

Background: NOD2 belongs to the NLR family. JNKBP1, a scaffold protein, is characterized by an N-terminal WD-40 domain.
Results: JNKBP1 is co-expressed with NOD2 in epithelial and immune cells of intestine mucosa. Binding of JNKB1 to NOD2 attenuates NOD2 signaling by inhibiting its oligomerization.
Conclusion: JNKBP1 is a new NOD2 modulator.
Significance: JNKBP1, by down-regulating NOD2 signaling, could contribute to maintaining intestinal immune homeostasis.
Keywords: Immunology, Inflammatory Bowel Disease, NF-kappaB (NF-kappaB), Nod-like Receptors (NLR), Scaffold Proteins, Signal Transduction
Abstract
NOD2 is one of the best characterized members of the cytosolic NOD-like receptor family. NOD2 is able to sense muramyl dipeptide, a specific bacterial cell wall component, and to subsequently induce various signaling pathways leading to NF-κB activation and autophagy, both events contributing to an efficient innate and adaptive immune response. Interestingly, loss-of-function NOD2 variants were associated with a higher susceptibility for Crohn disease, which highlights the physiological importance of proper regulation of NOD2 activity. We performed a biochemical screen to search for new NOD2 regulators. We identified a new NOD2 partner, c-Jun N-terminal kinase-binding protein 1 (JNKBP1), a scaffold protein characterized by an N-terminal WD-40 domain. JNKBP1, through its WD-40 domain, binds to NOD2 following muramyl dipeptide activation. This interaction attenuates NOD2-mediated NF-κB activation and IL-8 secretion as well as NOD2 antibacterial activity. JNKBP1 exerts its repressor effect by disturbing NOD2 oligomerization and RIP2 tyrosine phosphorylation, both steps required for downstream NOD2 signaling. We furthermore showed that JNKBP1 and NOD2 are co-expressed in the human intestinal epithelium and in immune cells recruited in the lamina propria, which suggests that JNKBP1 contributes to maintain NOD2-mediated intestinal immune homeostasis.
Introduction
In animals, the detection of infectious agents relies on specific host pattern recognition receptors. Next to the well known family of membrane-bound Toll-like receptors (1), a new family of cytosolic pattern recognition receptors, called NOD (nucleotide-binding oligomerization domain)-like receptors (NLRs),3 was defined (2). In humans, the NLR family includes 23 members that are characterized by a tripartite structure: a central nucleotide-binding domain (NBD), C-terminal leucine-rich repeats (LRRs), and an N-terminal effector binding domain (2). Based on this effector domain, the NLR members are distributed in five subfamilies and mediate various signaling pathways (2).
NOD2 (nucleotide-binding oligomerization domain-containing protein 2) has been the most studied member of the NLR family since the identification of NOD2 variants associated with a higher susceptibility for Crohn disease (CD) (3, 4). NOD2 is characterized by two N-terminal caspase recruitment domains (CARDs) (5). Through its LRRs, NOD2 senses the muramyl dipeptide (MDP), a subunit of peptidoglycan present in the cell wall of Gram-negative and -positive bacteria and subsequently induces nuclear factor κ-light chain-enhancer of activated B cell (NF-κB) mediated inflammatory responses (6, 7). Upon MDP recognition, NOD2 oligomerizes via its NBD and recruits the serine-threonine-tyrosine kinase receptor-interacting protein 2 (RIP2) through homotypic CARD-CARD interactions (5). After autophosphorylation and polyubiquitination involving cellular inhibitor of apoptosis-1 and -2 (cIAP-1 and -2), RIP2 can recruit the kinase complexes TNF receptor-associated kinase 1 (TAK1) and IκB kinase (IKK), which in turn induce NF-κB activation and the expression of genes involved in inflammatory and immune responses (8–10). Only recently, it became clear that the function of the innate receptor NOD2 is far more complex than previously assumed (11). For example, NOD2 was also shown to mediate autophagy, allowing not only the bacterial clearance but also an efficient antigen presentation on dendritic cells, both events involved in innate and adaptive immune response, respectively (12, 13). Mechanisms by which NOD2 variants can lead to CD development are still under investigation. The most likely hypothesis suggests that the impaired function of NOD2 variants in intestinal epithelial and phagocytic cells results in deficiencies in epithelial barrier function, which subsequently lead to increased bacterial invasion and inflammation at intestinal sites (11).
It is now well accepted that NOD2 plays an important role in intestinal immune homeostasis (14, 15). Such a function has to be tightly controlled. However, although the NOD2-mediated NF-κB activation pathway has been extensively studied, little is known about its regulation. In an attempt to bring new insights on the NOD2 regulation mechanisms, we co-purified NOD2 binding partners in HEK293 cells stably expressing NOD2. The characterization of a new NOD2 partner, JNK-binding protein 1 (JNKBP1), is described in this work.
Mouse JNKBP1, also named mitogen-activated protein kinase-binding protein 1 (MAPKBP1), was initially identified in a yeast two-hybrid system used to search for proteins interacting with JNK3 (16). JNKBP1 contains 12 WD-40 repeats in the N-terminal part and a JNK-binding region located in the C-terminal region (16). JNKBP1 appears to be ubiquitously expressed in mouse tissues and to play a scaffold function in JNK signaling. An association of mouse JNKBP1 with TAK1 and TNF-receptor associated factor 2 (TRAF2) has been recently shown together with an up-regulating effect of JNKBP1 on NF-κB activation induced by both TAK1 and TRAF2 (17). Human JNKBP1 shares 85% homology with mouse JNKBP1. A scaffold function of human JNKBP1, however, has never been investigated.
We demonstrate here that human JNKBP1 attenuates NOD2-mediated proinflammatory and antibacterial signaling. This is mediated through physical interaction with NOD2 involving the WD-40 domain in JNKBP1. We further provide details of the molecular mechanism underlying JNKBP1 function in NOD2 signaling.
EXPERIMENTAL PROCEDURES
Cell Culture
Human embryonic kidney 293 (HEK293), HEK293T, Jurkat, U937, T98G, HCT116, and THP1 cells were obtained from the American Type Culture Collection (Manassas, VA). LN18 cells were provided by P. Robe (Laboratory of Human Genetics, GIGA-Cancer, University of Liege, Liege, Belgium). Stably transfected cell lines were generated by lentiviral transduction of HEK293 cells followed by blasticidin selection (10 μg/ml; InvivoGen). These cells were divided by fluorescence-activated cell sorting (FACS) into four cellular populations called GNV-1, -2, -3, and -4 for HEK293 cells stably expressing GFP-NOD2-V5 (supplemental Fig. S1A). This work was performed with the GNV-3 cell population expressing a NOD2 intermediate expression level and allowing an MDP-inducible NF-κB activation (supplemental Fig. S1, B and C). HEK293T cells and GNV cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum, 1% glutamine (Invitrogen). HCT116 cells were cultured in McCoy's 5A medium (Lonza, Belgium) supplemented with 10% fetal bovine serum, 1% glutamine (Invitrogen). THP1 cells were cultured in RPMI 1640 medium (Lonza, Belgium) supplemented with 10% fetal bovine serum, 1% glutamine (Invitrogen), and 1% antibiotics (streptomycin and penicillin). The cells were maintained at 37 °C in a 5% CO2 atmosphere.
Antibodies and Reagents
MDP (A9519) was purchased from Sigma-Aldrich. Tumor necrosis factor α (TNFα) (300-01A) was obtained from PeproTech EC. The protease inhibitor set (Complete) was obtained from Roche Applied Science. The reporter construct (κB)5-LUC was purchased from Stratagene (Agilent Technologies, Santa Clara, CA). Antibodies used in this study were mouse monoclonal anti-β-tubulin antibody (T4026, Sigma-Aldrich), mouse monoclonal anti-FLAG M2 antibody (F3165, Sigma-Aldrich), mouse monoclonal anti-V5 antibody (Invitrogen), mouse monoclonal anti-HA antibody (16B12, Covance), mouse anti-GFP antibody (Roche Applied Science), mouse monoclonal anti-phosphotyrosine (4G10, Millipore), mouse anti-phospho-IκBα (Ser32/36) antibody (5A5, Cell Signaling), rabbit monoclonal anti-phospho-IKKα (Ser176)/IKKβ (Ser177) antibody (C84E11, Cell Signaling), rabbit polyclonal anti-phospho-NF-κB p65 (Ser536) antibody (3031, Cell Signaling), rabbit polyclonal anti-NOD2 antibody (2513, ProSci Inc.), mouse anti-NOD2 antibody (2D9, Millipore), rabbit anti-MAPKBP1 antibody (HPA030832, Sigma), and rabbit polyclonal anti-CD68 antibody (KP1, Dako). Gentamicin was obtained from InvivoGen.
