

Background: Infants of mothers carrying the H54R mutation in ZnT-2 develop transient neonatal zinc deficiency (TNZD).
Results: Transfection of a novel heterozygous G87R mutant ZnT-2 resulted in its mislocalization, impaired zinc transport, and negative dominance.
Conclusion: G87R is an inactivating mutation inflicting a dominant negative effect via homodimer formation.
Significance: This study significantly advances our understanding regarding the molecular mechanism underlying TNZD.
Keywords: Metabolic Diseases, Metal Homeostasis, Mutant, Nutrition, Transport Metals, Zinc, Dominant Negative Mutation, Homodimer, Neonatal Zinc Deficiency, Zinc-deficient Milk
Abstract
Zinc is an essential mineral, and infants are particularly vulnerable to zinc deficiency as they require large amounts of zinc for their normal growth and development. We have recently described the first loss-of-function mutation (H54R) in the zinc transporter ZnT-2 (SLC30A2) in mothers with infants harboring transient neonatal zinc deficiency (TNZD). Here we identified and characterized a novel heterozygous G87R ZnT-2 mutation in two unrelated Ashkenazi Jewish mothers with infants displaying TNZD. Transient transfection of G87R ZnT-2 resulted in endoplasmic reticulum-Golgi retention, whereas the WT transporter properly localized to intracellular secretory vesicles in HC11 and MCF-7 cells. Consequently, G87R ZnT-2 showed decreased stability compared with WT ZnT-2 as revealed by Western blot analysis. Three-dimensional homology modeling based on the crystal structure of YiiP, a close zinc transporter homologue from Escherichia coli, revealed that the basic arginine residue of the mutant G87R points toward the membrane lipid core, suggesting misfolding and possible loss-of-function. Indeed, functional assays including vesicular zinc accumulation, zinc secretion, and cytoplasmic zinc pool assessment revealed markedly impaired zinc transport in G87R ZnT-2 transfectants. Moreover, co-transfection experiments with both mutant and WT transporters revealed a dominant negative effect of G87R ZnT-2 over the WT ZnT-2; this was associated with mislocalization, decreased stability, and loss of zinc transport activity of the WT ZnT-2 due to homodimerization observed upon immunoprecipitation experiments. These findings establish that inactivating ZnT-2 mutations are an underlying basis of TNZD and provide the first evidence for the dominant inheritance of heterozygous ZnT-2 mutations via negative dominance due to homodimer formation.
Introduction
Maintenance of adequate zinc blood levels is crucial as zinc is an essential metal that plays a key role in the activities of numerous enzymes and proteins that are involved in a multitude of physiological processes including normal growth and development, cellular integrity, protein synthesis, nucleic acid metabolism, and apoptosis (1, 2). The initial main symptoms of zinc deficiency are dermatitis, diarrhea, alopecia, and loss of appetite. Prolonged zinc deficiency is often expressed in growth impairment and neuropsychological changes such as emotional instability, irritability, and depression (3). Infants are particularly vulnerable to zinc deficiency as they require large amounts of zinc for their normal growth and development. Hence, zinc deficiency in infants can lead to less focused attention and decreased motor functions at 6 months of age. Zinc supplementation improved motor development in low birth weight infants and raised functional activity in infants and toddlers (4). Infants are born with hepatic zinc storage; however, this metal exists in a much higher relative concentration in breast milk, especially in the first 3 months of lactation (5). Therefore, adequate zinc nutrition of infants mostly depends on breast milk feeding or breast milk formula feeding, whereas premature infants are more vulnerable to zinc deficiency as they have sparse hepatic zinc storage (5).
