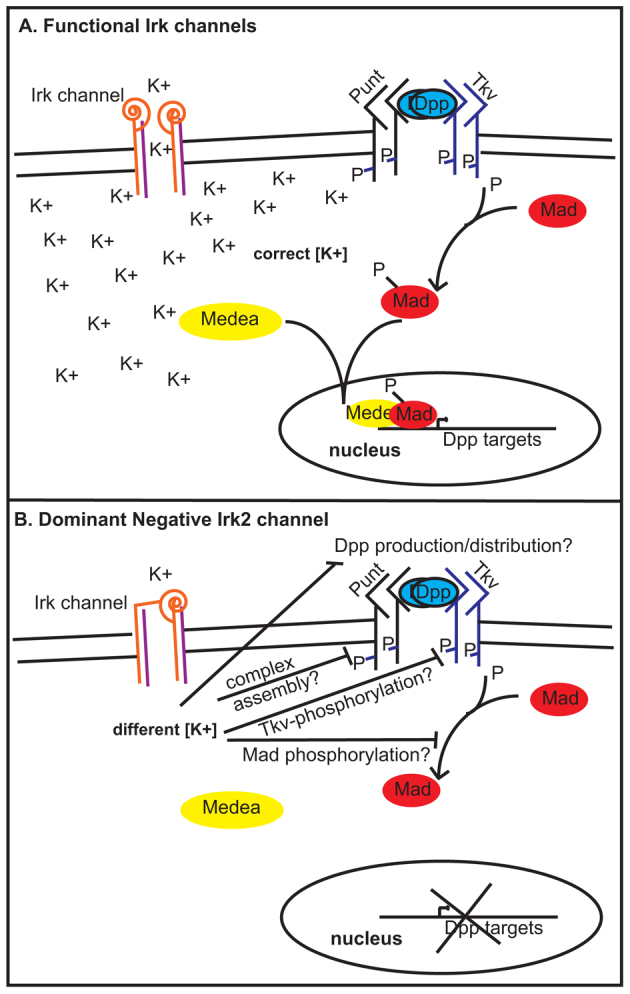
Model for Irk channels in Dpp signaling. (A) With functional Irk channels, Dpp binds type 1 (Thickveins) and type 2 (Punt) kinase receptors that are stabilized by proteoglycans (Dally, not shown). Upon Dpp binding, activated type 1 receptors phosphorylate Mad. P-Mad binds Medea and enters the nucleus to affect transcription. (B) Blocking Irk channels hinders Mad phosphorylation. Irk channels could be necessary for Dpp production/distribution, receptor complex stabilization or Tkv/Mad phosphorylation.
If inwardly rectifying K+ channels are necessary for Dpp signaling to designate wing patterning, how could Irk subunits have been missed in Dpp modifier screens? The partial redundancy of the Irk subunits make severe phenotypes unlikely unless all of the subunits are compromised. Phenotypes are not severe at 25°C, the temperature at which development occurs for most screens. Third, the necessity of Irk2 seems to be tissue specific. Dpp is required for patterning of multiple structures, but in screening multiple GAL4 drivers, expression of Irk2DN causes defects in only a few of these.
Many BMP/TGFβ-dependent processes go awry when Kir2.1 is not functional in humans and in mice. The phenotypes of Kir2.1 knockout mice are reminiscent of the morphological defects of ATS: cleft palate, incomplete dentition and digit abnormalities. All of these phenotypes have also been associated with defects in TGFβ superfamily signaling. For example, TGFβ2 and TGFβ3 knockout animals have cleft palate (Sanford et al., 1997; Taya et al., 1999). Loss of BMP4 impedes proper tooth development (Neubüser et al., 1997; Jernvall et al., 1998; Tucker et al., 1998a; Tucker et al., 1998b). Deletion of Pax9, which activates BMP4 transcription, causes cleft palate, incomplete dentition, and extra digits in mice (Peters et al., 1998). A conditional knockout of BMP2 and BMP4 in the forepaw causes extra digits like the Kir2.1 knockout (Bandyopadhyay et al., 2006). BMP signaling is responsible for inhibiting growth of extra digits and initiating apoptosis to separate digits in mice (Schaller et al., 2001; Schaller and Muneoka, 2001; Zuzarte-Luis and Hurle, 2005). Although the developmental defects associated with Kir2.1 knockout are incompletely characterized in the mouse, these BMP-like phenotypes support the hypothesis that disruption of Kir2.1 interferes with TGFβ/BMP signaling in mammals.