Expression Plasmids
Expression plasmids encoding wild-type (WT) NOD2 or HA- or FLAG-tagged NOD2 forms (WT, ΔCARDs, ΔLRRs, CARD1, CARD2, NBD, LRRs, R702, G908, and L1007fs) were obtained from G. Nuñez (University of Michigan Medical School and Comprehensive Cancer Center, Ann Harbor, MI). Expression plasmids encoding RIP2-FLAG were a kind gift from Mathieu Bertrand (Department for Molecular Biomedical Research, VIB = Vlaams Instituut voor Biotechnólógie, Ghent, Belgium). Expression plasmid encoding FLAG-NOD1 is described elsewhere (18). FLAG-NLRC4 and FLAG-NLRP3 were kind gifts from Feng Shao (National Institute of Biological Sciences, Beijing, China) and Greta Guarda (Department of Biochemistry, University of Lausanne, Switzerland), respectively. GFP-NOD2 was generated by PCR using the GFP Fusion TOPO® TA expression kit (Invitrogen) from HA-NOD2. GFP-NOD2-V5 was generated by PCR using the pLenti6/V5 Directional TOPO® cloning kit (Invitrogen) from GFP-NOD2. JNKBP1-V5 was generated by PCR using the pcDNA3.1 topo/D kit (Invitrogen) from 40083182 vector (IRCMp5012C0132D, pCR Blunt II topo MAPKBP1, Source BioScience). JNKBP1 mutants were generated by PCR using Pfu Turbo DNA polymerase according to the manufacturer's protocol (Stratagene, Agilent Technologies). The used primers are described in supplemental Table 1). All expression vectors were sequenced, and the protein expression was confirmed by immunoblot.
NOD2 Complex Purification and Mass Spectrometry Identification of Purified Proteins
The cell lysates were made from HEK293 and GNV cells using modified RIPA buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Igepal, 0.25% sodium deoxycholate, 1 mm PMSF) in the presence of protease inhibitors set and phosphatase inhibitors (25 mm β-glycerol phosphate, 1 mm NaVO4, 3 mm Na3F, 5 mm sodium pyrophosphate). The cell lysates (100 mg of total proteins) were bound to 100 μl of an anti-V5-antibody agarose affinity gel (Sigma-Aldrich) for 90 min at 4 °C, followed by washing with 50 ml of modified RIPA lysis buffer. We eluted NOD2 complexes with V5 peptides (Sigma-Aldrich), and we added one volume of loading buffer (Invitrogen). After separation by SDS-PAGE, in-gel digestion was performed by the addition of modified trypsin (Promega, Madison, WI) in 50 mm ammonium bicarbonate at 37 °C overnight. The tryptic digests were air-dried and then dissolved in formic acid (0.1%) for further MS-MS analysis. Each in-gel digest of an individual band was analyzed by nano-high-performance liquid chromatography (HPLC) on a chip coupled with MS-MS using an XCT ion trap mass spectrometer (Agilent Technologies). The HPLC separations were performed on an RP C18 Zorbax column (Agilent Technologies). The mobile phase was a 90-min gradient mixture formed as follows: mixture A, water/acetonitrile/formic acid (97:3:0.1, v/v/v); mixture B, acetonitrile/water/formic acid (90:10:0.1, v/v/v). The flow rate was fixed at 300 nl/min. The collision energy was set automatically depending on the mass of the parent ion. Each MS full scan was followed by MS-MS scans of the first four most intense peaks detected in the prior MS scan. A list of peptide masses was subsequently introduced into the database for protein identification searches using MASCOT (Matrix Sciences).
Protein Extraction
Cells were washed with cold PBS, scraped, and centrifuged. The cells were lysed in modified RIPA buffer with protease and phosphatase inhibitors and incubated for 15 min on ice and then centrifuged for 20 min at 14,000 rpm at 4 °C. The amount of protein in each sample was determined using the Bio-Rad protein assay, and bovine serum albumin (BSA, Sigma-Aldrich) was used as a standard.
Immunoblot
One volume of 2× loading buffer (60 mm Tris-HCl, pH 6.8, 2.5% SDS, 10% glycerol, 5% β-mercaptoethanol, and 0.03% bromphenol blue) was added to protein samples. After boiling for 5 min, SDS-PAGE was performed, and proteins were electrotransferred to PVDF membranes (Roche Applied Science). After probing with primary and secondary antibodies, membranes were finally analyzed by chemiluminescence with an ECL system (Amersham Biosciences).
Immunoprecipitation (IP)
For IP with anti-V5 or -HA antibodies, cells were lysed on ice for 15 min in modified RIPA buffer with protease inhibitors and phosphatase inhibitors (25 mm β-glycerol phosphate, 1 mm NaVO4, 3 mm Na3F, 5 mm sodium pyrophosphate). Lysates were precleared with 10 μl of protein G-agarose slurry (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) for 1 h at 4 °C. Clarified lysates were incubated with anti-V5 or -HA antibodies along with 10 μl of protein G-agarose slurry. After 2 h of incubation, bead-linked immune complexes were washed four times with corresponding IP buffer, boiled in 2× loading buffer, and analyzed by immunoblot. For IP with an anti-FLAG antibody, cells were lysed on ice for 15 min in RIPA buffer (10 mm Tris-HCl pH 8, 150 mm NaCl, 1% Igepal, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm PMSF) with protease inhibitors and phosphatase inhibitors. Lysates were precleared with 30 μl of protein A-agarose beads (Thermo Fisher Scientific) for 2 h at 4 °C. Clarified lysates were incubated with an anti-FLAG antibody that was coupled with protein A-agarose. After 3 h of incubation, bead-linked immune complexes were washed four times with corresponding IP buffer, boiled in 2× loading buffer, and analyzed by immunoblot. For endogenous NOD2 IP, rabbit polyclonal anti-NOD2 antibody (2513, ProSci Inc.) was coupled overnight with 50 μl of protein A-agarose slurry (Santa Cruz Biotechnology, Inc.). The cells were lysed on ice for 15 min in Nonidet P-40 buffer (10 mm Tris-HCl pH 8, 150 mm NaCl, 1 mm EDTA, 1% Igepal, 1 mm PMSF) with protease inhibitors and phosphatase inhibitors. Lysates were precleared with 30 μl of protein A-agarose beads for 2 h at 4 °C. Clarified lysates were incubated with the anti-NOD2 antibody coupled with protein A-agarose. After overnight incubation, bead-linked immune complexes were washed five times with Nonidet P-40 buffer, boiled in loading buffer, and analyzed by immunoblots.
Immunohistochemistry
Sections from intestine biopsies and surgical tissue specimens from 20 patients with CΔ or from 20 healthy patients were selected and reviewed by a pathologist. The tissues were been routinely fixed in 10% neutral formalin and embedded in paraffin. 4-μm-thick sections were deparaffinized and rehydrated in graded alcohol. Endogenous peroxidases were blocked with 4.5% hydrogen peroxide in absolute methyl alcohol. For heat-induced epitope retrieval, the sections were subjected to 10 mm citrate buffer (pH 6.0) in a microwave oven at 95 °C for 23 min. The sections were blocked with serum-free protein block (X0909, Dako) for 10 min and were subsequently incubated overnight with primary antibody anti-MAPKBP1 (1:25) or incubated for 1 h with mouse anti-NOD2 antibody (2D9, Millipore; 1:500) or incubated for 30 min with anti-CD68 antibody (1:1800). The slides then were washed and incubated with LSAB2 System-HRP (Dako, Denmark) or the EnVision+ System-HRP (Dako, Denmark), respectively. The reaction products were then stained with diaminobenzidine and counterstained with hematoxylin.
siRNA Transfection
Two specific siRNAs against human JNKBP1 were synthesized by Eurogentec (siRNA 4) and by Qiagen (siRNA 6) (supplemental Table 3). Control siRNA was purchased from Applied Biosystems (Silencer® Select Negative Control 2 siRNA). siRNAs were used at a concentration of 20 nm for GNV cells and 60 nm for HCT116 cells, and transfection was performed using calcium phosphate (ProFection mammalian transfection system, Promega). The depletion of endogenous JNKBP1 expression by siRNA was confirmed by Western blot and by qRT-PCR.
Luciferase Assay
The cells were plated in 12-well plates and were transfected with reporter (κB)5-LUC and expression plasmids using the calcium phosphate method. After 24–48 h, cells were treated for 16 h with TNFα (100 units/ml) or MDP (1 μg/ml) or left untreated. The cells were lysed and assayed for luciferase activity according to the manufacturer's protocol (Luciferase Reporter Gene Assay, Roche Applied Science). The κB-dependent luciferase activity was normalized to protein concentration measured by the Bio-Rad protein assay (Bio-Rad). All samples were measured in triplicates.