Zinc deficiency in mammalian newborns is a result of deficient nutrition due to consumption of food that is poor in zinc bioavailability or has been associated with three distinct genetic disorders in zinc metabolism. The first is associated with mutations in SLC39A4/Zip-4 (6, 7), which is responsible for zinc absorption in the small intestine and when mutated leads to a rare, autosomal recessive disease called acrodermatitis enteropathica (AE)3 (OMIM 201100). AE manifests in impaired intestinal zinc absorption; hence, patients harboring AE require lifelong zinc supplementation (8, 9). The second is associated with a mutation in SLC30A4/ZnT-4 (10) that is associated with reduced zinc incorporation into milk (11). Dams homozygous for a ZnT-4 mutation are known as lethal milk mice because they produce less milk and milk that is zinc-deficient (OMIM 602095). Consequently, pups nursed by lethal milk mice died in neonatal life unless they received oral zinc supplementation (12). The third was found to be associated with a mutation in SLC30A2/ZnT-2 (NP_001004434.1), which resulted in transient neonatal zinc deficiency (TNZD) (OMIM 608118) in breast-fed infants; TNZD was described in some previous reports (13–15). A heterozygous H54R mutation in ZnT-2 that we recently identified in two related women resulted in zinc-deficient milk. Consequently, their infants developed TNZD that was resolved after oral zinc supplementation (16). Moreover, gene knockdown of ZnT-2 in mammary epithelial cells reduced zinc secretion, suggesting a role for this transporter in zinc secretion from this cell type (16). ZnT-2 was also found to be up-regulated and relocalized to vesicles after exposure of mammary epithelial cells to prolactin. Similarly, mammary gland ZnT-2 was up-regulated and relocalized to the luminal membrane in lactating rats when plasma zinc increased during lactation (17, 18). Unlike AE patients, infants affected with TNZD absorb zinc normally and require zinc supplementation only during their nursing period; likewise, their mothers did not carry mutations in ZnT-4 (19) or develop zinc deficiency or any other clinical signs characteristic of the lethal milk mice disorder (20). Additionally, maternal zinc supplementation could not correct TNZD (14, 16), whereas it improved survival of lethal milk mice pups (10).
Here we identified and characterized a novel heterozygous G87R mutation in ZnT-2 leading to production of zinc-deficient milk in two Israeli mothers from distinct Jewish Ashkenazi families; as a result, their exclusively breast-fed infants developed TNZD with low zinc blood levels that resolved upon zinc supplementation. We show that the G87R mutation is a loss of function mutation and provide the first evidence for the dominant inheritance of heterozygous ZnT-2 mutations via negative dominance due to homodimer formation.
EXPERIMENTAL PROCEDURES
Clinical Data of Subjects
The study was approved by the Institutional Review Board of the Sheba Medical Center (SMC-7786-10). Written consent was obtained from all subjects. Serum and milk zinc levels of mothers and patients were retrieved from their clinical records. Zinc deficiency was first diagnosed by the attending pediatrician based on clinical presentation and supported by determination of serum zinc levels. Information regarding dietary content of the infants including breast-feeding duration and supplementation with nutritional formula was obtained by mothers' questionnaires. The medical history of the family was established based upon the mothers' interview.
Sequence Analysis of ZnT-2 (SLC30A2) and Zip-4 (SLC39A4)
After informed consent, blood samples were obtained from two women with low milk zinc concentration and their available family members. DNA was extracted from blood using ArchivePure DNA Blood Base Kit (5 Prime, Hamburg, Germany). Primers were designed to amplify each one of the 8 or 12 exons of ZnT-2 or Zip4, respectively, including their flanking introns (supplemental Tables S1 and S2). Genomic DNA was amplified by PCR using the primers described above (Red Load Taq master mix, Larova GmbH, Teltow, Germany). PCR products were analyzed by nucleotide sequencing using Big Dye Termination chemistry and an ABI Prism 3100 sequencer (V1.1, Applied Biosystems, Foster City, CA). Sequence analysis was performed using Bioedit software, and sequence chromatograms were visually analyzed for mutations.
Restriction Enzyme Assay
DNA sequencing results were used for designing a restriction enzyme assay. The G87R mutation created a new BclI restriction site in the mutant PCR amplicon that could distinguish between homozygous, heterozygous, and healthy individuals. PCR products were subjected to enzymatic digestion by BclI according to the manufacturer's specifications (New England Biolabs, Ipswich, MA). The resulting products were electrophoresed on 2% agarose gels. Two hundred and six control alleles from 103 unrelated anonymous healthy individuals of Ashkenazi descent were tested to rule out polymorphism and to estimate the prevalence of the G87R mutation in this population.