The finding that inwardly rectifying K+ channels are necessary for BMP signaling in the Drosophila wing alters the landscape of current research by demonstrating that K+ channels contribute significantly to development. How broadly can these findings be applied to other K+ channels? A gain-of-function GYG to SYG change in the selectivity filter of the G protein-coupled Irk2 (GIRK2) channel allows it to pass Na+ and Ca2+, which alters cerebellar development in the weaver mouse (Rakic and Sidman, 1973; Hatten et al., 1984; Hatten et al., 1986; Patil et al., 1995; Silverman et al., 1996; Tong et al., 1996). However, deletion of GIRK2 allows mice to develop normally (Signorini et al., 1997). Therefore, developmental defects of weaver mice are presumed to be due to changes in Na+ and Ca2+, rather than changes in K+, levels. By contrast, the mutations that are associated with ATS cause loss of Kir2.1 function (Bendahhou et al., 2003; Hibino et al., 2010) and the Kir2.1 knockout mouse has severe developmental defects. Our data presents a rationale for inquiry into the putative contribution of other K+ channels to developmental signaling.
Altering Irk channels interferes with BMP signaling to contribute to morphological abnormalities in Drosophila. It is likely that the developmental defects associated with ATS and the Kir2.1 knockout mouse are similarly due to defective TGFβ/BMP signaling. If Kir2.1 channel function is necessary for TGFβ/BMP signaling in mammals, the Kir channels could represent new potential therapeutic targets for slowing tumor growth and metastasis.
Supplementary Material
Acknowledgments
We thank Dr Allen Buskirk, Dr Anthea Letsou, and Drs Peter and Alex Bates for editing. We thank the Bloomington Drosophila Stock Center, the Vienna Drosophila Stock Center, Dr Anthea Letsou and Dr Sally Kornbluth for fly stocks. We also thank Adi Salzberg for anti-Spalt. We thank Ree Lu, Phoebe Lin, Devon Kinghorn, Alex Johnson, Elise Wilson and Abigail Gehrett for wing disc dissections.
Footnotes
Funding
This work was funded by the Howard Hughes Medical Institute, the Muscular Dystrophy Association and the Sandler Neurogenetics Fund (to L.J.P.), and by the Brigham Young University Cancer Research Center (to E.A.B. and G.D.). Deposited in PMC for release after 6 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.078592/-/DC1
References
- Adachi-Yamada T., O’Connor M. B. (2002). Morphogenetic apoptosis: a mechanism for correcting discontinuities in morphogen gradients. Dev. Biol. 251, 74–90 [DOI] [PubMed] [Google Scholar]
- Adachi-Yamada T., Fujimura-Kamada K., Nishida Y., Matsumoto K. (1999). Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature 400, 166–169 [DOI] [PubMed] [Google Scholar]
- Andersen E. D., Krasilnikoff P. A., Overvad H. (1971). Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome? Acta Paediatr. Scand. 60, 559–564 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A., Tsuji K., Cox K., Harfe B. D., Rosen V., Tabin C. J. (2006). Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2, e216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendahhou S., Donaldson M. R., Plaster N. M., Tristani-Firouzi M., Fu Y. H., Ptácek L. J. (2003). Defective potassium channel Kir2.1 trafficking underlies Andersen-Tawil syndrome. J. Biol. Chem. 278, 51779–51785 [DOI] [PubMed] [Google Scholar]
- Bichet D., Haass F. A., Jan L. Y. (2003). Merging functional studies with structures of inward-rectifier K(+) channels. Nat. Rev. Neurosci. 4, 957–967 [DOI] [PubMed] [Google Scholar]
