

Abstract
Oxidative stress is frequently implicated in the pathology of neurodegenerative disease. The chief source of this stress is from mitochondrial respiration, via the passage of reducing equivalents through the respiratory chain resulting in a small but potentially pathological production of superoxide. The superoxide that is produced during normal respiration is primarily detoxified within the mitochondria by superoxide dismutase 2 (Sod2), a key protein for maintaining mitochondrial function. Mitochondria are distributed throughout the soma of neurons, as well as along neuronal processes and at the synaptic terminus. This distribution of potentially independent mitochondria throughout the neuron, at distinct subcellular locations, allows for the possibility of regional subcellular deficits in mitochondrial function. There has been increasing interest in the quantification and characterization of messages and proteins at the synapse, due to its importance in neurodegenerative disease, most notably Alzheimer’s disease. Here, we report the transcriptomic and proteomic changes that occur in synaptosomes from frontal cortices of Sod2 null mice. Constitutively null Sod2 mice were differentially dosed with the synthetic catalytic antioxidant EUK-189, which can extend the lifespan of these mice, as well as uncover or prevent neurodegeneration due to endogenous oxidative stress. This approach facilitated insight into quantification of trafficked messages and proteins to the synaptosome. We used two complementary methods to investigate the nature of the synaptosome under oxidative stress; either whole genome gene expression microarrays or mass spectrometry-based proteomics using isobaric tagging for relative and absolute quantitation (iTRAQ) of proteins. We have characterized the relative enrichments of gene ontologies at both gene and protein expression that occur due to mitochondrial oxidative stress in the synaptosome, which may lead to new avenues of investigation in understanding the regulation of the synaptic function in normal and diseased states. As a result of using these approaches, we report for the first time an activation of the mTOR pathway in synaptosomes isolated from Sod2 null mice, confirmed by an upregulation of the phosphorylation of 4E-BP1.
Introduction
The synapse is a functionally distinct region of the neuron that is dynamically involved in learning and memory [1,2]. Impaired synaptic function is associated with a number of neurodegenerative diseases and disorders [3–10]. Both coding and non-coding RNA transcripts expressed in the nucleus are trafficked to the synaptic bouton for either local translation to support synaptic function, or storage in a number of nascent formats such as processing bodies (PB) or stress granules (SG) for later use in differing synaptic functional states [1,11–15]. The “synaptosome” is functionally distinct region of a neuronal process, and contains all the presynaptic machinery for synaptic transmission, including mitochondria to supply ATP, endoplasmic reticulum for on-site protein synthesis using trafficked mRNAs, and synaptic vesicles containing neurotransmitters [16].
Synaptosomes are advantageous because they can be prepared from specific dissected regions of the brain and allow for the study of a relatively pure biological sample that can be prepared in ample quantities to perform very detailed studies on the function of the synapse [17–19]. Synaptosomes are ~1–2 microns in size, contain 1–2 individual mitochondria [17], are bioenergetically competent, and act in vitro as self-contained membrane-bound “cells”, facilitating the study of many aspects of neurotransmission [17]. During the isolation of the synaptosome, the post-synaptic density can also become closely associated, and so some proteins from this region can become inextricably linked to the isolated synaptosomes. However, for the sake of clarity, we will refer to this complex purified subcellular compartment as the “synaptosome”, incorporating elements of pre-synaptic nerve terminals and junctions, as well as elements of the post-synaptic density.
Synaptosomes can be prepared from intact brain using density gradient centrifugation, and the basic methodology has been used for more than 30 years in bulk preparations to study the bioenergetics of synaptic function [20,21]. More recently, this general approach has been used to catalog synaptosomal RNAs and proteins [1,11,13,14,18,22–31]. There have been several studies emphasizing how synaptic function and energetics in bulk synaptosome preparations change in response to energy demand [17,32], and we have previously reported synaptic bioenergetic changes in response to endogenous mitochondrial oxidative stress [33]. In a prior study using identical an methodology to isolate synaptosomes employed in this report, we compared synaptosomes isolated from healthy mice versus those suffering a neurodegenerative disorder from endogenous mitochondrial oxidative stress due to lack of mitochondrial superoxide dismutase (Sod2). Synaptosomes from Sod2 null mice had a profound bioenergetic deficit, demonstrating significant consequences to energy metabolism as a function of endogenous mitochondrial oxidative stress [33].
Obtaining a better understanding of trafficked gene transcripts and proteins at the synapse in normal and diseased states is therefore a critical step towards a mechanistic understanding and treatment of neurodegenerative disease. In this study, we examined the transcriptome and proteome from synaptosomes derived from mouse forebrain undergoing endogenous mitochondrial oxidative stress due to lack of Sod2. Both genomic and proteomic datasets were derived from synaptosomes in parallel, and these experiments provide the opportunity to gain greater insight into the composition and function of this specific component of neuronal tissue. This overall approach was recently discussed in a review by Geschwind and Konopka in which they specifically point out the need for such studies in the field of neuroscience [34].
However, a complicating factor in execution of these types of experiments in the central nervous system is purification of one sub-cellular region or cell type without contamination of other biological material. This is often further confounded by the minimum amount of material needed for various “-omics” level assays. In this study, we resolve some of these technical limitations, and carry out both genomic and proteomic profiling using robust and sensitive assays. Bioinformatics analysis of the resultant data sets, indicated that the mTOR pathway (mammalian target of rapamycin), involved in growth and metabolism was being differentially affected in the synaptosomal compartment due to mitochondrial oxidative stress. This analysis also revealed that the transcriptional response of mTOR signaling components was dependant on the severity of the oxidative stress. We confirmed this prediction by Western blotting for increased levels of phosphorylation of 4E-BP1, a key component of the mTOR signaling pathway, demonstrating the utility of our overall approach.
Materials and Methods
Animals
Constitutive Sod2 null homozygous mice on a CD1 background ranging in age from 17 to 21 days old and age matched wild type siblings were used for all experiments as previously described [35–37]. Genotyping was carried out at 3 days of age, as previously described [35–37] and either 1 mg/kg or 30 mg/kg of the synthetic antioxidant EUK-189 (Dalton Pharma Services, Toronto, ON, Canada) was injected daily via intraperitoneal injection from the 3rd day after birth until the day before the animals were euthanized. All mouse procedures were carried out under approved Buck Institutional IACUC protocols.
Isolation and preparation of cortical synaptosomes
Mice were euthanized by CO2 overdose & decapitation, and cortices were removed and subjected to a synaptosome isolation method as described in our prior work [33], and is similar to that reported by Dunkley et al. [38]. The basic methodology has been used in hundreds of studies [20,21], is relatively straightforward, but has more recently been optimized [38].
Briefly, frontal cortex was rapidly isolated (about 100 mg/brain) and rinsed with ice-cold sucrose medium (320 mM sucrose, 1 mM EDTA, 0.25 mM dithiothreitol, pH 7.4) to remove excess blood, transferred to a pre-chilled Dounce glass homogenizer containing 3 ml sucrose medium, and homogenized gently by ten strokes. The homogenate was then centrifuged at 1,000g for 10 minutes at 4°C. The supernatant was carefully layered on top of a discontinuous Percoll gradient (3, 10 and 23% Percoll in sucrose medium) in a 15ml centrifuge tube, and centrifuged at 32,500g for 10 minutes at 4°C in a JA-25.50 fixed angle rotor in a Beckman Avanti J-26 XPI centrifuge. Synaptosomes were isolated and recovered from the band between 10% and 23% Percoll. The synaptosomes isolated from the 10–23% interface were diluted into ‘ionic medium’ (20 mM HEPES, 10 mM D-Glucose, 1.2 mM Na2HPO4, 1 mM MgCl2, 5 mM NaHCO3, 5 mM KCl, 140 mM NaCl, pH 7.4) to maintain bioenergetic function [39]. The final synaptosome pellet was then resuspended in ionic medium, and protein concentration was determined by the Bradford method (Bio-Rad). The yield of synaptosomes isolated from the cortex of Sod2 mice was not significantly different from that of the wild-type controls.
Microarray of RNA from Synaptosomes
To examine the whole genome gene expression profile of synaptosomes generated from individual mice, mRNA was extracted from synaptosome samples of individual mice from each genotype/treatment group (N=9–11 per group/treatment). Total RNA was extracted from synaptosomes using the miRNeasy kit (Qiagen, Valencia, CA, USA) on a QiaCube robot (Qiagen) according to the manufacturer’s instructions. RNA samples were then QC’d using a Nanodrop Spectrophotometer and the Agilent Bioanalyzer Nano Chip System (Cat #5065-4476). The concentration (ng/μl) and the OD 260/280 ratios were obtained by loading 1μl of Total RNA sample onto the Nanodrop instrument for measurement. Passing OD 260/280 values were around 2.0. Validated RNA samples were then further evaluated by Bioanalyzer on an Agilent Nano Chip (1μl per sample), and checked for RNA integrity by determining the presence of characteristic non-degraded 18S and 28S ribosomal peaks.