Quantitative RT-PCR
Total RNA was extracted from the cells using the RNeasy minikit (Qiagen, Germany) according to the manufacturer's instructions. RNAs were treated with DNase I (Roche Applied Science) for 20 min at 37 °C in DNase I buffer (100 mm Tris, 100 mm MgCl2, pH 7.6). 1 μg of RNA was used for reverse transcription with Moloney murine leukemia virus reverse transcriptase (Invitrogen). For quantitative real-time RT-PCR, each cDNA was analyzed, in triplicate, with the SYBR Green Master Mix (Applied Biosystems) in the ABI sequence detection system. The results were normalized to β2-microglobulin or hypoxanthine-guanine phosphoribosyltransferase (HPRT1). Relative mRNA expression of samples was calculated using the standard curve method and the 2−ΔCt method (19). p values were calculated using the GraphPad Quickcalcs software (t test). The primers used to analyze the different transcripts were designed with the software Primer ExpressTM (Applied Biosystems) (supplemental Table 2).
IL-8 ELISA
HCT116 cells (105cells/ml) were transfected with Ctrl or JNKBP1 siRNA (60 nm). After 48 h, the cells were stimulated with MDP (20 μg/ml) or left untreated. Supernatants were collected 6 h after treatment, and the amount of human IL-8 was quantified by an enzyme-linked immunosorbent assay (human IL-8 ELISA ready-set-go, eBioscience). The results were expressed in pg/ml.
Gentamicin Protection Assay
Listeria monocytogenes strain 10403s was obtained from E. Muraille (Laboratory of Parasitology, University of Brussels, Belgium). Bacteria were cultured in brain heart infusion (BHI) broth (Fluka Analytical, Sigma-Aldrich). Single colonies of bacteria were used for inoculation of medium and expanded by incubation at 37 °C for 18 h. Overnight cultures were diluted 1:100 and grown for 2–3 h to reach an A600 equal to 0.1. Bacteria were washed and resuspended in cell culture medium. The GNV cells were seeded in 12-well tissue culture plate with 2 × 105 cells/well and transfected by the calcium phosphate method with JNKBP1-V5 or empty vector. After 48 h, the cells were infected with L. monocytogenes using a multiplicity of infection (MOI) of 80 bacteria/cell. After 1 h of infection, extracellular bacteria were killed by adding fresh medium containing gentamicin (100 μg/ml) for 90 min. The cells were washed and lysed with 0.1% Triton X-100 PBS. Lysates were diluted and plated into BHI agar plates to determine the number of colony-forming units (cfu). All experiments were performed in triplicate. The relative infection was expressed as the percentage of cfu from cells transfected with empty vector compared with cells transfected with JNBKP1-expressing vector.
RESULTS
Identification of JNKBP1 as a NOD2-interacting Protein
To identify new NOD2 binding partners that could modulate NOD2 signaling, we performed a biochemical purification of NOD2 co-immunoprecipitating proteins in a HEK293 cell line that stably expresses GFP-NOD2-V5 (GNV). NOD2-containing protein complexes were immunoprecipitated with an anti-V5 antibody from lysates of GNV cells, and NOD2 co-purifying proteins were identified by liquid chromatography tandem mass spectroscopy. Lysates of the parental HEK293 cell line were processed in the same manner as a negative control (Fig. 1A).
FIGURE 1.

JNKBP1 interacts with NOD2. A, visualization of NOD2 binding partners on a one-dimensional gel by silver staining. NOD2-containing protein complexes were immunoprecipitated with an anti-V5 antibody from lysates of GNV and HEK293 cells. The NOD2 co-purifying proteins were eluted by V5 peptides. After protein separation by SDS-PAGE, we performed silver staining on the gel. B, co-immunoprecipitation of ectopically expressed HA-NOD2 and JNKBP1-V5 in HEK293T cells. HEK293T cells were transfected with HA-NOD2 (+) or empty vector (−) along with JNKBP1-V5 (+). Samples were immunoprecipitated with anti-HA or anti-V5 antibodies, and Western blot analysis was performed with either anti-V5 or anti-HA antibodies. The presence of each tagged protein was checked in whole lysates by anti-HA or -V5 immunoblots (bottom). C, co-immunoprecipitation of JNKBP1-V5 with other NLRs in HEK293T cells. HEK293T cells were transfected with the indicated expression constructs (+) or empty vector (−) along with JNKBP1-V5 (+). Samples were immunoprecipitated with anti-V5 antibody, and Western blot analysis was performed with either anti-V5, anti-FLAG, or anti-GFP antibodies. The presence of each tagged protein was checked in whole lysates through anti-FLAG or -GFP immunoblots (bottom). D–E, co-immunoprecipitation of both endogenous NOD2 and JNKBP1 in THP1 and HCT116 cells. THP1 (D) and HCT116 cells (E) were treated with MDP (100 and 50 μg/ml, respectively) for the indicated periods. Lysates were immunoprecipitated with an anti-NOD2 antibody or with an IgG control (Irr). Immunoblotting was performed with an anti-JNKBP1 antibody. IκBα phosphorylation in response to MDP was monitored by an anti-phospho-Ser32 and -Ser36 IκBα immunoblot in whole cell lysates (bottom).
As expected, mass spectrometry analysis identified NOD2 in each immunoprecipitate from GNV cells. Moreover, we identified several previously unreported NOD2 binding partners, one of these being c-Jun N-terminal kinase-binding protein 1 (JNKBP1), on which we focus here.
The interaction between NOD2 and JNKBP1 was first confirmed by immunoprecipitation in HEK293T cells transiently expressing HA-NOD2 and JNKBP1-V5. NOD2 and JNKBP1 were immunoprecipitated with either an anti-HA or anti-V5 antibody and analyzed by Western blot. Fig. 1B demonstrates that JNKBP1-V5 co-immunoprecipitates with HA-NOD2 and, reciprocally, HA-NOD2 is pulled down with JNKBP1-V5. The interaction between NOD2 and JNKBP1 was shown to be specific because no binding of JNKBP1 was observed with other NLR proteins, including NOD1, NLRC4, and NLRP3 (Fig. 1C). Next, we assessed the interaction between both endogenous proteins in the human monocyte line THP1 and in the intestinal epithelial cells HCT116. The lysates of untreated or MDP-stimulated cells were immunoprecipitated with either an anti-NOD2 antibody or an IgG control and analyzed with an antibody against human JNKBP1. In THP1 cells, a significant co-immunoprecipitation between both proteins appeared after 15 min of MDP treatment and decreased at later times (Fig. 1D). MDP stimulation also induced the co-immunoprecipitation between endogenous NOD2 and JNKBP1 in HCT116 cells but in a less quick and less transient way than in THP1 cells (Fig. 1E). The efficiency of MDP treatment was demonstrated by immunoblot against IκBα phosphorylated on Ser32 and Ser36 in whole cell lysates (Fig. 1, D and E). In both cell types, the kinetics of MDP-stimulated IκBα phosphorylation correlates with the time course of MDP-induced interaction between NOD2 and JNKBP1. Conclusively, these results show that JNKBP1 physically interacts with NOD2 and that this interaction is increased by MDP treatment.
Expression of JNKBP1 in Human Cell Lines and in the Intestine
Little is known about the tissue distribution of JNKBP1 in humans, whereas it has been shown to be ubiquitously expressed in mouse tissues and principally in the brain (16). To assess the expression of human JNKBP1 in various kinds of cells, we determined the levels of JNKBP1 mRNA in different human cell lines by qRT-PCR. The JNKBP1 expression levels for each cell line were compared with those obtained for GNV cells (Fig. 2A). JNKBP1 seems to be ubiquitously expressed with higher levels in T lymphocytes (Jurkat), human glioblastoma cells (LN18), and intestinal epithelial cells (HCT116). Consequently, HCT116 cells, which express both endogenous NOD2 (20) and JNKBP1, were chosen as a relevant model to study the role of JNKBP1 in NOD2 signaling.
FIGURE 2.

Expression of JNKBP1 in various cell lines and in intestine mucosa. A, JNKBP1 expression in different cell lines. mRNA from the indicated cell lines was extracted, and JNKBP1 mRNA levels were measured by quantitative RT-PCR. The HPRT1 transcript was used as an internal standard. *, JNKBP1 and NOD2 expression and localization in human intestine mucosa. I, immunohistochemical staining of JNKBP1 in healthy intestine (magnification ×100). EC, epithelial cells; V, endothelial cells; SM, smooth muscle; I, immune cells. II, healthy intestine (magnification ×100), negative control without anti-JNKBP1 antibody. III, immunohistochemical staining of NOD2 in healthy intestinal tissue (magnification ×100). IV, negative control without anti-NOD2 antibody. V, staining of JNKBP1 in intestinal epithelial cells and crypts from colonic epithelium of a CD patient (magnification ×200). VI, NOD2 staining in crypts from colonic epithelium of a CD patient (magnification ×400). VII–IX, JNKBP1 and NOD2 are co-expressed in monocytes-macrophages (M). Shown is immunohistochemical labeling of JNKBP1 (magnification ×200) (VII), NOD2 (VIII), and CD68 (IX) in colonic mucosa of a CD patient. X, immunohistochemical staining of JNKBP1 in lymphocytes of CD patient colonic mucosa (magnification ×400). All sections were counterstained with hematoxylin. Representative pictures are shown from 20 healthy and CD patients, values represent the means ± S.D. (error bars) of duplicate cultures.