- Bjork B. C., Turbe-Doan A., Prysak M., Herron B. J., Beier D. R. (2010). Prdm16 is required for normal palatogenesis in mice. Hum. Mol. Genet. 19, 774–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair S. S. (2007). Wing vein patterning in Drosophila and the analysis of intercellular signaling. Annu. Rev. Cell Dev. Biol. 23, 293–319 [DOI] [PubMed] [Google Scholar]
- Brook W. J., Cohen S. M. (1996). Antagonistic interactions between wingless and decapentaplegic responsible for dorsal-ventral pattern in the Drosophila Leg. Science 273, 1373–1377 [DOI] [PubMed] [Google Scholar]
- Canún S., Pérez N., Beirana L. G. (1999). Andersen syndrome autosomal dominant in three generations. Am. J. Med. Genet. 85, 147–156 [DOI] [PubMed] [Google Scholar]
- Cao J., Pellock B. J., White K., Raftery L. A. (2006). A commercial phospho-Smad antibody detects endogenous BMP signaling in Drosophila tissues. Dros. Inf. Serv. 89, 131–135 [Google Scholar]
- Casey L. M., Lan Y., Cho E. S., Maltby K. M., Gridley T., Jiang R. (2006). Jag2-Notch1 signaling regulates oral epithelial differentiation and palate development. Dev. Dyn. 235, 1830–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe S. (2002). Potassium channel structures. Nat. Rev. Neurosci. 3, 115–121 [DOI] [PubMed] [Google Scholar]
- Cook O., Biehs B., Bier E. (2004). brinker and optomotor-blind act coordinately to initiate development of the L5 wing vein primordium in Drosophila. Development 131, 2113–2124 [DOI] [PubMed] [Google Scholar]
- de Celis J. F. (1997). Expression and function of decapentaplegic and thick veins during the differentiation of the veins in the Drosophila wing. Development 124, 1007–1018 [DOI] [PubMed] [Google Scholar]
- del Alamo Rodríguez D., Terriente Felix J., Díaz-Benjumea F. J. (2004). The role of the T-box gene optomotor-blind in patterning the Drosophila wing. Dev. Biol. 268, 481–492 [DOI] [PubMed] [Google Scholar]
- Derynck R., Jarrett J. A., Chen E. Y., Eaton D. H., Bell J. R., Assoian R. K., Goeddel D. V. (1985). Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature 316, 701–705 [DOI] [PubMed] [Google Scholar]
- Dietzl G., Chen D., Schnorrer F., Su K. C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S., et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156 [DOI] [PubMed] [Google Scholar]
- Döring F., Wischmeyer E., Kühnlein R. P., Jäckle H., Karschin A. (2002). Inwardly rectifying K+ (Kir) channels in Drosophila. A crucial role of cellular milieu factors Kir channel function. J. Biol. Chem. 277, 25554–25561 [DOI] [PubMed] [Google Scholar]
- Dudas M., Sridurongrit S., Nagy A., Okazaki K., Kaartinen V. (2004). Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech. Dev. 121, 173–182 [DOI] [PubMed] [Google Scholar]
- Eivers E., Demagny H., De Robertis E. M. (2009a). Integration of BMP and Wnt signaling via vertebrate Smad1/5/8 and Drosophila Mad. Cytokine Growth Factor Rev. 20, 357–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eivers E., Fuentealba L. C., Sander V., Clemens J. C., Hartnett L., De Robertis E. M. (2009b). Mad is required for wingless signaling in wing development and segment patterning in Drosophila. PLoS ONE 4, e6543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eivers E., Demagny H., Choi R. H., De Robertis E. M. (2011). Phosphorylation of Mad controls competition between wingless and BMP signaling. Sci. Signal. 4, ra68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti E., Li B., Zewdu R., Wells V., Hebert J. M., Karner C., Anderson M. J., Williams T., Dixon J., Dixon M. J., et al. (2011). A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev. Cell 21, 627–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujise M., Takeo S., Kamimura K., Matsuo T., Aigaki T., Izumi S., Nakato H. (2003). Dally regulates Dpp morphogen gradient formation in the Drosophila wing. Development 130, 1515–1522 [DOI] [PubMed] [Google Scholar]