200ng of the purified total RNA was then amplified one round using Ambion’s Illumina RNA Amplification Kit, to prepare cRNA for labeling and hybridization to Illumina’s MouseRef-8 v2.0 expression bead chips as per the manufacturers instructions (Illumina, San Diego, CA, USA). Gene expression filtering was performed using standard multiple testing procedures and clustering, based on packages available in Bioconductor (www.bioconductor.org). For each dataset comparison, we choose “significantly” differentially expressed genes based on a false discovery rate (FDR) q-value <0.05.
Protein Sample Preparation
Synaptosome pellets were dissolved in 0.2% SDS and 1.0% NP-40 and quantified using BCA proteins assay. A subset of synaptosome protein pellets from the same animals used in the gene expression profiling experiments were used for evaluating the proteomics of synaptosomes (N= 8 for both Sod2−/− animals and wild type, with and without EUK-189 high and low dose treatments, 50 μg of each) were then prepared by diluting to equal volumes (25 μl) with pH 8.0 250 mM tris-ethyl ammonium bicarbonate buffer, bringing the final detergent concentrations to ~0.1% SDS and ~0.5% NP-40. Samples were then labeled with iTRAQ 8-plex reagents according to the manufacturer’s guidelines (Applied Biosystems, Foster City, CA). Briefly, samples were reduced (5 tris(2-carboxyethyl)phosphate at 60°C for 60 min), blocked (10mM methyl methanethiosulfonate for 10 min at RT) and digested with 2.5 μg trypsin (Protégé Madison, WI) at 37°C overnight. Samples were then iTRAQ labeled for two hours and 4 wild-type and 4 Sod2 null samples were combined for each of the two iTRAQ 8-plex trials. After combining, the samples were fractionated using a SCX cartridge (Applied Biosystems) and eluted with 1ml solutions (pH 4.0, 4.25, 4.5, 4.75, 5.0, 5.25, 5.5, 6.0, 6.5, 7.0, and 8.0) of 10mM citric acid adjusted to pH with NH4OH followed by 1 ml of high salt elute (350 mM KCl, 25% acetonitrile). The fractionated peptides were then dried in a speed-vac concentrator (Savant) at 25°C.
Liquid Chromatography and Mass Spectrometry
Samples were re-dissolved in 2% acetonitrile (0.1% formic acid) and then desalted on a Dionex (Sunnyvale, CA) Acclaim LC Pickings μ-Precolumn Cartridge (5mm × 300μm RP-C18 trap column, 5μm beads, 100 Å pore size). Chromatographic separation of iTRAQ labeled peptides was performed on a Dionex Acclaim Pepmap column (75μm × 150 mm RP-C18, 3um beads, 100 Å pore size) with a 2–80% acetonitrile (0.1% formic acid) 120 min gradient at a flow rate of 300 nL/min. A QSTAR Elite QqTOF (AB Sciex, Foster City, CA) operating in the positive ion mode (2.3 kV) performed the analysis, with the five most intense ions between 350 and 1600 m/z (over 20 counts) and with charge states of z=2–5 in the TOF-MS scan were subjected to information-dependent acquisition MS-MS with Smart Exit function employed (setting 8). Ions within +/− 100 ppm of a former sampled ion were dynamically excluded for 120s. Each SCX fraction was run twice.
Mass Spectrometry Data Processing
Data was processed using Protein Pilot 3.0 software (AB Sciex) with background correction and bias correction employed. Searching was conducted with Swissprot Database 56.1 (mus musculus: 15,915 entries) and only peptides of confidence greater than 50% were used for protein quantification. A 95% confidence (unused prot score >1.3) threshold for protein identification established by Protein Pilot 3.0 was the criteria for inclusion into the final protein report. Within experiment analysis, a single replicate was used as a reference, and all other samples were compared to the reference; this sample served as an internal normalization method. Replicate peptides, were used to derive the average expression across observations within a biological sample that represent the same peptide. The final list contained 3,486 peptides that were detected representing 982 proteins. A modified t-test statistic is thus used to compare the difference in the relative expression (difference in these ratios) in Sod2 null vs. wild-type controls. We report the average difference in relative expression as well as the p-value and the q-value, based on a multiple testing adjustment (controlling the false discovery rate).
Pathway and Ontology Analysis
Data sets were analyzed using Ingenuity pathway analysis software (Ingenuity Systems, Redwood City, CA, USA). All protein and gene expression data was computed as the ratio between the wild-type and Sod2 null animals and matched to their p-value of statistical significance. The functional ontology and canonical pathway analysis was generated in Ingenuity pathway analysis using the core analysis function. The protein network analysis was then performed only considering the data points that were significantly detected from proteins, and also filtered for only the significant network connections. Significant expression changes occurring between the wild-type and Sod2 null animals are color coded according to the convention described in the legend accompanying supplemental Figure 2.
Analysis of mitochondrial functional changes in the synaptic proteome
A modified t-test statistic was used to compare the difference in the relative expression (difference in these ratios each sample to arbitrary reference sample) in Sod2 null mice vs. wild-types, where the modification is derived from the empirical Bayes estimates of the variance of these ratios using the so-called Limma method [40], adjusting for experiment, as implemented, and is part of the bioconductor package [41]. The final summary measure was the average difference in relative expression (to the “standard”) as well as the p-value and the q-value, and based on a multiple testing adjustment (controlling the false discovery rate). Finally, the peptides were grouped by the protein complexes that comprise the respiratory chain to see whether there are systematic downregulation in groups defined by being a member of these complexes (http://www.genenames.org/genefamilies/mitocomplex#com) was used to link specific proteins to the various complexes). Given the linear model for each peptide is mean(Yp|Exp,KO)=bp0+bp1Strain+bp2Exp, with KO an indicator that the sample was from an Sod2 null mouse, WT is wild-type, and Exp is the experiment, and we expressed the ratio of relative expression for peptide p in KO vs. WT by the Ratiop = (bp0+ bp1)/bp0
Evaluation of mTOR pathway component 4E-BP1 phosphorylation in synaptosomes
Synaptosomal proteins were examined for activation of signaling pathways with sandwich ELISA kits specific for phosphorylated proteins. mTOR activity was assay with Cell Signaling Technology’s Pathscan® Phospho-mTOR (Ser2448) sandwich ELISA kit. 4E-BP-1 activation was assayed with Cell Signaling Technology’s Pathscan® Phospho-4E-BP1 (Thr37/Thr46) sandwich ELISA kit. The ELISA was performed according to the manufactures protocol using the recommended amount of lysate input. Western blots were also performed to determine the amount of ULK1 and the synaptosomal marker SNAP25. The blots were probed with anti-ULK1 (D8H5) (Cell Signaling Technology, #8054) at 1:1000 and anti-SNAP-25 (Sigma, #S9684) at 1:1000. The blots were visualized with Amersham ECL advance (Amersham, #RPN2135) and imaged in Alpha Innotech’s AlphaImager EC. The resulting images were then quantified for protein abundance using Adobe Photoshop CS5.
Microarray Data
All gene expression data has been submitted to the GEO, using the accession number (in process).
Results
Differential Gene Expression Analysis in synaptosomes from frontal cortices of wild-type and Sod2 null mice
Animals were genotyped at three days of age, and were treated daily by IP injection with either a high dose (30 mg/kg EUK-189) or a low dose (1 mg/kg EUK-189) of SOD mimetic, resulting in 4 experimental groups (low dose treated wild-type, high dose treated wild-type, low dose treated Sod2 −/−, or high dose treated Sod2 −/−). Synaptosomes were isolated from neurons of the frontal cortex from age matched wild-type or Sod2 null mice (n = 10 mice per treatment). RNA was then isolated from the synaptosomal fraction, and labeled and processed for microarray hybridization using Illumina gene expression bead-chips.
Daily high and low dose antioxidant treatment was employed because we have previously demonstrated either the prevention or recovery of a neurodegenerative phenotype in this model in response to such treatment [42]. The mRNAs isolated from cortical synaptosomes of the Sod2 null mice in drug treatment groups have a number of genes with significantly changed expression from wild-type control synaptosomes (Figure 1). These significantly differentially expressed genes in synaptosomes were used to create a heat map of gene expression, representing each experimental condition. This heatmap demonstrates that treatment with a low dose of EUK-189, results in a distinct expression profile that largely clusters separately from all other treatments, similar to that we previously reported from cortical homogenates comprised of all the different cell types that comprise whole cortex in the brain [42]. The expression profiles of high dose treated Sod2 −/− mice are interspersed with wild-type controls, indicating a similarity of expressed genes trafficked to the synaptosome between these two groups. This lends support to the notion that the expression changes observed in messages isolated from synaptosomes in response to high or low dose antioxidant treatment are related to neurodegeneration, as differential treatment with antioxidant dose results in greatly diminished levels of neurodegeneration [35].