Because NOD2 is known to be mainly expressed in epithelial and dendritic cells as well as in monocytes-macrophages from intestine mucosa (21, 22), where it plays an important role in immune homeostasis, we analyzed the expression of JNKBP1 by immunohistochemical analyses in intestine mucosa from healthy and CD patients. A positive staining, using an anti-JNKBP1 antibody, was observed, at various levels, in epithelial (EC) and endothelial cells (V) but also in the smooth muscle (SM) and immune cells (I), such as lymphocytes from healthy intestinal mucosa (Fig. 2B, panel I). No staining was observed in healthy mucosa incubated without primary antibody (Fig. 2B, panel II). As previously shown (21, 22), immunohistochemical experiments with an antibody raised against NOD2 demonstrated a much lower ubiquitous NOD2 expression with a positive staining mainly in Paneth cells and, at lower extent, in the glandular epithelium from healthy intestine (Fig. 2B, panel III, black arrow). No staining was detected without primary antibody (Fig. 2B, panel IV). We did not observe any difference in the JNKBP1 expression and localization between healthy and Crohn patients. In epithelial cells, JNKBP1 was observed in cytosol and, in some cells, also in the nucleus (Fig. 2B, panel V), whereas NOD2 was restricted to the cytosol (Fig. 2B, panel VI). Interestingly, in the surface epithelium, JNKBP1 staining seemed to be more pronounced with a reinforcement in the apical cytoplasm (Fig. 2B, panel V). Immunoreactivity for JNKBP1 was also detected in inflammatory cells infiltrating the intestine mucosa of CD patients (Fig. 2B, panels I, VII, and X). Notably, staining for both NOD2 and JNKBP1 was also observed within monocyte-macrophage, as shown in panels VII–IX. Most lymphocytes had a positive immunohistochemical staining for JNKBP1 (Fig. 2B, panel X, black arrows).
These immunohistochemistry experiments on intestinal tissue confirm the results obtained in qRT-PCR assays on human cell lines (Fig. 2A). We also showed that JNKBP1 mRNA is well expressed in intestine biopsies by qRT-PCR assays (data not shown). However, JNKBP1 expression within colon and ileum mucosa does not seem to be modulated in the CD patients compared with healthy individuals (data not shown).
Mapping of the Interaction Domains
To map the NOD2 domain(s) involved in the interaction with JNKBP1, we tested various NOD2 deletion mutants (Fig. 3A). HEK293T cells were transfected with these constructs encoding for the HA- or FLAG-tagged NOD2 mutants and with the JNKBP1-V5 expression plasmid. The lysates of transfected HEK293T cells were immunoprecipitated with an anti-V5 antibody and analyzed with an anti-HA or -FLAG antibody. The deletion of both CARDs or the LRRs did not prevent the interaction of NOD2 with JNKBP1 (Fig. 3B). When the four domains (CARD1, CARD2, NBD, and LRRs) were separately analyzed, the N-terminal CARD1 (Fig. 3C) and, to a lesser extent, the C-terminal LRRs (Fig. 3E) were able to co-immunoprecipitate with JNKBP1, whereas the N-terminal CARD2 (Fig. 3C) and the central region NBD could not (Fig. 3D). These results suggest that CARD1 and LRRs could cooperate for efficient interaction with JNKBP1. The three main CD-associated NOD2 mutants, R702W, G908R, and L1007fs, interacted with JNKBP1 as efficiently as WT NOD2 (Fig. 3F).
FIGURE 3.

Mapping of the domains of interaction. A, schematic representation of full-length and truncated NOD2 proteins used in co-immunoprecipitation studies. B, the deletion of both CARDs or LRRs still allows NOD2 to interact with JNKBP1. HEK293T cells were transfected with JNKBP1-V5 and HA-NOD2 WT or the truncated form (ΔCARD and ΔLRR). Lysates were immunoprecipitated with an anti-V5 antibody, and co-immunoprecipitated NOD2 proteins were revealed with an anti-HA antibody. The presence of each tagged protein was checked in whole lysates through anti-HA immunoblot (bottom). C, the CARD1 domain is sufficient to maintain the interaction of NOD2 with JNKBP1. HEK293T cells were transfected with JNKBP1-V5 and HA-CARD1 or HA-CARD2. Lysates were immunoprecipitated with an anti-V5 antibody, and Western blot analysis was performed with an anti-HA antibody. The presence of each tagged protein was checked in whole lysates through anti-HA or -V5 immunoblots (bottom). D, the NBD domain is not involved in the interaction between NOD2 and JNKBP1. HEK293T cells were transfected with JNKBP1-V5 and FLAG-NBD. Lysates were immunoprecipitated with anti-V5 antibody, and Western blot analysis was performed with an anti-FLAG antibody. The presence of each tagged protein was checked in whole lysates through anti-FLAG or -V5 immunoblots (bottom). E, the LRR domain is still able to bind JNKBP1. HEK293T cells were transfected with JNKBP1-V5 and HA-LRR. Lysates were immunoprecipitated with an anti-V5 antibody, and Western blot analysis was performed with an anti-HA antibody. The presence of each tagged protein was checked in whole lysates through anti-HA or -V5 immunoblots (bottom). F, the three main CD-associated NOD2 mutants (R702W, G908R, and L1007fs) still interact with JNKBP1. JNKBP1-V5 was cotransfected into HEK293T cells with HA-NOD2 WT, HA-R702, HA-G908, or HA-L1007fs. After 48 h, cells were harvested for protein extraction. Samples were immunoprecipitated with an anti-V5 antibody, and the NOD2 co-immunoprecipitation was analyzed by an anti-HA immunoblot. Total lysates were immunoblotted with anti-HA and -V5 antibodies. G, schematic representation of full length and truncated JNKBP1 proteins used in co-immunoprecipitation studies. H, the N-terminal WD-40 domain of JNKBP1 is essential for NOD2 binding. WT JNKBP1-V5, ΔC JNKBP1-V5, or ΔN JNKBP1-V5 was cotransfected in HEK293T cells with HA-NOD2. Lysates were immunoprecipitated with an anti-V5 antibody, and immunoblotting was performed using the indicated antibodies. The presence of each tagged protein was checked in whole lysates through anti-HA or -V5 immunoblots (bottom).
Two domains can be identified in JNKBP1: the N-terminal part containing 12 WD-40 repeats referred as the WD-40 domain hereafter and the C-terminal part with several predicted phosphorylation sites and putative protein-binding sequences (Fig. 3G). Plasmids allowing the expression of V5-tagged JNKBP1 lacking the N-terminal or the C-terminal part were generated (ΔN JNKBP1 and ΔC JNKBP1, respectively; Fig. 3G). Although both mutants have the same size (757 amino acids), ΔN JNKBP1 migrates much more slowly than ΔC JNKBP1, suggesting that the C-terminal part of JNKBP1 undergoes post-translational modifications (Fig. 3H). The full-length protein, whose expected molecular mass is 168 kDa, also behaves like a heavier protein (∼180 kDa) (data not shown). Each JNKBP1 form (WT, ΔC, and ΔN) was transfected in HEK293T cells with HA-NOD2. JNKBP1 lacking the C-terminal part was able to co-immunoprecipitate NOD2 as efficiently as the WT form, whereas the ΔN JNKBP1 mutant could not (Fig. 3H). These results revealed that the N-terminal WD-40 domain of JNKBP1 is necessary for the interaction with NOD2.
JNKBP1 Attenuates NOD2-mediated NF-κB Activation
To investigate a putative role of JNKBP1 in regulating NOD2-mediated signaling, NF-κB luciferase reporter assays were conducted. First, the effect of JNKBP1 overexpression was studied on NF-κB activity in HEK293T cells (Fig. 4A). Although the overexpression of JNKBP1 had very little effect on basal NF-κB activity, it significantly down-regulated NOD2-mediated NF-κB activation in a dose-dependent manner (Fig. 4A). MDP stimulation of NOD2-expressing cells induced a robust NF-κB activation, which was also strongly inhibited by JNKBP1. The TNFα-induced NF-κB activation was only very slightly affected by the presence of JNKBP1, thereby showing that JNKBP1 is not a general NF-κB inhibitor but seems to be specific to the NOD2 pathway. (Fig. 4B). Next, the effect of the endogenous JNKBP1 knockdown was assessed on NOD2-dependent NF-κB activation using siRNA. The efficiency of the selected siRNA 4 to induce a strong decrease of JNKBP1 expression is demonstrated in the Fig. 4C. siRNA 4-mediated JNKBP1 knockdown had a positive impact on MDP-stimulated NF-κB activity estimated by NF-κB luciferase reporter assays in GNV cells (Fig. 4D), corroborating the previous results obtained by overexpression experiments.
FIGURE 4.