- Gelbart W. M. (1982). Synapsis-dependent allelic complementation at the decapentaplegic gene complex in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 79, 2636–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisaru S., Cano-Gauci D., Tee J., Filmus J., Rosenblum N. D. (2001). Glypican-3 modulates BMP- and FGF-mediated effects during renal branching morphogenesis. Dev. Biol. 231, 31–46 [DOI] [PubMed] [Google Scholar]
- Gullaud M., Delanoue R., Silber J. (2003). A Drosophila model to study the functions of TWIST orthologs in apoptosis and proliferation. Cell Death Differ. 10, 641–651 [DOI] [PubMed] [Google Scholar]
- Halachmi N., Schulze K. L., Inbal A., Salzberg A. (2007). Additional sex combs affects antennal development by means of spatially restricted repression of Antp and wg. Dev. Dyn. 236, 2118–2130 [DOI] [PubMed] [Google Scholar]
- Hatten M. E., Liem R. K., Mason C. A. (1984). Defects in specific associations between astroglia and neurons occur in microcultures of weaver mouse cerebellar cells. J. Neurosci. 4, 1163–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten M. E., Liem R. K., Mason C. A. (1986). Weaver mouse cerebellar granule neurons fail to migrate on wild-type astroglial processes in vitro. J. Neurosci. 6, 2676–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay B. A., Wolff T., Rubin G. M. (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121–2129 [DOI] [PubMed] [Google Scholar]
- He F., Xiong W., Wang Y., Li L., Liu C., Yamagami T., Taketo M. M., Zhou C., Chen Y. (2011). Epithelial Wnt/β-catenin signaling regulates palatal shelf fusion through regulation of Tgfβ3 expression. Dev. Biol. 350, 511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., Kurachi Y. (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366 [DOI] [PubMed] [Google Scholar]
- Huh J. R., Guo M., Hay B. A. (2004). Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr. Biol. 14, 1262–1266 [DOI] [PubMed] [Google Scholar]
- Irish V. F., Gelbart W. M. (1987). The decapentaplegic gene is required for dorsal-ventral patterning of the Drosophila embryo. Genes Dev. 1, 868–879 [DOI] [PubMed] [Google Scholar]
- Jackson S. M., Nakato H., Sugiura M., Jannuzi A., Oakes R., Kaluza V., Golden C., Selleck S. B. (1997). dally, a Drosophila glypican, controls cellular responses to the TGF-beta-related morphogen, Dpp. Development 124, 4113–4120 [DOI] [PubMed] [Google Scholar]
- Jernvall J., Aberg T., Kettunen P., Keränen S., Thesleff I. (1998). The life history of an embryonic signaling center: BMP-4 induces p21 and is associated with apoptosis in the mouse tooth enamel knot. Development 125, 161–169 [DOI] [PubMed] [Google Scholar]
- Jiang R., Lan Y., Chapman H. D., Shawber C., Norton C. R., Serreze D. V., Weinmaster G., Gridley T. (1998). Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 12, 1046–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y. R., Turcotte T. J., Crocker A. L., Han X. H., Yoon J. K. (2011). The canonical Wnt signaling activator, R-spondin2, regulates craniofacial patterning and morphogenesis within the branchial arch through ectodermal-mesenchymal interaction. Dev. Biol. 352, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Johnson K., Chen H. J., Carroll S., Laughon A. (1997). Drosophila Mad binds to DNA and directly mediates activation of vestigial by Decapentaplegic. Nature 388, 304–308 [DOI] [PubMed] [Google Scholar]
- Lécuyer E., Parthasarathy N., Krause H. M. (2008). Fluorescent in situ hybridization protocols in Drosophila embryos and tissues. Methods Mol. Biol. 420, 289–302 [DOI] [PubMed] [Google Scholar]