Figure 1. Synaptic gene expression levels in Sod2 −/− animals and wild-type mice reveal expression changes related to the level of oxidative stress.
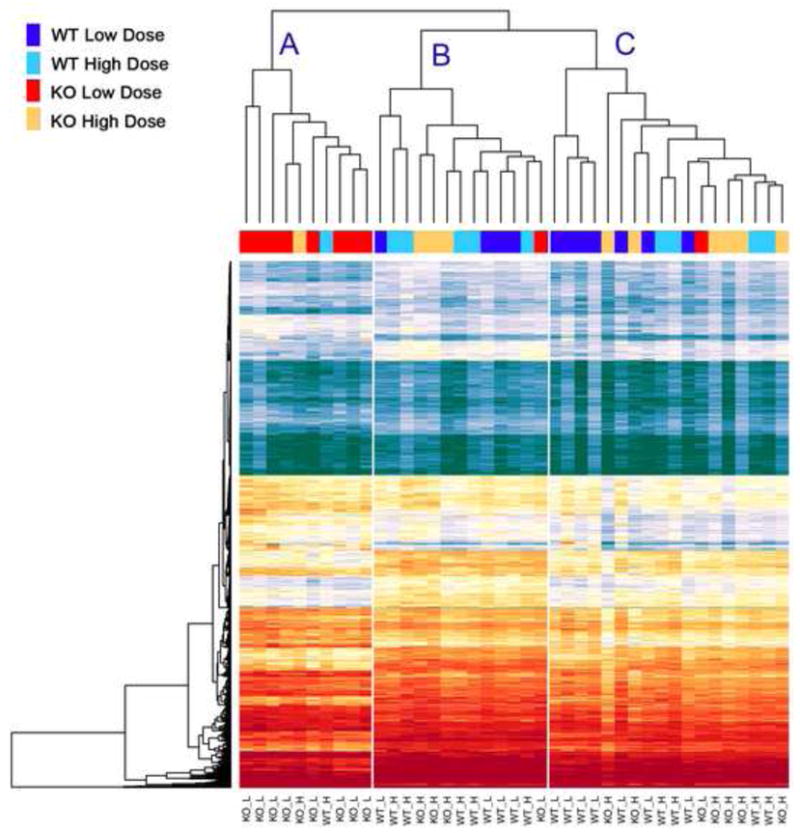
Heatmap of significant changes in genome expression changes reveal that the expression changes in the mice are most apparent in the animals that are treated with a low dose of EUK-189 synthetic antioxidant. Gene expression upregulation is colored in red/yellow, while downregulated genes are shown in blue/green. Using hierarchical clustering of each animal based upon these changes results in 8 of 10 low dose Sod2 −/− mice (KO_L, Red) to cluster into branch A. This expression of the transcriptome shifts to a pattern more akin to wild-type animals (WT_L and WT_H, Light Blue and Dark Blue) with the administration of a high dose of EUK-189 in the Sod2 −/− mice (KO_H, Yellow) and clusters among them in branches B and C.
In order to better understand functional categorization of the significantly differentially expressed genes trafficked to the synaptosomes between treatment groups, we applied a number of additional analyses, including Ingenuity pathway analysis. The whole transcriptome dataset was input into Ingenuity pathway analysis where the cutoff for significantly differentially expressed genes and the fisher’s exact test for significant overrepresentation was set at p < 0.05. The high dose treated (30 mg/kg EUK-189) Sod2 null mice as expected had a number of disease-related pathology categories overrepresented relative to wild-type (Figure 2A) including cell death and metabolic disease.
Figure 2. Analysis of Sod2 −/− and wild-type animals which are treated with a high dose of EUK-189 (30 mg/kg) antioxidant compound.

Pathway analysis for animals receiving High dosage (30 mg/kg) of the antioxidant treatment. (A) Gene function analysis of the most significant gene functions where the red line indicates the significance cut off of -log(0.05) for the fisher’s exact t-test. (B) Analysis of the top signaling and metabolic pathways which are associated with the differential expression in the Sod2 −/− animals. The red line again indicates the significance cut off of p < 0.05 for the fisher’s exact test. The black line indicates the ratio of signal values for the wild-type to the Sod2 −/− knockout animals which is represented on the lower axis.
The genes that are specifically differentially expressed are also overrepresented in a number of categories that are related to canonical signaling pathways. Figure 2B shows these categories based upon their order of significance. Also plotted is the ratio of the genes within each given category that are differentially expressed. From this data it is clear that the synthesis and degradation of ketone bodies is significantly overrepresented as a consequence of loss of functional SOD2. This is consistent with the biochemical detection of elevated blood ketones in Sod2 null mice we had previously reported [43]. However, we were unable to detect ketones present in any fraction from synaptosomes of either wild-type or Sod2 null mice, possibly due to washout during the isolation procedure (data not shown).
We also carried out IPA analysis with the low dose treated Sod2 null and wild-type animals, and observed significant differences from the analysis of Sod2 null animals treated with high dose antioxidant. Neurological disease moves from the 33rd most significant category in the high-dose paradigm, to the 2nd most significant in the disease categories in the more severely affected low dose paradigm, indicating that the increasing mitochondrial oxidative stress due to low-dose antioxidant treatment causes a shift in the gene expression profile which more closely associates with neurological disease (Figure 3A). Examination of the overrepresented canonical pathways in the low dose treated animals also reveals an increase in the overall number of pathways represented, as well as the number of genes that fall within these pathways (Figure 3B). Consistent with the model, oxidative stress and mitochondrial damage related pathways at both the gene expression and protein levels are revealed through this analysis (Figure 3). Interestingly, there are also unexpected pathways uncovered such as mTOR signaling, HMGB1, or Glioma signaling within the dataset. HMGB1 has been associated with an oxidative stress response in a number of disease contexts [44–46], and may therefore also be activated in synaptosomes through endogenous oxidative stress due to mitochondrial dysfunction.
Figure 3. Analysis of Sod2 −/− animals which are low dose of EUK-189 (1 mg/kg) antioxidant compound.

Ingenuity pathway gene expression analysis for animals undergoing neurodegeneration, while receiving a low dose of antioxidant treatment. (A) Gene function analysis of the most significant gene functions where the red line indicates the significance cut off of -log(0.05) for the fisher’s exact t-test. (B) Analysis of the top signaling and metabolic pathways that are associated with the differential expression in the Sod2 −/− animals. The red line again indicates the significance cut off of p < 0.05 for the fisher’s exact test. The black line indicates the ratio of signal values for the wild-type to the Sod2 −/− knockout animals.
iTRAQ proteomic analysis of the cortical mouse synapse
The microarray data implicates a number of new potential targets for exploring oxidative stress mediated changes in the synaptosome. However, it is important to move beyond single “omic” studies, as biological insights will be somewhat limited via this focused approach. Therefore, we wished to identify the proteomic profile of the synaptosome on the same samples we carried out gene expression studies on, to better understand interactions between gene and protein expression. Investigation of protein expression and gene expression in the same samples is uncommonly done, likely due to technological or biological limitations pertaining to sample prep.
For assessment of both RNA and protein in the pre-synaptic nerve terminal, a number of outcomes are possible. Each biological situation is unique; some samples will have highly convergent message and protein profiles, while as others may have little concordance. In our case, the technologies we used permitted us to investigate both mRNA and protein in a sub-cellular region of the brain of an animal model of mitochondrial mediated neurodegeneration, a somewhat rare situation. To estimate the proteome of the pre-synaptic nerve terminal, we isolated synaptosomes from both wild-type and Sod2 null mice treated with either a low-dose of the antioxidant (n=6 biological replicates per treatment). Proteins isolated from these synaptosome samples were subjected to iTRAQ isobaric labeling after tryptic digestion [47] to measure the differences in expression between the two treatments. The analysis identified 982 unique proteins that were significantly detected from their corresponding iTRAQ labeled peptide fragments in the synaptic samples. This analysis identified only a small set of proteins that were present at significantly different levels in the Sod2 −/− mice as compared to their wild-type controls (Supplemental Table 1).