JNKBP1 attenuates NOD2-mediated NF-κB activation. A, JNKBP1 in a dose-dependent manner inhibits NOD2-mediated NF-κB activation. HEK293T cells were transfected with the reporter plasmid (κB)5-LUC and increasing amounts of JNKBP1-V5 along with (black bars) or without (white bars) NOD2. 24 h post-transfection, cells were harvested for LUC assays. Values represent the means ± S.D. (error bars) of triplicate cultures. Immunoblot with an anti-V5 antibody shows the JNKBP1-V5 expression level. B, effect of JNKBP1 on TNFα- and MDP- induced NF-κB activation. HEK293T cells were transfected with the reporter plasmid (κB)5-LUC and increasing amounts of JNKBP1-V5 in the presence (white and black bars) or absence (gray bars) of NOD2. 24 h post-transfection, cells were left untreated (white bars) or were stimulated for 16 h with MDP (1 μg/ml) (black bars) or with TNFα (100 units/ml) (gray bars) before being harvested for LUC assays. Values represent the means ± S.D. of triplicate cultures. Immunoblot with an anti-V5 antibody shows the JNKBP1-V5 expression level. C, efficiency of specific siRNA 4 to target JNKBP1 expression. HEK293T cells were transfected with JNKBP1-V5 along with either the siRNA 4 JNKBP1 or a siRNA control. After 48 h, cell lysates were analyzed by immunoblotting with an anti-V5 antibody. Equal loading was monitored by concomitant detection of β-tubulin. D, knockdown of JNKBP1 increases MDP-induced NF-κB activation. GNV cells were transfected with the siRNA 4 JNKBP1 or the siRNA control. After 24 h, the cells were transfected with the reporter plasmid (κB)5-LUC, and following another period of 24 h, cells were treated with MDP (1 μg/ml) for 16 h before being harvested for LUC assays (mean ± S.D. (n = 3); *, p < 0.04). E, knockdown of JNKBP1 up-regulates MDP-induced IKKα/β, IκBα, and p65 phosphorylation. HCT116 cells were transfected with JNKBP1 siRNA 4 or 6 or the siRNA control. After 48 h, cells were treated with MDP (60 μg/ml) for the indicated periods. The lysates were analyzed by immunoblotting with the following antibodies: anti-NOD2, anti-JNKBP1, anti-phospho-Ser176/177 IKKα/β, anti-phospho-Ser32/36 IκBα, and anti-phospho-Ser536 p65. Equal loading was monitored by concomitant detection of β-tubulin. RLU, relative light units.
To further confirm the down-regulation exerted by JNKBP1 on NOD2-dependent NF-κB activation, we tested the effect of JNKBP1 knockdown through two different siRNAs (siRNAs 4 and 6) on earlier steps of the NF-κB pathway, such as IKKα/β, IκBα, and p65 NF-κB phosphorylation in response to MDP. HCT116 cells transfected with siRNA Ctrl or siRNA JNKBP1 (siRNA 4 or 6) for 48 h were stimulated with MDP for the indicated time. Lysates were collected and immunoblotted for phosphorylated IκBα on Ser32 and Ser36, IKKα/β on Ser176 and Ser177, and p65 on Ser536 (Fig. 4E). Efficient knockdown of JNKBP1 with two different siRNA duplexes (siRNA 4 or 6), as demonstrated by the anti-JNKBP1 immunoblot, significantly enhanced IKKα/β, IκBα, and p65 phosphorylation at various times of MDP treatment (Fig. 4E). The MDP-stimulated IκBα and p65 phosphorylation even seemed to be sped up in JNKBP1-silenced HCT116 cells. Altogether, these results show that the binding of JNKBP1 to NOD2 dampens the NOD2-dependent downstream events, such as IKKα/β, IκBα, and p65 NF-κB phosphorylation, leading to decreased NF-κB activation.
JNKBP1 Negatively Regulates NOD2-mediated IL-8 Secretion
Because NOD2-induced NF-κB activation has previously been shown to induce the expression and secretion of chemotactic cytokine IL-8 (23), we tested the influence of JNKBP1 on NOD2-mediated IL-8 expression. As expected, JNKBP1 significantly inhibited MDP-stimulated IL-8 expression in GNV cells in a dose-dependent manner (Fig. 5A). The ΔC JNKBP1 mutant, which still interacts with NOD2, had the same down-regulating effect on MDP-induced IL-8 expression as the WT form. By contrast, the ΔN JNKBP1 mutant, unable to bind to NOD2, did not affect IL-8 mRNA levels after MDP treatment (Fig. 5B). Conversely, knockdown of JNKBP1 enhanced IL-8 expression in response to MDP in GNV cells (Fig. 5C). We also tested the effect of siRNA-mediated JNKBP1 depletion on MDP-stimulated IL-8 expression in intestinal epithelial HCT116 cells that endogenously express both NOD2 (20) and JNKBP1 (Figs. 1E, 2A, and 4E). The efficiency of both siRNAs (siRNA 4 or 6) to knock down JNKBP1 in HCT116 cells was demonstrated by qRT-PCR (Fig. 5D). Interestingly, knockdown of JNKBP1 through both target siRNAs in HCT116 cells had a strong positive impact on MDP-induced IL-8 mRNA levels (Fig. 5E). As expected, IL-8 release measured by ELISA in culture supernatants of MDP-stimulated HCT116 cells was also up-regulated when JNKBP1 was depleted (Fig. 5F).
FIGURE 5.

JNKBP1 down-regulates NOD2-induced IL-8 secretion. A, JNKBP1 dose-dependently decreases MDP-induced IL-8 mRNA levels. GNV cells were transfected with increasing amounts of JNKBP1. After 48 h, cells were left untreated or were stimulated with MDP (10 μg/ml) for 4 h before being harvested for RNA extraction. IL-8 mRNA levels were measured by qRT-PCR using the HPRT1 transcript as an internal standard (mean ± S.D. (error bars) (n = 3); **, p = 0.003; ***, p < 0.0001). Immunoblot with an anti-V5 antibody shows the JNKBP1-V5 expression level. B, the N-terminal WD-40 domain of JNKBP1 is essential to exert its down-regulator effect. GNV cells were transfected with empty vector (pcDNA3.1) or plasmids encoding either the WT JNKBP1 (JNKBP1) or the deletion mutant lacking the C-terminal (ΔC JNKBP1) or the N-terminal part (ΔN JNKBP1). After 48 h, cells were left untreated or were stimulated with MDP (10 μg/ml) for 4 h before being harvested for RNA extraction. IL-8 mRNA levels were measured by qRT-PCR using the β2-microglobulin transcript as an internal standard (mean ± S.D. (n = 3); ***, p < 0.0001). Immunoblot with an anti-V5 antibody shows the expression level of different JNKBP1 mutants. C, knockdown of JNKBP1 has a positive impact on MDP-induced IL-8 mRNA levels in GNV cells. GNV cells were transfected with the siRNA 4 JNKBP1 or the siRNA control. After 48 h, cells were left untreated or were stimulated with MDP (50 μg/ml) for 4 h before being harvested for RNA extraction. IL-8 mRNA expression was measured by qRT-PCR using HPRT1 mRNA as an internal standard (mean ± S.D. (n = 3); **, p < 0.0015). D, efficiency of JNKBP1 knockdown in intestinal epithelial HCT116 cells. HCT116 cells were transfected with the siRNA JNKBP1 4 or 6 or the siRNA control. 48 h post-transfection, RNA was extracted, and JNKBP1 mRNA was monitored by qRT-PCR using HPRT1 mRNA as an internal standard. E, knockdown of JNKBP1 increases MDP-induced IL-8 mRNA levels in intestinal epithelial HCT116 cells. HCT116 cells were transfected with the siRNA JNKBP1 4 or 6 or the siRNA control. After 48 h, the cells were left untreated (NT) or stimulated with MDP (30 μg/ml) for 2 h before being harvested for RNA extraction. IL-8 mRNA expression was measured by qRT-PCR using HPRT1 mRNA as an internal standard. Values represent the means + S.D. of triplicate cultures (mean ± S.D. (n = 3); ***, p < 0.0001). F, knockdown of JNKBP1 primes MDP-induced IL-8 secretion by intestinal epithelial HCT116 cells. HCT116 cells were transfected with the siRNA JNKBP1 4 or the siRNA control. After 48 h, the cells were left untreated (NT) or stimulated with MDP (50 μg/ml) for 6 h. IL-8 levels in supernatants were measured by ELISA (mean ± S.D. (n = 3); **, p < 0.0002).
JNKBP1 Reduces NOD2-induced Bacterial Killing
NOD2 was shown to protect the cells from infection with invasive bacteria, such as L. monocytogenes (12, 24). A gentamicin protection assay was performed to determine the effect of JNKBP1 on NOD2-induced Listeria killing. GNV cells transfected with JNKBP1 for 48 h were infected with L. monocytogenes (MOI 80). 1 h postinfection, GNV cells were washed and incubated with gentamicin for 90 min before being harvested to monitor the number of viable intracellular Listeria. JNKBP1 overexpression (Fig. 6B) significantly increased the number of intracellular bacteria (Fig. 6A). The results indicate that JNKBP1 is also able to negatively regulate the antibacterial activity of NOD2.