- Letsou A., Arora K., Wrana J. L., Simin K., Twombly V., Jamal J., Staehling-Hampton K., Hoffmann F. M., Gelbart W. M., Massagué J., et al. (1995). Drosophila Dpp signaling is mediated by the punt gene product: a dual ligand-binding type II receptor of the TGF beta receptor family. Cell 80, 899–908 [DOI] [PubMed] [Google Scholar]
- Lin C., Fisher A. V., Yin Y., Maruyama T., Veith G. M., Dhandha M., Huang G. J., Hsu W., Ma L. (2011). The inductive role of Wnt-β-Catenin signaling in the formation of oral apparatus. Dev. Biol. 356, 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Selever J., Murali D., Sun X., Brugger S. M., Ma L., Schwartz R. J., Maxson R., Furuta Y., Martin J. F. (2005). Threshold-specific requirements for Bmp4 in mandibular development. Dev. Biol. 283, 282–293 [DOI] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(–Delta Delta C(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- Lu R., Niesen M. J., Hu W., Vaidehi N., Shively J. E. (2011). Interaction of actin with carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) receptor in liposomes is Ca2+- and phospholipid-dependent. J. Biol. Chem. 286, 27528–27536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean S. J., Andrews B. C., Verheyen E. M. (2002). Characterization of Dir: a putative potassium inward rectifying channel in Drosophila. Mech. Dev. 116, 193–197 [DOI] [PubMed] [Google Scholar]
- Marquez R. M., Singer M. A., Takaesu N. T., Waldrip W. R., Kraytsberg Y., Newfeld S. J. (2001). Transgenic analysis of the Smad family of TGF-beta signal transducers in Drosophila melanogaster suggests new roles and new interactions between family members. Genetics 157, 1639–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín F. A., Pérez-Garijo A., Moreno E., Morata G. (2004). The brinker gradient controls wing growth in Drosophila. Development 131, 4921–4930 [DOI] [PubMed] [Google Scholar]
- Mason A. J., Hayflick J. S., Ling N., Esch F., Ueno N., Ying S. Y., Seeburg P. H. (1985). Complementary DNA sequences of ovarian follicular fluid inhibin show precursor structure and homology with transforming growth factor-beta. Nature 318, 659–663 [DOI] [PubMed] [Google Scholar]
- McLerie M., Lopatin A. N. (2003). Dominant-negative suppression of I(K1) in the mouse heart leads to altered cardiac excitability. J. Mol. Cell. Cardiol. 35, 367–378 [DOI] [PubMed] [Google Scholar]
- Menezes R., Letra A., Kim A. H., Küchler E. C., Day A., Tannure P. N., Gomes da Motta L., Paiva K. B., Granjeiro J. M., Vieira A. R. (2010). Studies with Wnt genes and nonsyndromic cleft lip and palate. Birth Defects Res. A Clin. Mol. Teratol. 88, 995–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E., Basler K., Morata G. (2002). Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 416, 755–759 [DOI] [PubMed] [Google Scholar]
- Nellen D., Affolter M., Basler K. (1994). Receptor serine/threonine kinases implicated in the control of Drosophila body pattern by decapentaplegic. Cell 78, 225–237 [DOI] [PubMed] [Google Scholar]
- Neubüser A., Peters H., Balling R., Martin G. R. (1997). Antagonistic interactions between FGF and BMP signaling pathways: a mechanism for positioning the sites of tooth formation. Cell 90, 247–255 [DOI] [PubMed] [Google Scholar]
- O’Connor M. B., Umulis D., Othmer H. G., Blair S. S. (2006). Shaping BMP morphogen gradients in the Drosophila embryo and pupal wing. Development 133, 183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta M., Greenberger J. S., Anklesaria P., Bassols A., Massague J. (1987). Two forms of transforming growth factor-beta distinguished by multipotential haematopoietic progenitor cells. Nature 329, 539–541 [DOI] [PubMed] [Google Scholar]