Despite the relatively few proteins that showed clear expression differences between Sod2 null mice versus controls, the overall results provided a catalog of proteins that are robustly identified in the synaptosome in this model. These data allowed for a better understanding of what proteins comprise the synaptosome from frontal cortex in this mouse model (Supplemental Table 2). The majority of the significantly different proteins between genotypes were related to the mitochondrial compartment of the cell including Sod2, which was not unexpected. We established that the loss of Sod2 directly affects respiratory chain components, including loss of succinate dehydrogenase complex, subunits A and B, components of Complex II of the electron transport chain. The observed changes in protein levels confirms and refines to the level of the synaptosome our earlier findings which reported a significant loss of complex II function in cortical homogenates derived from multiple cell types of Sod2 −/− mice [33].
The significantly differentially expressed proteins in low dose treated animals were then processed in Ingenuity pathway analysis (wild-type vs. Sod2 −/−) to determine the major related functional categories that are over represented in synaptic proteins (Figure 4). Major features of the analysis are an over-representation of cell function and maintenance, and free radical scavenging, as well as lipid metabolism and cell death. Figure 4A shows the over represented categories in the low dose treated animals. Figure 4B shows the major canonical pathways that are affected at the protein level in the low dose treated animals undergoing neurodegeneration. Most of the key categories relate to mitochondrial function, including the TCA cycle, mitochondrial dysfunction, NRF2 oxidative stress response, and ketone body metabolism. Interestingly, there are also some surprising pathways uncovered via our approach, such as Huntington’s disease signaling and an over representation of cancer, and cell cycle related proteins at the synapse. The list of significantly differentially expressed proteins as well as the entire protein dataset was examined using the DAVID bioinformatic database which revealed similar ontologies of this subset (Supplemental Table 3) [48,49].
Figure 4. Analysis of the proteomic changes in Sod2 −/− knockout mice.
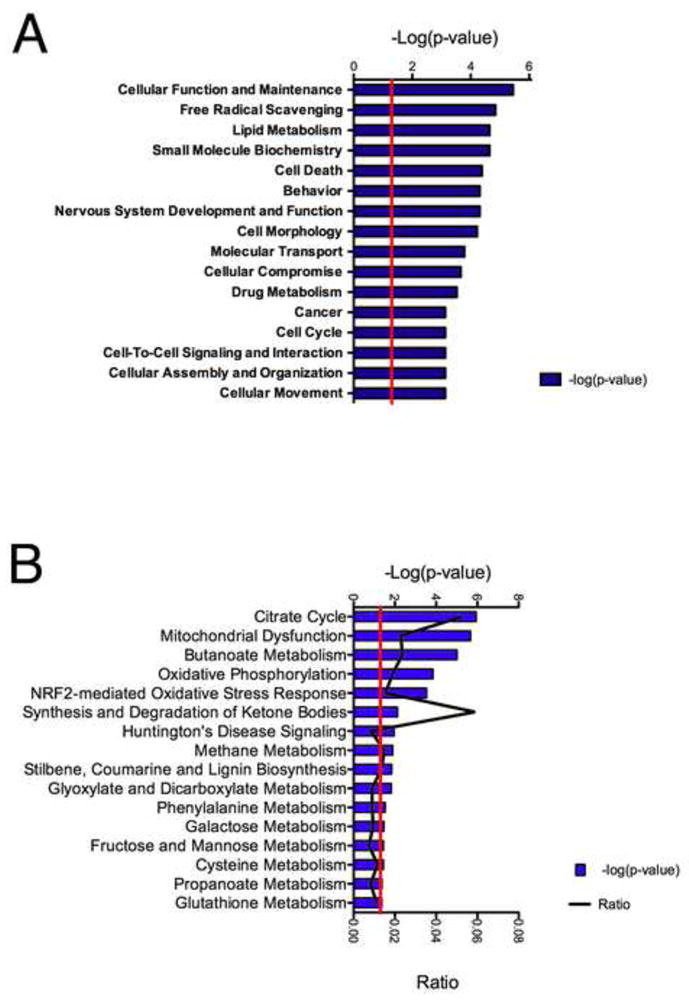
Using the subset of proteins which demonstrated significantly different expression in the Sod2 −/− synaptosomes against their wild-type controls a proteomic profile was generate using Ingenuity pathway analysis. These significantly altered proteins were used as the basis for the list of the top gene functions (A) and canonical pathways (B). Analysis of proteins which reveals a number of specific protein function categories related to metabolic function with the citrate cycle and oxidative phosphorylation score highly. All Listed categories were identified as significant through if they reached p value less than 0.05 from a fisher’s exact t-test.
From our iTRAQ studies, we wished to investigate whether there was differential sensitivity amongst the respiratory chain complexes to the endogenous oxidative stress due to lack of SOD2 in the low dose treated animals (most affected by loss of SOD2). Therefore, we identified all subunits that were mitochondrial via their amino acid sequence, and associated them to their respective respiratory chain complex (Complexes I–V). Two separate experiments were performed and we combined them via a regression model to derive an overall measure of expression per unique peptide identified. A pre-screening was done to eliminate peptides with low levels of confidence (score of less than 50 from the LC-MS/MS experiments). For experiment one, this resulted in 5,107 unique peptides, and in experiment two, 5,407. When we combine these two experiments, 3,486 peptides were common to both experiments. The results are displayed in Figure 5, where it appears there is a systematic down regulation of proteins in Complex I, and a profound and clear deficit in Complex II, relative to all other detected proteins (“other”, which represents proteins not in the respiratory chain).
Figure 5. Specific respiratory chain proteomic alterations.
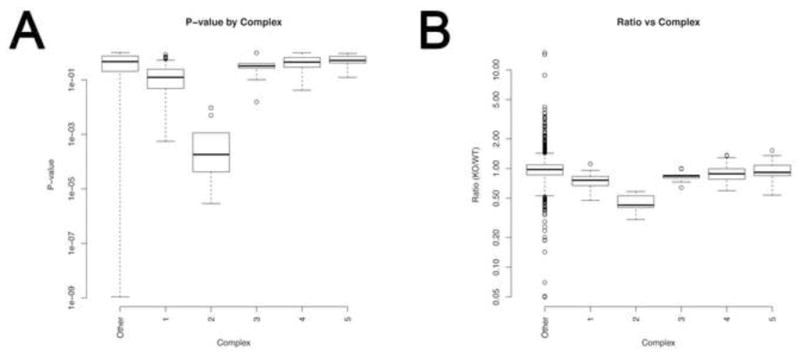
Plots of respiratory chain complexes differentially affected by endogenous oxidative stress. Both the p-values (A) and KO/WT ratios (B) are derived via Limma as described in text. The other category represents peptides not identified to be part of respiratory chain.
Integrative analysis of transcriptomic and proteomic effects of mitochondrial oxidative stress
To further examine the interrelated nature of the synaptic proteome and the synaptic gene expression changes present in our model system, we matched the detected proteins present at the synapse to the expression of each gene from the microarray data. This data was then used to examine the differential gene expression of the wild-type and Sod2 −/− mice within the high and low dose antioxidant treated groups. This data is represented as a volcano plot of the gene expression ratio between the wild-type and Sod2 −/− animals for each identified protein within a given group (Figure 6). On the perpendicular axis, the significance of the gene expression changes is plotted for each target. This plot demonstrates that despite not detecting a large subset of significant changes at the proteome level between controls and Sod2−/− mice (for both high and low dose treated), there are a number of significant changes at the gene expression level. These changes are related to the level of oxidative stress as the low dose animals (Red) have a much greater dynamic range in gene expression than the high dose treated animals (Blue). Additionally, many more genes are significantly differentially expressed (447 low dose treatment versus 94 genes for the high dose, cutoff p = 0.05). This also supports the data shown in Figure 1 in which the low dose knockout animals clustered together and had the most significant expression profiles.
Figure 6. Analysis of gene expression of proteins localized to the synapse.