FIGURE 6.
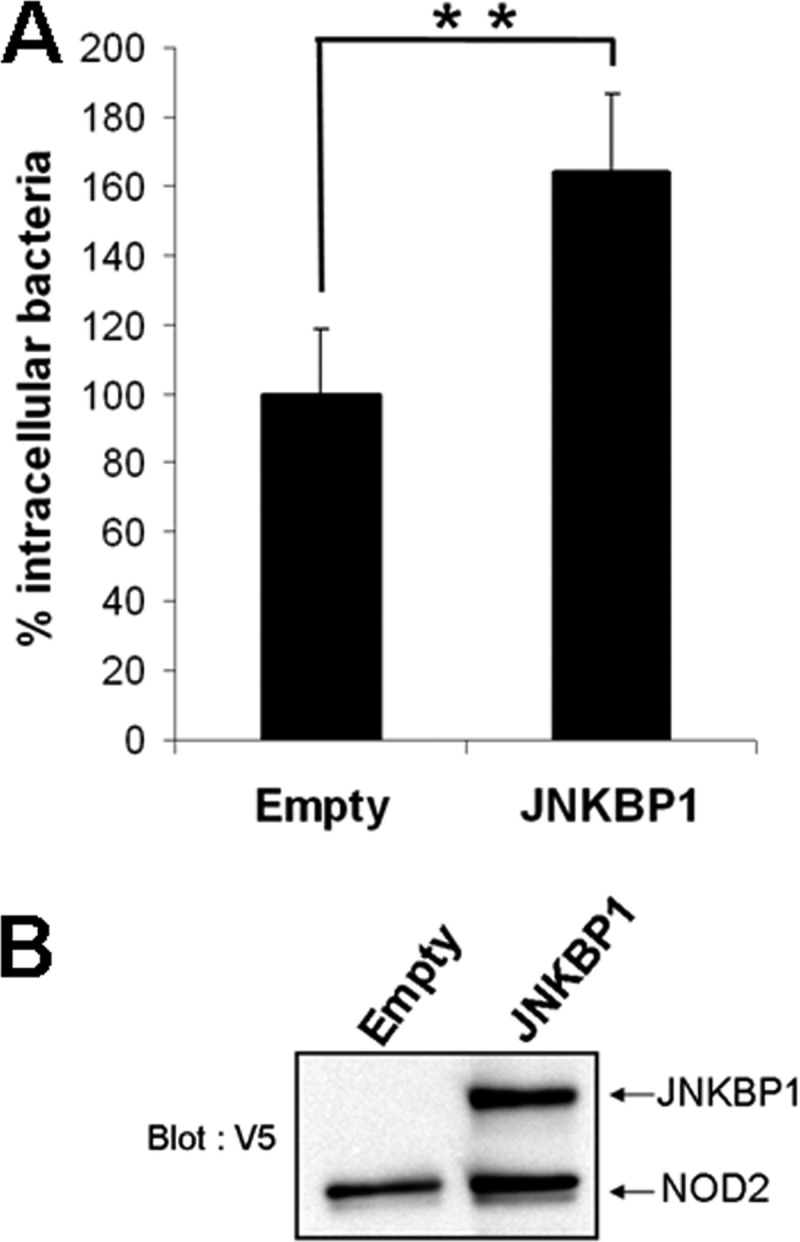
JNKBP1 disturbs NOD2-induced Listeria killing. A, gentamicin protection assay. GNV cells were transfected with empty vector or plasmid, allowing the expression of JNKBP1-V5. After 48 h, the cells were infected with L. monocytogenes (MOI 80). 1 h after infection, fresh medium with gentamicin was added for 90 min to kill extracellular bacteria. Then the cells were lysed, and samples were plated onto BHI agar plates to determine the number of cfu. Results are expressed as intracellular bacteria number relative to those obtained with cells transfected with empty vector taken as 100%. Data are representative of three independent experiments (mean ± S.D. (error bars) (n = 3); **, p < 0.001). B, JNKBP1 and NOD2 expression in GNV cells is shown by immunoblotting using an anti-V5 antibody.
JNKBP1 Interferes with NOD2 Oligomerization and RIP2 Tyrosine Autophosphorylation
The first steps of the signaling pathway triggered by NOD2 activation following its overexpression or MDP treatment consist of NOD2 oligomerization through the central NBD and RIP2 recruitment via homotypic CARD-CARD interactions (5). RIP2 is then ubiquitinated and autophosphorylates. Although originally identified as a serine-threonine kinase, RIP2 was recently shown to also have tyrosine kinase activity, RIP2 undergoes autophosphorylation on Tyr474, which is required for effective NOD2 signaling (8). In order to dissect the molecular mechanism by which JNKBP1 negatively modulates NOD2 signaling, we investigated the effect of JNKBP1 on NOD2 oligomerization and RIP2 tyrosine phosphorylation.
To determine whether JNKBP1 affects the assembly of the NODosome, HEK293T cells were transfected with both GFP-NOD2 and HA-NOD2 together or not with JNKBP1-V5. 48 h post-transfection, cells were treated with MDP for 30 min before being harvested for immunoprecipitation assays. Lysates were immunoprecipitated with an anti-HA antibody and analyzed by Western blotting with anti-GFP and -V5 antibodies. A significant amount of GFP-NOD2 was co-immunoprecipitated with HA-NOD2, reflecting the NOD2 oligomerization (Fig. 7A). Interestingly, when JNKBP1 was expressed, it interacted with NOD2, as revealed by its co-immunoprecipitation with HA-NOD2, and prevented the self-association of NOD2, as demonstrated by the lack of GFP-NOD2 recovery in anti-HA immunoprecipitate (Fig. 7A).
FIGURE 7.

JNKBP1 interferes with NOD2 oligomerization and NOD2-mediated RIP2 tyrosine phosphorylation. A, JNKBP1 interferes with NOD2 oligomerization. GFP-NOD2 and HA-NOD2 were transfected in HEK293T cells with an empty or JNKBP1-V5 expression vector. After 48 h, cells were treated with MDP (10 μg/ml) for 30 min before being harvested for protein extraction. Samples were immunoprecipitated with an anti-HA antibody. The self-association of NOD2 and the presence of JNKBP1 in the NOD2-containing complex were analyzed by anti-GFP and anti-V5 immunoblot, respectively. Total lysates were immunoblotted with anti-GFP, -HA, and -V5 antibodies. B, JNKBP1 interferes with NOD2-mediated RIP2 tyrosine phosphorylation. FLAG-RIP2 and GFP-NOD2 were transfected in HEK293T cells with an empty or JNKBP1 expression vector. After 48 h, cells were harvested for protein extraction. Proteins were immunoprecipitated with an anti-FLAG antibody. The RIP2 tyrosine phosphorylation and the presence of NOD2 in the RIP2-containing complex were analyzed by anti-phosphotyrosine and anti-GFP immunoblots, respectively. Total lysates were immunoblotted with anti-GFP, -FLAG, and -V5 antibodies.
Overexpression of NOD2 itself causes RIP2 tyrosine phosphorylation and NF-κB activation (8). To study the effect of JNKBP1 on RIP2 tyrosine phosphorylation, we transfected HEK293T cells with FLAG-RIP2 and GFP-NOD2 together or not with JNKBP1-V5. Both lysates were immunoprecipitated with an anti-FLAG antibody and analyzed by an anti-FLAG, anti-GFP, and anti-phosphotyrosine immunoblot. Although the same amount of RIP2 was immunoprecipitated in both samples, RIP2 was tyrosine-phosphorylated only in the absence of JNKBP1 (Fig. 7B). The inhibiting effect of JNKBP1 on NOD2-mediated RIP2 tyrosine autophosphorylation could also be observed by immunoblotting anti-FLAG on whole cell extracts, allowing us to distinguish unphosphorylated RIP2 (lower band) from phosphorylated RIP2 (upper band) (Fig. 7B). Surprisingly, the presence of JNKBP1 did not interfere with the interaction between NOD2 and RIP2, as demonstrated by the same amount of NOD2 co-immunoprecipitated with RIP2 in both lysates (Fig. 7B). Altogether, these results suggest that JNKBP1 disturbs NOD2 oligomerization but not RIP2 recruitment. The lack of NOD2 oligomerization would prevent the NODosome assembly, which would be required for the RIP2 tyrosine autophosphorylation.