- Padgett R. W., St Johnston R. D., Gelbart W. M. (1987). A transcript from a Drosophila pattern gene predicts a protein homologous to the transforming growth factor-beta family. Nature 325, 81–84 [DOI] [PubMed] [Google Scholar]
- Paine-Saunders S., Viviano B. L., Economides A. N., Saunders S. (2002). Heparan sulfate proteoglycans retain Noggin at the cell surface: a potential mechanism for shaping bone morphogenetic protein gradients. J. Biol. Chem. 277, 2089–2096 [DOI] [PubMed] [Google Scholar]
- Parks A. L., Cook K. R., Belvin M., Dompe N. A., Fawcett R., Huppert K., Tan L. R., Winter C. G., Bogart K. P., Deal J. E., et al. (2004). Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 36, 288–292 [DOI] [PubMed] [Google Scholar]
- Patil N., Cox D. R., Bhat D., Faham M., Myers R. M., Peterson A. S. (1995). A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat. Genet. 11, 126–129 [DOI] [PubMed] [Google Scholar]
- Pérez-Garijo A., Martín F. A., Morata G. (2004). Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 131, 5591–5598 [DOI] [PubMed] [Google Scholar]
- Pérez-Garijo A., Martín F. A., Struhl G., Morata G. (2005). Dpp signaling and the induction of neoplastic tumors by caspase-inhibited apoptotic cells in Drosophila. Proc. Natl. Acad. Sci. USA 102, 17664–17669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters H., Neubüser A., Kratochwil K., Balling R. (1998). Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 12, 2735–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P., Sidman R. L. (1973). Weaver mutant mouse cerebellum: defective neuronal migration secondary to abnormality of Bergmann glia. Proc. Natl. Acad. Sci. USA 70, 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R. J., Dixon J., Jiang R., Dixon M. J. (2009). Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum. Mol. Genet. 18, 2632–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruberte E., Marty T., Nellen D., Affolter M., Basler K. (1995). An absolute requirement for both the type II and type I receptors, punt and thick veins, for dpp signaling in vivo. Cell 80, 889–897 [DOI] [PubMed] [Google Scholar]
- Ryoo H. D., Gorenc T., Steller H. (2004). Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 7, 491–501 [DOI] [PubMed] [Google Scholar]
- Sampath T. K., Rashka K. E., Doctor J. S., Tucker R. F., Hoffmann F. M. (1993). Drosophila transforming growth factor beta superfamily proteins induce endochondral bone formation in mammals. Proc. Natl. Acad. Sci. USA 90, 6004–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford L. P., Ormsby I., Gittenberger-de Groot A. C., Sariola H., Friedman R., Boivin G. P., Cardell E. L., Doetschman T. (1997). TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124, 2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone V., Griggs R. C., Meola G., Ptácek L. J., Barohn R., Iannaccone S., Bryan W., Baker N., Janas S. J., Scott W., et al. (1997). Andersen’s syndrome: a distinct periodic paralysis. Ann. Neurol. 42, 305–312 [DOI] [PubMed] [Google Scholar]
- Schaller S. A., Muneoka K. (2001). Inhibition of polarizing activity in the anterior limb bud is regulated by extracellular factors. Dev. Biol. 240, 443–456 [DOI] [PubMed] [Google Scholar]
- Schaller S. A., Li S., Ngo-Muller V., Han M. J., Omi M., Anderson R., Muneoka K. (2001). Cell biology of limb patterning. Int. Rev. Cytol. 203, 483–517 [DOI] [PubMed] [Google Scholar]