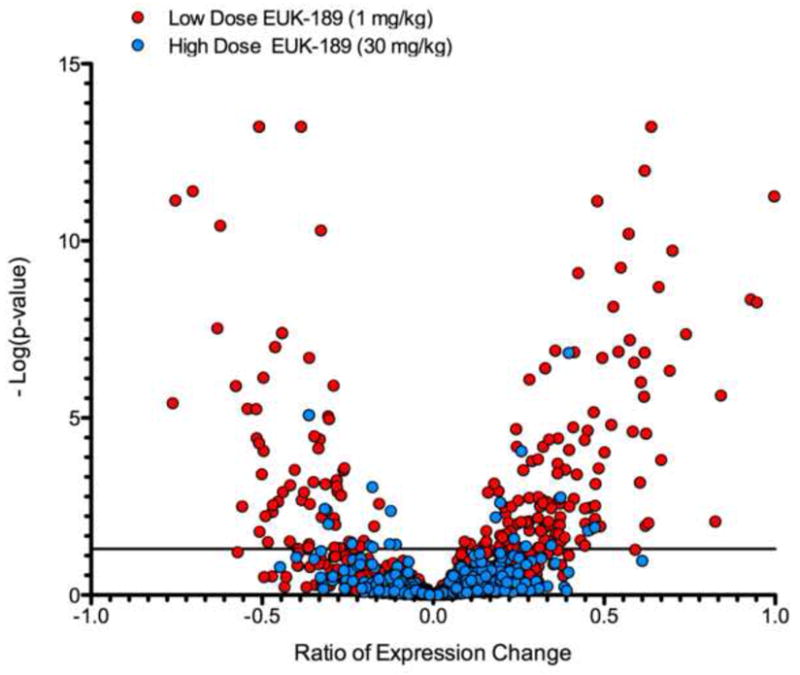
This volcano plot displays the gene expression data of the LC-MS identified proteins in the low dose treated Sod2 −/− as compared to their wild-type controls are shifted in their expression levels in both their magnitude and significance of change. The significance cutoff (p < 0.05) is represented by the horizontal line; points above this line are significantly changed as compared to their respective wild-type controls. Treatment of the Sod2 −/− animals with a higher dose of EUK-189 effectively normalizes the expression of most but not all proteins at the synapse
To gain a better understanding of how the synaptic proteins identified by mass spectrometry interact with each other, we compared the significantly differentially expressed proteins to their corresponding gene expression (Figure 7). The mean expression ratio for each gene and protein is plotted demonstrating that for some proteins the expression ratio is correlated to the gene expression such as Actin (Actn1) and neural cell adhesion molecule 1 (Ncam1). Other proteins did not correlate with their expression level such as Sod2, which in the constitutive Sod2 null mouse produces no detectable activity of the native protein [43]. Surprisingly, the bioinformatic analysis of transcriptome data suggested that mTOR signaling was critically affected due to mitochondrial oxidative stress. This inference was supported by increased alteration of the mTOR pathway components in response to higher levels of mitochondrial oxidative stress (Supplemental Figure 1). In order to validate this bioinformatics prediction, we therefore directly measured mTOR phosphorylation in the synaptosomes we had carried out transcriptomic and proteomic studies on. Phosphorylation of mTOR and 4E-BP1 were tested via sandwich ELISA. While the overall level of mTOR phosphorylation was unchanged; phosphorylation of 4E-BP1, a key downstream target of mTOR, is significantly increased in the Sod2 −/− synaptosomes (n=7, p=0.0023) (Figure 8A). We also examined alternative targets in the mTOR pathway such as UNC-51-like kinase (ULK1) that modulates the autophagy response in cells and is involved in neuronal development (Figure 8B). The expression level of this kinase was unchanged in synaptosomes from the Sod2 −/− mice, however this does not rule out the activation or inhibition of the kinase activity. Further studies of this paradigm are required to fully appreciate the signaling changes as a result of mitochondrial dysfunction.
Figure 7. Correlation of the proteomic and gene expression analysis.

To examine the correlation between the expression changes at the gene expression level to the protein levels detected with mass spectrometry we have expressed this data as a chart displaying the ratio between the wild-type and Sod2 −/− synaptosome samples. This chart identifies that some proteins levels do correlate with the gene expression data, while others do not match their gene expression levels. This reveals that the gene expression and protein level does not necessarily correlate which suggests that a multilevel approach to examination of a disease state is potentially more powerful then examining gene expression or proteins alone.
Figure 8. Increased levels of phosphorylation of 4E-BP1 due to endogenous oxidative stress in synaptosomes.

(A) levels of phosphorylated mTOR do not change as a consequence of endogenous oxidative stress. However, levels of phosphorylated 4E-BP1 are significantly increased in synaptosomes from (p=0.0023) Sod2 null mice. (B). Levels of ULK1, a protein involved in autophagy are not changed in synaptosomes from Sod2 null mice. Equivalent amounts of synaptosomal proteins between wild-type controls and Sod2 null mice (SNAP25 synaptosomal marker) were loaded, and then levels of ULK1 (Unc 51 kinase like 1, a key protein involved in autophagy mediated through TOR) were estimated. No significant differences were seen in synaptosomal levels of ULK1.
Lastly, to explore the interactions among the significantly differentially expressed proteins, we used Ingenuity pathway analysis to create a directed network to map associated nodes of proteins, miRNAs and chemicals (Supplemental Figure 2). The network was created by only mapping interconnected nodes with experimentally validated interacting nodes in the Ingenuity pathway analysis program. Genes with significantly changed expression (p < 0.05) in response to oxidative stress (low-dose EUK-189) were labeled up regulated proteins, and are indicated in green and down regulated nodes are in red; all other nodes are labeled in blue. From this network it is clear that the proteome of the synapse is a complex sub-cellular compartment and that mitochondrial oxidative stress affects expression in a diverse set of genes affecting more than mitochondria related processes.
Discussion
We previously demonstrated in the Sod2 null mouse model that chronic antioxidant drug treatment allows the development (Low dose EUK-189, 1 mg/kg) or prevention (high dose EUK-189, 30 mg/kg) of neurodegeneration and a spongiform encephalopathy [35,36] with altered transcriptome profiles in whole cortical homogenates [42]. A key component of this approach was a bioinformatic analysis on the gene expression data that suggested that Akt (a serine/threonine protein specific kinase involved in cell growth and stress response) signaling, was being differentially affected. We subsequently confirmed this activation in cortical homogenates from Sod2 null mice (differential phosphorylation via Western blotting) in Akt. While that dataset provides a rich resource for analyzing bulk cortical gene expression profiles derived from many cell types and subcellular regions due to endogenous mitochondrial oxidative stress, there are clearly inherent disadvantages in examining homogenates of such a complex tissue as the brain. By sub-fractionating the brain into a relatively pure sub-cellular compartment such as the synaptosome using well established methodologies [20,21,39], one can remove much of the brains complex cellular makeup, facilitating the study of a distinct sub-cellular region of the brain. We had previously used such an approach to demonstrate the consequences to synaptic bioenergetic function by contrasting the spare respiratory capacity of equivalent amounts of synaptosomes (with equivalent mitochondrial content) isolated from Sod2 null mice with those from wild-type, and demonstrated a profound bioenergetic deficit [33]. Therefore, it was important to maintain the same purification procedure in isolating synaptosomes, to allow comparison of the two data sets. Moreover, as the synaptosome is by definition derived from the neuron, our current study focused our analyses to that specific cell type. This overall strategy is highly desirable in studying neurodegenerative phenotypes in the brain.
As in a previous study by us [42], the vast majority of studies on the brain rely on bulk cellular preparations. This means that not only are multiple cell types pooled, potentially obscuring important cell specific changes, but neuronal architecture is completely disrupted, eliminating any insight into subcellular variation relating to neurological phenotypes. As there is a potential for selective loss of synaptosomal sub-populations during the isolation procedure, it was important to measure the yields of synaptosomal protein between the two groups. We found no significant differences in yield between synaptosomes isolated from Sod2 null mice versus wild types. However, there may still be some selective loss despite these observations. In addition, there is some contamination through use of the Percoll gradient [38], and any approach that relies on gradients per se will not be 100% pure. It is also important to note that as we had significant differences in bioenergetic function between synaptosomes of wild type and Sod2 null mice, this implies that if there were significant losses of synaptosomes due to endogenous oxidative stress in the Sod2 null mice synaptosomes (from oxidative damage to mitochondria or synaptosomes), any significant differences we detect here are likely under-reporting the magnitude of possible differences due to endogenous oxidative stress. Mitigating the possibility of selective loss of synaptosomes are measures we previously reported as being equivalent in synaptosomes from Sod2 null mice including resting metabolic rate, and stimulated glutamate release. Only under conditions of energetic load do we see significant differences in bioenergetic function [33].
Arguably the most optimal method for measuring synaptosomal function, whilst simultaneously ascertaining their purity would be at the level of the single synaptosome, where many individual parameters can be evaluated either in parallel, or serially for hundreds to thousands of synaptosomes. Such an approach is feasible, and has been carried out in the context of a mouse model of neurodegeneration [17], but is currently not compatible with large scale genomic and proteomic technologies due to the amount of material required for these assays. Here, we have simultaneously examined both proteomic and genomic outcomes on a relatively pure subcellular preparation from the brains of wild type mice compared to a well-characterized neurodegenerative animal model of mitochondrial oxidative stress, and provided a powerful new approach to evaluate changes in the synaptosome that may be vitally important in improving our understanding of early events in the breakdown of the synaptic network in neurodegenerative disease.