DISCUSSION
In this work, we identified a new NOD2-interacting protein, JNKBP1, by a proteomic approach. This protein is characterized by an N-terminal WD-40 domain and a C-terminal part carrying putative phosphorylation sites and protein-binding sequences. We confirmed the interaction between both ectopically expressed proteins in HEK293T cells and between both endogenous proteins in human THP1 monocytes and HCT116 intestinal epithelial cells. This interaction was enforced after MDP treatment. The N-terminal WD-40 domain of JNKBP1 is sufficient for its recruitment to NOD2, whereas both the N-terminal CARD1 and, to a lesser extent, the C-terminal LRRs mediate the NOD2 binding to JNKBP1. The interaction of JNKBP1 with NOD2 seems specific because JNKBP1 does not interact with other proteins of the NLR family (Fig. 1C). The NOD2-mediated NF-κB activation is significantly inhibited by JNKBP1, whereas the TNFα-induced NF-κB activation is only slightly affected, thereby showing that JNKBP1 is not a general NF-κB inhibitor but seems to be specific to the NOD2 pathway. As expected, JNKBP1 also down-regulates NOD2-dependent IL-8 expression. Again, the N-terminal WD-40 domain of JNKBP1 (ΔC JNKBP1) is sufficient to exert its down-regulating effect on NOD2-mediated IL-8 expression. Interestingly, JNKBP1 has also a negative impact on the NOD2 anti-bacterial activity. When we investigated the molecular mechanism underlying the JNKBP1 repressor effect, we observed that JNKBP1 is able to inhibit two early steps of the NOD2 signaling, such as NOD2 oligomerization and RIP2 tyrosine phosphorylation, whereas it does not seem to affect the RIP2 recruitment to NOD2.
It has been proposed that, in resting cells, NOD2 (like other NLR proteins) stays in an autoinhibited form through intramolecular inhibition of the NBD domain by LRRs (25, 26). Upon MDP recognition through LRRs, NOD2 undergoes conformational modifications, which, via the release of NBD and both CARDs, allow NOD2 oligomerization and RIP2 recruitment. Both events are required to induce the cross-activation and tyrosine autophosphorylation of RIP2 (27, 28). We propose that conformational modifications of NOD2 induced by MDP stimulation might allow recruitment of JNKBP1 through CARD1 and the LRRs, which could be a hindrance to NOD2 oligomerization and would prevent RIP2 tyrosine autophosphorylation. Consequently, the downstream steps, such as TAK1 and IKK complex activation, would be down-modulated. This hypothesis is reinforced by the observation that the JNKBP1 recruitment to NOD2 in MDP-treated HCT116 cells after 30 and 60 min (Fig. 1E) is concomitant with the down-modulation of IκBα and p65 phosphorylation (Fig. 4E).
The interaction between NOD2 and JNKBP1 seems to be transient. Indeed, in THP1 cells, after 15 min of MDP treatment, the interaction between NOD2 and JNKBP1 increases but after 30 min, it decreases (Fig. 1D). In HCT116 cells, the interaction between NOD2 and JNKBP1 becomes more important after 60 min (Fig. 1E). The reason for such a transient interaction between NOD2 and JNKBP1 is currently unknown. We can guess that JNKBP1 is either removed by other NOD2 partners or post-translationally modified. Actually, the C-terminal part of JNKBP1 contains several putative phosphorylation sites. Moreover, we observed an important electrophoretic mobility shift of this C-terminal fragment (Fig. 3H) and, to a lesser extent, of the full-length protein, suggesting that JNKBP1 undergoes post-translational modifications in its C-terminal part. It would be interesting to further investigate the nature and the role of these post-translational modifications.
We showed that the N-terminal WD-40 domain of JNKBP1 is sufficient to mediate its interaction with NOD2 and its repressor effect on NOD2 signaling. This is not the first report demonstrating an interaction of NOD2 with a WD-40 repeat-containing protein. Indeed, AAMP (angio-associated migratory cell protein), containing six WD-40 repeats spanning the full-length sequence, was identified as a binding partner of NOD2, able to down-regulate NOD2-mediated NF-κB activation (29). WD-40 repeats typically fold into propellers, which have several distinct surfaces available for interactions (30). Consequently, WD-40 proteins often act as a scaffold. It is tempting to speculate that JNKBP1 interacts not only with NOD2 but also with other partners, inducing the assembly of a large complex, which, in turn, would hinder the NODosome formation. Obviously, structural studies of JNKBP1 would be required to confirm the propeller structure of its WD-40 domain.
JNKBP1 was initially identified in mice as a protein interacting with JNK and playing a scaffold role in the JNK-dependent pathway (16). This function is independent on N-terminal WD-40 domain but involves a region in the C-terminal part (Fig. 3G). Human and mouse JNKBP1 share 85% homology with the most conserved sequences in the WD-40 domain. NOD2 activation also leads to the activation of MAPKs, including p38, ERK, and JNK (31). The NOD2-mediated MAPK activation pathway also involves the early steps, such as NOD2 oligomerization, RIP2 recruitment, and TAK1 activation, but the downstream effectors are less well characterized. We tested the effect of human JNKBP1 on NOD2-mediated MAPK activation by immunoblot experiments, which allowed the detection of the phosphorylated forms of ERK, p38, and JNK in GNV or HCT116 cells after MDP treatment (data not shown). We did not observe any significant effect of JNKBP1 on NOD2-mediated MAPK activation (data not shown). It is possible that the down-regulator effect of JNKBP1 on the triggering of the NOD2 pathway counteracts its up-regulator effect on JNK signaling. The same team also showed an interaction of mouse JNKBP1 with TAK1 and TRAF2, but they did not map interaction domains (17). When they overexpressed mouse JNKBP1, they observed an increase of NF-κB activation induced by overexpression of TAK1 or TRAF2. They did not confirm these results with the knockdown of JNKBP1. Moreover, they did not test the effect of JNKBP1 on NF-κB activation in response to proinflammatory stimuli, such as TNFα. We also observed an interaction between JNKBP1 and TAK1 involving the N-terminal WD-40 domain (data not shown). Consequently, we cannot rule out the hypothesis that the JNKBP1-TAK1 interaction, in addition to that between JNKBP1 and NOD2, could affect NOD2-mediated NF-κB activation. Anyway, the resulting effect of JNKBP1 on NOD2 signaling is a down-regulation.
In addition to repressing the NOD2-mediated proinflammatory signaling, JNKBP1 also induces a decrease of NOD2-mediated bacteria killing. The NOD2-mediated antibacterial response involves two kinds of mechanisms: direct bacterial killing mechanisms, such as those involving autophagy and reactive oxygen species production (12, 13, 24, 32), and indirect mechanisms, which depend on the expression of NF-κB-target proinflammatory and antibacterial genes (14, 33, 34). The NOD2 anti-bacterial effect monitored by the gentamicin protection assay after L. monocytogenes infection for 2 h results more probably from a direct killing, such as autophagy. Several studies demonstrated a critical role for RIP2 in NOD2-mediated Listeria autophagy (12, 35). It would be interesting to test the effect of JNKBP1 on MDP- or Listeria-induced autophagy in GNV cells by employing the autophagy marker, LC3 (microtubule-associated protein 1A/1B-light chain 3), as a readout (36).
We showed by immunohistochemical staining that JNKBP1 is expressed in human intestinal mucosa, mainly in epithelial and immune cells. Moreover, we confirmed JNKBP1 expression by qRT-PCR in human ileum and colon biopsies (data not shown).
Because JNKBP1 and NOD2 are co-expressed in crypts and in immune cells of the lamina propria, JNKBP1 could contribute to NOD2-mediated intestinal immune homeostasis by regulating NOD2-dependent proinflammatory and antibacterial signaling.
Supplementary Material
Acknowledgments
We thank N. Renotte, C. Lassence, G. Cobraiville, and E. Dortu for technical assistance. We also thank the Viral Vector Platform, GIGA-R, University of Liege. We thank S. Ormenese and R. Stephan for FACS analysis (Imaging GIGA-R technological platform) and A. Delga (Biothèque Université de Liége, University of Liege, Centre Hospitalier Universitaire of Liege, Belgium). The results of sequencing were obtained thanks to the Genomic-Sequencing Platform, GIGA-R, University of Liege.
This work was supported by the “Fonds Spéciaux de l'Université de Liège” (Crédit d'Impulsion), by the “Fonds pour la Recherche dans l'Industrie et l'Agriculture” (FRIA, Brussels, Belgium), by the “Fonds National pour la Recherche Scientifique” (FNRS, Brussels, Belgium), by the Leon Fredericq Foundation (University of Liege, Belgium), and by the Inter-University Attraction Poles (IAP6/18) (Brussels, Belgium).

This article contains supplemental Tables 1–3 and Fig. S1.
- NLR
- NOD (nucleotide-binding oligomerization domain)-like receptor
- NBD
- nucleotide binding domain
- LRR
- leucine-rich repeat
- CD
- Crohn disease
- CARD
- caspase recruitment domain
- MDP
- muramyl dipeptide
- HEK
- human embryonic kidney
- GNV
- HEK293 cells expressing GFP-NOD2-V5
- RIPA
- radioimmunoprecipitation assay
- IP
- immunoprecipitation
- qRT-PCR
- quantitative RT-PCR
- MOI
- multiplicity of infection
- BHI
- brain heart infusion.