- Segal D., Gelbart W. M. (1985). Shortvein, a new component of the decapentaplegic gene complex in Drosophila melanogaster. Genetics 109, 119–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signorini S., Liao Y. J., Duncan S. A., Jan L. Y., Stoffel M. (1997). Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc. Natl. Acad. Sci. USA 94, 923–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman S. K., Kofuji P., Dougherty D. A., Davidson N., Lester H. A. (1996). A regenerative link in the ionic fluxes through the weaver potassium channel underlies the pathophysiology of the mutation. Proc. Natl. Acad. Sci. USA 93, 15429–15434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeath J. B., Carroll S. B. (1991). Regulation of achaete-scute gene expression and sensory organ pattern formation in the Drosophila wing. Genes Dev. 5, 984–995 [DOI] [PubMed] [Google Scholar]
- Spencer F. A., Hoffmann F. M., Gelbart W. M. (1982). Decapentaplegic: a gene complex affecting morphogenesis in Drosophila melanogaster. Cell 28, 451–461 [DOI] [PubMed] [Google Scholar]
- Tabata T., Kornberg T. B. (1994). Hedgehog is a signaling protein with a key role in patterning Drosophila imaginal discs. Cell 76, 89–102 [DOI] [PubMed] [Google Scholar]
- Tawil R., Ptacek L. J., Pavlakis S. G., DeVivo D. C., Penn A. S., Ozdemir C., Griggs R. C. (1994). Andersen’s syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 35, 326–330 [DOI] [PubMed] [Google Scholar]
- Taya Y., O’Kane S., Ferguson M. W. (1999). Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development 126, 3869–3879 [DOI] [PubMed] [Google Scholar]
- Teleman A. A., Cohen S. M. (2000). Dpp gradient formation in the Drosophila wing imaginal disc. Cell 103, 971–980 [DOI] [PubMed] [Google Scholar]
- Tong Y., Wei J., Zhang S., Strong J. A., Dlouhy S. R., Hodes M. E., Ghetti B., Yu L. (1996). The weaver mutation changes the ion selectivity of the affected inwardly rectifying potassium channel GIRK2. FEBS Lett. 390, 63–68 [DOI] [PubMed] [Google Scholar]
- Tucker A. S., Al Khamis A., Sharpe P. T. (1998a). Interactions between Bmp-4 and Msx-1 act to restrict gene expression to odontogenic mesenchyme. Dev. Dyn. 212, 533–539 [DOI] [PubMed] [Google Scholar]
- Tucker A. S., Matthews K. L., Sharpe P. T. (1998b). Transformation of tooth type induced by inhibition of BMP signaling. Science 282, 1136–1138 [DOI] [PubMed] [Google Scholar]
- Xu J., Krebs L. T., Gridley T. (2010). Generation of mice with a conditional null allele of the Jagged2 gene. Genesis 48, 390–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yapici N., Kim Y. J., Ribeiro C., Dickson B. J. (2008). A receptor that mediates the post-mating switch in Drosophila reproductive behaviour. Nature 451, 33–37 [DOI] [PubMed] [Google Scholar]
- Yoon G., Oberoi S., Tristani-Firouzi M., Etheridge S. P., Quitania L., Kramer J. H., Miller B. L., Fu Y. H., Ptáček L. J. (2006a). Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am. J. Med. Genet. 140A, 312–321 [DOI] [PubMed] [Google Scholar]
- Yoon G., Quitania L., Kramer J. H., Fu Y. H., Miller B. L., Ptácek L. J. (2006b). Andersen-Tawil syndrome: definition of a neurocognitive phenotype. Neurology 66, 1703–1710 [DOI] [PubMed] [Google Scholar]
- Zaritsky J. J., Eckman D. M., Wellman G. C., Nelson M. T., Schwarz T. L. (2000). Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ. Res. 87, 160–166 [DOI] [PubMed] [Google Scholar]
- Zecca M., Basler K., Struhl G. (1995). Sequential organizing activities of engrailed, hedgehog and decapentaplegic in the Drosophila wing. Development 121, 2265–2278 [DOI] [PubMed] [Google Scholar]
- Ziv O., Suissa Y., Neuman H., Dinur T., Geuking P., Rhiner C., Portela M., Lolo F., Moreno E., Gerlitz O. (2009). The co-regulator dNAB interacts with Brinker to eliminate cells with reduced Dpp signaling. Development 136, 1137–1145 [DOI] [PubMed] [Google Scholar]
- Zuzarte-Luis V., Hurle J. M. (2005). Programmed cell death in the embryonic vertebrate limb. Semin. Cell Dev. Biol. 16, 261–269 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.