For whole transcriptome gene expression profiling we examined four groups of 10 or more mice, representing wild-type and Sod2 −/− mice, treated with either a low (1 mg/kg) or a high (30 mg/kg) dose of antioxidant. This whole genome expression profiling approach of mRNAs trafficked to synaptosomes resulted in the identification of 1,101 significantly differentially expressed genes, due to endogenous oxidative stress in the synaptosome. These targets were then hierarchically clustered and displayed in a heat-map that shows the interactions among both the genes and samples. Our cluster analysis found that the low dose Sod2 null mice closely associate together, while the other treatments are interspersed amongst each other. This demonstrates that the animals undergoing neurodegeneration due to endogenous mitochondrial oxidative stress are experiencing a different gene expression “space” in the synaptosome than those animals that lack Sod2, yet are protected with a higher dose of antioxidant. Interestingly, while the higher dosage of antioxidant can normalize much of the differential gene expression to wild-type levels, it does not preserve the mitochondrial enzyme function or the bioenergetic capacity at the synapse [33]. This likely means that loss of neurons due to mitochondrial oxidative stress is occurring at multiple levels throughout the neuron, and is not a purely bioenergetic problem at the synapse.
We discovered that there are a number of gene functional categories that are significantly overrepresented and are not immediately related to mitochondrial function. Some of these functional categories such as “cell death” are dramatically increased in the significance of the category in the low dose treated vs. the high dose treated group. It is possible that the magnitude of the altered expression within these categories is what determines cell fate between survival in the high dose treated animals, versus death in the low dose treated animals. The use of pathway analysis is particularly useful because it narrows the ontological functions and gene suites that might prove most important to the response to mitochondrial oxidative stress. The analysis also provides the overrepresented canonical signaling pathways in the two treatments (Figure 2B and 3B). In Figure 2B, the response to oxidative stress in the high dose treated mice has a number of biochemical pathways related to the metabolism of the cell including “synthesis and degradation of ketone bodies.” We were unable to detect ketones in synaptosomes from any animals in our study, likely due to washout of ketones during purification of the synaptosomes. Further support for the ketone pathway being relevant is an initial observation made in the sera of Sod2 null mice showing elevated ketone bodies [43]. This ketone synthesis/degradation pathway has been suggested to be protective against the oxidative stress associated with Alzheimer’s disease by fortifying mitochondrial function [50–52]. In the low dose treated animals, this category is less strongly represented in the transcriptomic data indicating that modulation of this pathway could be linked to the level of oxidative stress incurred by neurons. This pattern is also seen with the over-representation of inflammation through Interlukin-8 (IL-8) signaling which has been associated with age related neurodegenerative diseases [53–55]. Under conditions of mild oxidative stress that would occur normally throughout the aging process, these pathways may modulate synaptic function to avoid the progression of neurodegeneration.
The over-representation of the mTOR pathway we show in Figure 2B suggested that signaling in this pathway was being differentially affected due to endogenous mitochondrial oxidative stress. One of the key proteins in this pathway is 4E-BP1 (Eukaryotic translation initiation factor 4E-binding protein 1), which interacts with mTOR and is involved in mRNA translation. Phosphorylation of 4E-BP1 results in increased levels of mRNA translation, typically in response to insulin signaling and stress. We therefore tested whether or not the phosphorylation of 4E-BP1 was being differentially phosphorylated as a consequence of endogenous mitochondrial oxidative stress in synaptosomes. We showed that consistent with our prior studies on activation of Akt [42] in cortical homogenates, 4E-BP1 was being differentially phosphorylated due to mitochondrial oxidative stress. This result implies that in response to mitochondrial oxidative stress at the synaptic junction, there is increased demand for protein synthesis.
The proteome of the synapse has been examined in a number of paradigms [56,57], however these studies are rarely combined with other high dimensional datasets such as metabolomics or transcriptomics which can provide more depth to the analysis than a single method alone [58,59]. Here we report the proteome of the mouse cortical synapse (Supplemental Table 1) consisting of proteins detectable in synaptosome samples in the wild-type or Sod2 −/− mice as well as the significantly differentially expressed proteins between these genotypes (Supplemental Table 2). The detected proteins in the low dose treated samples were analyzed to examine significant changes in the proteome between the two genotypes. Very few proteins appeared to be significantly altered in their relative expression levels, but the ones that are significantly different provide insight into the effects of mitochondrial oxidative stress. Although the differential expression results in our proteomic approach were modest, it allows us to localize to the level of the synaptosome specific functional deficits due to endogenous oxidative stress, something which our prior approaches were unable to resolve. Further, this protein data also adds a new dimension to this analysis, as the protein level is not concordant with the gene expression data. There could be several reasons for this lack of concordance, including a high degree of biological variation among the wild-type and Sod2 −/− mice, an inherent lack of concordance between various messages and their respective proteins, or an under sampling of the proteome using the iTRAQ methodology despite extensive fractionation at the peptide level. Alternatively, it is also possible that only modest proteomic expression changes take place in the Sod2−/− mice despite a higher level of changes seen at the transcriptome level. It is also possible that the proteomic changes involved primarily posttranslational modifications, such as changes in phosphorylation and acetylation, or oxidative modifications such as cysteine oxidation [60], carbonylation [61], or tyrosine nitration [62]. We are currently pursuing studies that would target changes in the posttranslational status of the proteome in the Sod2 −/− mouse model, especially with respect to oxidative damage.
Prior to the studies we present here, it was formally possible that there may have been a high concordance between message and protein trafficked to the synaptosome in our model, but without measuring both message and protein, it is impossible to generalize whether or not mRNA/protein levels are concordant, as they vary with each biological situation. The list of detected proteins provides useful data for additional bioinformatic analysis. The total list of proteins was input into the DAVID bioinformatic database (Supplemental Table 3) and Ingenuity pathway analysis to determine the overrepresented functions of the proteins independent of the presence or absence of oxidative stress. These two subsets were then examined for concordance of ontological functions. The results of this analysis showed a number of ontological categories that regulate nervous system development, and metabolic functions. Despite a general scarcity of data showing proteomic variation between the groups being compared in our experiments, this data serves as a useful baseline, where we can examine the gene expression changes of proteins in different models that are pre-validated as being present at the cortical pre-synaptic nerve terminal.
To accomplish a combined analysis for gene expression and proteins, all gene expression probes were matched to their respect proteins identified by mass spectrometry. This expression data was then examined to view the effects of oxidative stress in the low dose and high dose treated mice. Figure 5 shows a volcano plot of this data demonstrating that the expression of the proteins at the synapse are more effected in both significance and magnitude by increased oxidative stress in the low dose treated mice, compared to those mice treated with a higher dose of antioxidant. Combined analysis of the transcriptomic data and the proteomic data allows for the creation of a network of interacting proteins at the synapse (Supplemental Figure 2).
It is also worth commenting on the specific vulnerabilities of the respiratory chain to endogenous oxidative stress. It has long been suspected that Complex I is the most vulnerable respiratory chain component to endogenous insult, partly due to the prevalence of Complex I deficits in mitochondrial disease, but also due to a number of reports implicating Complex I deficiencies in age related neurodegenerative disease such as Alzheimer’s or Parkinson’s disease. Although we do see Complex I deficits in the synaptosome via our iTRAQ approach, Complex II is far more severely affected, consistent with our prior studies in other tissues. We previously reported deficits in aconitase, as well as Complex II in different tissues and treatments of the Sod2 null mouse [35,42,63,64]. The TCA cycle enzyme aconitase in particular is well recognized as being vulnerable to oxidative stress through oxidation of its vulnerable iron-sulfur(Fe-S) cluster, which essentially inactivates the enzyme [65]. Mammalian mitochondria can serve as a site of synthesis to produce Fe-S clusters for use in other proteins throughout the cell [66]. However, Fe-S clusters are not exclusively produced within mitochondria. It is therefore possible that endogenous oxidative stress not only has the capacity to oxidize mitochondrial constituents, and directly damage macromolecules, but to propagate functional deficits throughout the cell by impairing proper assembly of iron-sulfur clusters in the mitochondria. Hence the Sod2 null mouse may serve as a model to better understand how endogenous oxidative stress can affect cell function through improper Fe-S cluster assembly.
The central nervous system is highly intricate, with many levels of complexity. There are regions of the brain that are composed of hundreds of cell types each with their own specific function. In this study, we have attempted to disentangle the complexity of the frontal cortex by isolating biological samples exclusive to neurons via isolation and characterization of the synaptosome. We examined these samples in the context of a well-established model of mitochondrial oxidative stress using proteomic and transcriptomic bioinformatic analysis, uncovering a novel response of protein synthesis due to mitochondrial oxidative stress in synaptosomes (increased levels of 4E-BP1). Overall, our analysis has generated a directed network of a portion of the synaptic proteome affected by oxidative stress, which may be valuable in framing new questions about the vulnerability of the synapse to endogenous oxidative stress.