REFERENCES
- 1. Kawai T., Akira S. (2005) Pathogen recognition with Toll-like receptors. Curr. Opin. Immunol. 17, 338–344 [DOI] [PubMed] [Google Scholar]
- 2. Chen G., Shaw M. H., Kim Y. G., Nuñez G. (2009) NOD-like receptors. Role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 4, 365–398 [DOI] [PubMed] [Google Scholar]
- 3. Hugot J. P., Chamaillard M., Zouali H., Lesage S., Cézard J. P., Belaiche J., Almer S., Tysk C., O'Morain C. A., Gassull M., Binder V., Finkel Y., Cortot A., Modigliani R., Laurent-Puig P., Gower-Rousseau C., Macry J., Colombel J. F., Sahbatou M., Thomas G. (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411, 599–603 [DOI] [PubMed] [Google Scholar]
- 4. Ogura Y., Bonen D. K., Inohara N., Nicolae D. L., Chen F. F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R. H., Achkar J. P., Brant S. R., Bayless T. M., Kirschner B. S., Hanauer S. B., Nuñez G., Cho J. H. (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411, 603–606 [DOI] [PubMed] [Google Scholar]
- 5. Ogura Y., Inohara N., Benito A., Chen F. F., Yamaoka S., Nunez G. (2001) Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J. Biol. Chem. 276, 4812–4818 [DOI] [PubMed] [Google Scholar]
- 6. Girardin S. E., Boneca I. G., Viala J., Chamaillard M., Labigne A., Thomas G., Philpott D. J., Sansonetti P. J. (2003) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 278, 8869–8872 [DOI] [PubMed] [Google Scholar]
- 7. Inohara N., Ogura Y., Fontalba A., Gutierrez O., Pons F., Crespo J., Fukase K., Inamura S., Kusumoto S., Hashimoto M., Foster S. J., Moran A. P., Fernandez-Luna J. L., Nuñez G. (2003) Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J. Biol. Chem. 278, 5509–5512 [DOI] [PubMed] [Google Scholar]
- 8. Tigno-Aranjuez J. T., Asara J. M., Abbott D. W. (2010) Inhibition of RIP2's tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. 24, 2666–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bertrand M. J., Doiron K., Labbé K., Korneluk R. G., Barker P. A., Saleh M. (2009) Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 30, 789–801 [DOI] [PubMed] [Google Scholar]
- 10. Kim J. Y., Omori E., Matsumoto K., Núñez G., Ninomiya-Tsuji J. (2008) TAK1 is a central mediator of NOD2 signaling in epidermal cells. J. Biol. Chem. 283, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lecat A., Piette J., Legrand-Poels S. (2010) The protein Nod2. An innate receptor more complex than previously assumed. Biochem. Pharmacol. 80, 2021–2031 [DOI] [PubMed] [Google Scholar]
- 12. Cooney R., Baker J., Brain O., Danis B., Pichulik T., Allan P., Ferguson D. J., Campbell B. J., Jewell D., Simmons A. (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97 [DOI] [PubMed] [Google Scholar]
- 13. Travassos L. H., Carneiro L. A., Ramjeet M., Hussey S., Kim Y. G., Magalhães J. G., Yuan L., Soares F., Chea E., Le Bourhis L., Boneca I. G., Allaoui A., Jones N. L., Nuñez G., Girardin S. E., Philpott D. J. (2010) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62 [DOI] [PubMed] [Google Scholar]
- 14. Kobayashi K. S., Chamaillard M., Ogura Y., Henegariu O., Inohara N., Nuñez G., Flavell R. A. (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 [DOI] [PubMed] [Google Scholar]
- 15. Kim Y. G., Kamada N., Shaw M. H., Warner N., Chen G. Y., Franchi L., Núñez G. (2011) The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 34, 769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koyano S., Ito M., Takamatsu N., Shiba T., Yamamoto K., Yoshioka K. (1999) A novel Jun N-terminal kinase (JNK)-binding protein that enhances the activation of JNK by MEK kinase 1 and TGF-β-activated kinase 1. FEBS Lett. 457, 385–388 [DOI] [PubMed] [Google Scholar]
- 17. Yamaguchi T., Miyashita C., Koyano S., Kanda H., Yoshioka K., Shiba T., Takamatsu N., Ito M. (2009) JNK-binding protein 1 regulates NF-κB activation through TRAF2 and TAK1. Cell Biol. Int. 33, 364–368 [DOI] [PubMed] [Google Scholar]
- 18. Kufer T. A., Kremmer E., Adam A. C., Philpott D. J., Sansonetti P. J. (2008) The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol. 10, 477–486 [DOI] [PubMed] [Google Scholar]
- 19. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 20. Barnich N., Aguirre J. E., Reinecker H. C., Xavier R., Podolsky D. K. (2005) Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-κB activation in muramyl dipeptide recognition. J. Cell Biol. 170, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ogura Y., Lala S., Xin W., Smith E., Dowds T. A., Chen F. F., Zimmermann E., Tretiakova M., Cho J. H., Hart J., Greenson J. K., Keshav S., Nuñez G. (2003) Expression of NOD2 in Paneth cells. A possible link to Crohn's ileitis. Gut 52, 1591–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lala S., Ogura Y., Osborne C., Hor S. Y., Bromfield A., Davies S., Ogunbiyi O., Nuñez G., Keshav S. (2003) Crohn's disease and the NOD2 gene. A role for paneth cells. Gastroenterology 125, 47–57 [DOI] [PubMed] [Google Scholar]
- 23. Rosenstiel P., Fantini M., Bräutigam K., Kühbacher T., Waetzig G. H., Seegert D., Schreiber S. (2003) TNF-α and IFN-γ regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology 124, 1001–1009 [DOI] [PubMed] [Google Scholar]
- 24. Lipinski S., Till A., Sina C., Arlt A., Grasberger H., Schreiber S., Rosenstiel P. (2009) DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J. Cell Sci. 122, 3522–3530 [DOI] [PubMed] [Google Scholar]
- 25. Duncan J. A., Bergstralh D. T., Wang Y., Willingham S. B., Ye Z., Zimmermann A. G., Ting J. P. (2007) Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 8041–8046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Faustin B., Lartigue L., Bruey J. M., Luciano F., Sergienko E., Bailly-Maitre B., Volkmann N., Hanein D., Rouiller I., Reed J. C. (2007) Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 25, 713–724 [DOI] [PubMed] [Google Scholar]
- 27. Tanabe T., Chamaillard M., Ogura Y., Zhu L., Qiu S., Masumoto J., Ghosh P., Moran A., Predergast M. M., Tromp G., Williams C. J., Inohara N., Núñez G. (2004) Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J. 23, 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Strober W., Murray P. J., Kitani A., Watanabe T. (2006) Signaling pathways and molecular interactions of NOD1 and NOD2. Nat. Rev. Immunol. 6, 9–20 [DOI] [PubMed] [Google Scholar]
- 29. Bielig H., Zurek B., Kutsch A., Menning M., Philpott D. J., Sansonetti P. J., Kufer T. A. (2009) A function for AAMP in Nod2-mediated NF-κB activation. Mol. Immunol. 46, 2647–2654 [DOI] [PubMed] [Google Scholar]
- 30. Stirnimann C. U., Petsalaki E., Russell R. B., Müller C. W. (2010) WD40 proteins propel cellular networks. Trends Biochem. Sci. 35, 565–574 [DOI] [PubMed] [Google Scholar]
- 31. Windheim M., Lang C., Peggie M., Plater L. A., Cohen P. (2007) Molecular mechanisms involved in the regulation of cytokine production by muramyl dipeptide. Biochem. J. 404, 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perez L. H., Butler M., Creasey T., Dzink-Fox J., Gounarides J., Petit S., Ropenga A., Ryder N., Smith K., Smith P., Parkinson S. J. (2010) Direct bacterial killing in vitro by recombinant Nod2 is compromised by Crohn's disease-associated mutations. PLoS One 5, e10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tattoli I., Travassos L. H., Carneiro L. A., Magalhaes J. G., Girardin S. E. (2007) The Nodosome. Nod1 and Nod2 control bacterial infections and inflammation. Semin. Immunopathol. 29, 289–301 [DOI] [PubMed] [Google Scholar]
- 34. Werts C., le Bourhis L., Liu J., Magalhaes J. G., Carneiro L. A., Fritz J. H., Stockinger S., Balloy V., Chignard M., Decker T., Philpott D. J., Ma X., Girardin S. E. (2007) Nod1 and Nod2 induce CCL5/RANTES through the NF-κB pathway. Eur. J. Immunol. 37, 2499–2508 [DOI] [PubMed] [Google Scholar]
- 35. Anand P. K., Tait S. W., Lamkanfi M., Amer A. O., Nunez G., Pagès G., Pouysségur J., McGargill M. A., Green D. R., Kanneganti T. D. (2011) TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation. J. Biol. Chem. 286, 42981–42991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.