Supplementary Material
The transcriptional modulation of mTOR signaling components was examined in response to both high and low doses of EUK189 antioxidant. The low dose treated mice had many more signaling components upregulated (yellow) or down regulated (blue) than the high dose treated animals.
To examine the relationship between the altered proteins and the intervening nodes Ingenuity pathway analysis was used to create a network. The analysis revealed a network that included a number of associated proteins, miRNAs and chemicals that are associated with the altered proteins. Green for up regulation, red for down regulation, and blue for related indirect nodes within the target network.
Mass spectrometry-based quantitative proteomic analysis using 8-plex iTRAQ isobaric chemical labeling of synaptosome proteins which are detected in Sod2 −/− animals as compared to the wild-type controls reveals a subset of altered proteins. Specifically identified proteins present at the synapse including 12 proteins that had significantly different expression ratios between the two genotypes.
The results of two independent experiments were combined to give a list of the detected peptides and their respective proteins. This table displays the results of the statistical analysis and the correction for the false discovery rate.
To further examine the identified protein set that was identified through the mass spectrometry proteomic experiments, the DAVID bioinformatics database was used to generate ontological analysis. The table lists the cell compartment, biological process, molecular function, and KEGG associated pathways. These lists were generated for the just the significantly altered proteins as well as the entire identified protein set. This analysis provides insight into the alterations created by the removal of Sod2 as well as the general composition and function of the mouse synapse.
Highlights.
Subcellular characterization of endogenous mitochondrial oxidative stress.
Proteogenomic analysis uncover novel interactions of oxidative stress.
Increased mTOR signaling due to endogenous mitochondrial oxidative stress.
Acknowledgments
This work was supported in part by the Glenn Foundation for Medical Research (SM), the Larry L. Hillblom Foundation (SM). Gene expression and mass spectrometry support was provided by the National Institutes of Health through the Nathan Shock Center [P30AG025708], Geroscience Mass Spectrometry and Imaging Core [PO1AG025901], and a [PO1AG025901](SM & BG), and through the NCRR shared instrumentation program [S10RR024615] (BG). JMF and SWC were supported by a [T32AG000266] awarded to the Buck Institute for Research on Aging. We thank Ken Beckman for key help in the gene expression studies.
Footnotes
Conflict of Interest statement. The authors declare there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fainzilber M, Budnik V, Segal RA, Kreutz MR. From Synapse to Nucleus and Back Again—Communication over Distance within Neurons. The Journal of Neuroscience. 2011;31:16045–16048. doi: 10.1523/JNEUROSCI.4006-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen S, Greenberg ME. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol. 2008;24:183–209. doi: 10.1146/annurev.cellbio.24.110707.175235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duric V, Banasr M, Stockmeier CA, Simen AA, Newton SS, Overholser JC, Jurjus GJ, Dieter L, Duman RS. Altered expression of synapse and glutamate related genes in postmortem hippocampus of depressed subjects. Int J Neuropsychopharmacol. 2012:1–14. doi: 10.1017/S1461145712000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Ann N Y Acad Sci. 2008;1147:53–60. doi: 10.1196/annals.1427.002. [DOI] [PubMed] [Google Scholar]
- 5.Fuhrmann M, Mitteregger G, Kretzschmar H, Herms J. Dendritic pathology in prion disease starts at the synaptic spine. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:6224–6233. doi: 10.1523/JNEUROSCI.5062-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hussain NK, Sheng M. Neuroscience. Making synapses: a balancing act. Science. 2005;307:1207–1208. doi: 10.1126/science.1110011. [DOI] [PubMed] [Google Scholar]
- 7.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. The American journal of pathology. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheff S, Price D. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Morrison J. Which synapses are affected in aging and what is the nature of their vulnerability? A commentary on “life span and synapses: will there be a primary senile dementia?”. Neurobiol Aging. 2001;22:349–350. doi: 10.1016/s0197-4580(00)00244-x. [DOI] [PubMed] [Google Scholar]
- 10.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 11.Kindler S, Kreienkamp H-J. Dendritic mRNA targeting and translation. Adv Exp Med Biol. 2012;970:285–305. doi: 10.1007/978-3-7091-0932-8_13. [DOI] [PubMed] [Google Scholar]
- 12.Thomas MG, Loschi M, Desbats MA, Boccaccio GL. RNA granules: the good, the bad and the ugly. Cellular signalling. 2011;23:324–334. doi: 10.1016/j.cellsig.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swanger SA, Bassell GJ. Making and breaking synapses through local mRNA regulation. Current opinion in genetics & development. 2011;21:414–421. doi: 10.1016/j.gde.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu-Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, Warren ST, Wolozin B. Local RNA translation at the synapse and in disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:16086–16093. doi: 10.1523/JNEUROSCI.4105-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 16.Nicholls DG. The glutamatergic nerve terminal. Eur J Biochem. 1993;212:613–631. doi: 10.1111/j.1432-1033.1993.tb17700.x. [DOI] [PubMed] [Google Scholar]
- 17.Choi SW, Gerencser AA, Lee DW, Rajagopalan S, Nicholls DG, Andersen JK, Brand MD. Intrinsic bioenergetic properties and stress- sensitivity in dopaminergic synaptosomes. Journal of Neuroscience. 2011 doi: 10.1523/JNEUROSCI.5817-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams C, Mehrian Shai R, Wu Y, Hsu Y-H, Sitzer T, Spann B, McCleary C, Mo Y, Miller CA. Transcriptome analysis of synaptoneurosomes identifies neuroplasticity genes overexpressed in incipient Alzheimer’s disease. PLoS ONE. 2009;4:e4936. doi: 10.1371/journal.pone.0004936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicholls DG, Sihra TS. Synaptosomes possess an exocytotic pool of glutamate. Nature. 1986;321:772–773. doi: 10.1038/321772a0. [DOI] [PubMed] [Google Scholar]
- 20.Whittaker VP. Thirty years of synaptosome research. J Neurocytol. 1993;22:735–742. doi: 10.1007/BF01181319. [DOI] [PubMed] [Google Scholar]
- 21.Nicholls DG. Bioenergetics and transmitter release in the isolated nerve terminal. Neurochem Res. 2003;28:1433–1441. doi: 10.1023/a:1025653805029. [DOI] [PubMed] [Google Scholar]
- 22.Meer EJ, Wang DO, Kim S, Barr I, Guo F, Martin KC. Identification of a cis-acting element that localizes mRNA to synapses. Proceedings of the National Academy of Sciences. 2012;109:4639–4644. doi: 10.1073/pnas.1116269109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dahlhaus M, Wan Li K, van der Schors RC, Saiepour MH, van Nierop P, Heimel JA, Hermans JM, Loos M, Smit AB, Levelt CN. The synaptic proteome during development and plasticity of the mouse visual cortex. Mol Cell Proteomics. 2011;10:M110. doi: 10.1074/mcp.M110.005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cajigas IJ, Will T, Schuman EM. Protein homeostasis and synaptic plasticity. The EMBO Journal. 2010;29:2746–2752. doi: 10.1038/emboj.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelman BB, Nguyen TP. Synaptic proteins linked to HIV-1 infection and immunoproteasome induction: proteomic analysis of human synaptosomes. J Neuroimmune Pharmacol. 2010;5:92–102. doi: 10.1007/s11481-009-9168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engmann O, Campbell J, Ward M, Giese KP, Thompson AJ. Comparison of a protein- level and peptide-level labeling strategy for quantitative proteomics of synaptosomes using isobaric tags. J Proteome Res. 2010;9:2725–2733. doi: 10.1021/pr900627e. [DOI] [PubMed] [Google Scholar]
- 27.Wang DO, Martin KC, Zukin RS. Spatially restricting gene expression by local translation at synapses. Trends in Neurosciences. 2010;33:173–182. doi: 10.1016/j.tins.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morciano M, Beckhaus T, Karas M, Zimmermann H, Volknandt W. The proteome of the presynaptic active zone: from docked synaptic vesicles to adhesion molecules and maxi-channels. Journal of Neurochemistry. 2009;108:662–675. doi: 10.1111/j.1471-4159.2008.05824.x. [DOI] [PubMed] [Google Scholar]
- 29.Etheridge N, Lewohl JM, Mayfield RD, Harris RA, Dodd PR. Synaptic proteome changes in the superior frontal gyrus and occipital cortex of the alcoholic brain. Proteomics Clin Appl. 2009;3:730–742. doi: 10.1002/prca.200800202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai F, Witzmann FA. Synaptosome proteomics. Subcell Biochem. 2007;43:77–98. doi: 10.1007/978-1-4020-5943-8_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barber DS, Stevens S, LoPachin RM. Proteomic analysis of rat striatal synaptosomes during acrylamide intoxication at a low dose rate. Toxicological sciences : an official journal of the Society of Toxicology. 2007;100:156–167. doi: 10.1093/toxsci/kfm210. [DOI] [PubMed] [Google Scholar]
- 32.Ch’ng TH, Martin KC. Synapse-to-nucleus signaling. Current opinion in neurobiology. 2011;21:345–352. doi: 10.1016/j.conb.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flynn JM, Choi SW, Day NU, Gerencser AA, Hubbard A, Melov S. Impaired spare respiratory capacity in cortical synaptosomes from Sod2 null mice. Free radical biology & medicine. 2011 doi: 10.1016/j.freeradbiomed.2010.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geschwind DH, Konopka G. Neuroscience in the era of functional genomics and systems biology. Nature. 2009;461:908–915. doi: 10.1038/nature08537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinerfeld D, Traini M, Weinberger R, Cochran B, Doctrow S, Harry J, Melov S. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. Journal of neurochemistry. 2004;88:657–667. doi: 10.1046/j.1471-4159.2003.02195.x. [DOI] [PubMed] [Google Scholar]
- 36.Melov S, Doctrow S, Schneider J, Haberson J, Patel M, Coskun P, Huffman K, Wallace D, Malfroy B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:8348–853. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melov S, Schneider J, Day B, Hinerfeld D, Coskun P, Mirra S, Crapo J, Wallace D. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 38.Dunkley PR, Jarvie PE, Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nature protocols. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- 39.Choi SW, Gerencser AA, Nicholls DG. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. Journal of Neurochemistry. 2009;109:1179–1191. doi: 10.1111/j.1471-4159.2009.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 41.Gentleman R, Carey V, Bates D, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini A, Sawitzki G, Smith C, Smyth G, Tierney L, Yang J, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golden T, Hubbard A, Morten K, Hinerfeld D, Melov S. Pharmacogenomic profiling of an oxidative stress-mediated spongiform encephalopathy. Free radical biology & medicine. 2005;39:152–163. doi: 10.1016/j.freeradbiomed.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Huang T, Carlson E, Melov S, Ursell P, Olson J, Noble L, Yoshimura M, Berger C, Chan P, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 44.Tang D, Kang R, Zeh HJ, Lotze MT. High-mobility group box 1, oxidative stress, and disease. Antioxidants & redox signaling. 2011;14:1315–1335. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annual Review of Immunology. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 46.Li W, Sama AE, Wang H. Role of HMGB1 in cardiovascular diseases. Current opinion in pharmacology. 2006;6:130–135. doi: 10.1016/j.coph.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 48.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 49.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 50.Kim DY, Davis LM, Sullivan PG, Maalouf M, Simeone TA, van Brederode J, Rho JM. Ketone bodies are protective against oxidative stress in neocortical neurons. Journal of neurochemistry. 2007;101:1316–1326. doi: 10.1111/j.1471-4159.2007.04483.x. [DOI] [PubMed] [Google Scholar]
- 51.Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 2007;145:256–264. doi: 10.1016/j.neuroscience.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Studzinski CM, MacKay WA, Beckett TL, Henderson ST, Murphy MP, Sullivan PG, Burnham WM. Induction of ketosis may improve mitochondrial function and decrease steady-state amyloid-beta precursor protein (APP) levels in the aged dog. Brain Res. 2008;1226:209–217. doi: 10.1016/j.brainres.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 53.Reale M, Iarlori C, Thomas A, Gambi D, Perfetti B, Di Nicola M, Onofrj M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav Immun. 2009;23:55–63. doi: 10.1016/j.bbi.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 54.Bonotis K, Krikki E, Holeva V, Aggouridaki C, Costa V, Baloyannis S. Systemic immune aberrations in Alzheimer’s disease patients. J Neuroimmunol. 2008;193:183–187. doi: 10.1016/j.jneuroim.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 55.Magaki S, Mueller C, Dickson C, Kirsch W. Increased production of inflammatory cytokines in mild cognitive impairment. Experimental gerontology. 2007;42:233–240. doi: 10.1016/j.exger.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schrimpf SP, Meskenaite V, Brunner E, Rutishauser D, Walther P, Eng J, Aebersold R, Sonderegger P. Proteomic analysis of synaptosomes using isotope-coded affinity tags and mass spectrometry. Proteomics. 2005;5:2531–2541. doi: 10.1002/pmic.200401198. [DOI] [PubMed] [Google Scholar]
- 57.Filiou MD, Bisle B, Reckow S, Teplytska L, Maccarrone G, Turck CW. Profiling of mouse synaptosome proteome and phosphoproteome by IEF. Electrophoresis. 2010;31:1294–1301. doi: 10.1002/elps.200900647. [DOI] [PubMed] [Google Scholar]
- 58.Gillardon F, Rist W, Kussmaul L, Vogel J, Berg M, Danzer K, Kraut N, Hengerer B. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics. 2007;7:605–616. doi: 10.1002/pmic.200600728. [DOI] [PubMed] [Google Scholar]
- 59.Bode M, Irmler M, Friedenberger M, May C, Jung K, Stephan C, Meyer HE, Lach C, Hillert R, Krusche A, Beckers J, Marcus K, Schubert W. Interlocking transcriptomics, proteomics and toponomics technologies for brain tissue analysis in murine hippocampus. Proteomics. 2008;8:1170–1178. doi: 10.1002/pmic.200700742. [DOI] [PubMed] [Google Scholar]
- 60.Held JM, Gibson BW. Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol Cell Proteomics. 2012;11:R111. doi: 10.1074/mcp.R111.013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Møller IM, Rogowska-Wrzesinska A, Rao RSP. Protein carbonylation and metal-catalyzed protein oxidation in a cellular perspective. J Proteomics. 2011;74:2228–2242. doi: 10.1016/j.jprot.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 62.Lee JR, Kim JK, Lee SJ, Kim KP. Role of protein tyrosine nitration in neurodegenerative diseases and atherosclerosis. Arch Pharm Res. 2009;32:1109–1118. doi: 10.1007/s12272-009-1802-0. [DOI] [PubMed] [Google Scholar]
- 63.Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun A, Zastawny T, Dizdaroglu M, Goodman S, Huang T, Miziorko H, Epstein C, Wallace D. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:846–51. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morten K, Ackrell B, Melov S. Mitochondrial Reactive Oxygen Species in Mice Lacking Superoxide Dismutase 2: ATTENUATION VIA ANTIOXIDANT TREATMENT. The Journal of biological chemistry. 2006;281:3354–3359. doi: 10.1074/jbc.M509261200. [DOI] [PubMed] [Google Scholar]
- 65.Imlay JA. Iron-sulphur clusters and the problem with oxygen. Mol Microbiol. 2006;59:1073–1082. doi: 10.1111/j.1365-2958.2006.05028.x. [DOI] [PubMed] [Google Scholar]
- 66.Ye H, Rouault TA. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry. 2010;49:4945–4956. doi: 10.1021/bi1004798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The transcriptional modulation of mTOR signaling components was examined in response to both high and low doses of EUK189 antioxidant. The low dose treated mice had many more signaling components upregulated (yellow) or down regulated (blue) than the high dose treated animals.
To examine the relationship between the altered proteins and the intervening nodes Ingenuity pathway analysis was used to create a network. The analysis revealed a network that included a number of associated proteins, miRNAs and chemicals that are associated with the altered proteins. Green for up regulation, red for down regulation, and blue for related indirect nodes within the target network.
Mass spectrometry-based quantitative proteomic analysis using 8-plex iTRAQ isobaric chemical labeling of synaptosome proteins which are detected in Sod2 −/− animals as compared to the wild-type controls reveals a subset of altered proteins. Specifically identified proteins present at the synapse including 12 proteins that had significantly different expression ratios between the two genotypes.
The results of two independent experiments were combined to give a list of the detected peptides and their respective proteins. This table displays the results of the statistical analysis and the correction for the false discovery rate.
To further examine the identified protein set that was identified through the mass spectrometry proteomic experiments, the DAVID bioinformatics database was used to generate ontological analysis. The table lists the cell compartment, biological process, molecular function, and KEGG associated pathways. These lists were generated for the just the significantly altered proteins as well as the entire identified protein set. This analysis provides insight into the alterations created by the removal of Sod2 as well as the general composition and function of the mouse synapse.