Introduction of the G87R and F134A/G135A/R138A/E140A Mutations into an Expression Vector Harboring an HA-tagged ZnT-2 (HA-ZnT-2)
The G87R mutation as well as the F134A/G135A/R138A/E140A mutations were introduced into pcDNA3.1/V5-His TOPO vector containing C-terminal HA-tagged ZnT-2 (16) by PCR mutagenesis (QuikChange kit, Stratagene, La Jolla, CA). The G87R mutation was introduced using the forward primer (5′-CTGCCTGTTGTTCATGATCAGAGAAGTCGTTGAGATC-3′) and reverse primer (5′-GATCTCAACGACTTCTCTGATCATGAACAACAGGCAG-3′). F134A and G135A mutations were introduced using the forward primer (5′-GCCACCAAGACCATGAACGCTGCCTGGCAGAGAGCTG-3′) and reverse primer (5′-CAGCTCTCTGCCAGGCAGCGTTCATGGTCTTGGTGGC-3′) at the first reaction of PCR. R138A and E140A mutations were introduced using the forward primer (5′-GAACGCTGCCTGGCAGGCAGCTGCGATCTTGGGAGCCCTGG-3′) and reverse primer (5′-CCAGGGCTCCCAAGATCGCAGCTGCCTGCCAGGCAGCGTTC-3′) in the second reaction of the PCR. Incorporation of the site-directed mutation as well as the fidelity of the insert and the epitope tag was verified by DNA sequencing (DNA Sequencing Facility, Technion-Israel Institute of Technology, Faculty of Medicine, Haifa, Israel).
Bioinformatics Analysis and Three-dimensional Model Prediction; Sequence Based Analysis
Close homologues of the long isoform of ZnT-2 (372 amino acids) were collected from the Sprot data base (UniProtKB/Swiss-Prot) (21) with the Protein BLAST program (22) implemented in the MPI Bioinformatics Toolkit server (23) using default parameters. The closest homologues of ZnT-2, which belong to the ZnT family (only their long isoforms), were aligned using a multiple sequence alignment applying the MAFFT program (24).
Helical Wheel
Helical wheel was generated using the internet site “helical wheel projection” created by Don Armstrong and Raphael Zidovetzki: Version Id: wheel.pl,v 1.4 2009-10-20 21:23:36 don Exp.
Structure-based Analysis
The sequence of the longer isoform of ZnT-2 (372 amino acids) was extracted from the Swissprot data base and searched against the Protein Data Bank using the HHpred engine (25). This search identified Protein Data Bank entry 3h90 chain A (zinc transporter YiiP) target as the closest homologue, with an E value = 0 and a global sequence identity of 21%, indicating a very probable match. The alignment included 248 amino acids of ZnT-2, hence leaving out the N-and C-terminal tails.
Transmembrane Helix Prediction and Three-dimensional Model of ZnT-2
To improve the quality of the alignment, several methods were used to predict the location of the six transmembrane helices (26). Then, a consensus location of each helix was derived (data not shown) and used to correct the alignment manually where necessary. This alignment was then used to create 10 three-dimensional models by Modeler (27).
Quality Estimation
The three-dimensional models were then studied using the server ConQuass (28), which assesses the quality of protein model structures using evolutionary conservation. ConQuass measures the compatibility between the conservation and accessibility patterns of a given structural model based on the observation that protein structural cores are composed mainly of conserved residues. The best ConQuass score for the monomers was 0.0277, indicating a structure of reasonable quality. The dimer structure was obtained by structural alignment between two monomers and chains A and C of the template 3h90 (YiiP). The dimer ConQuass score was slightly higher 0.0369, suggesting that the dimerization improves the conservation-exposure profile for this model.
Conservation and Hydrophobicity Calculations
Evolutionary conservation scores for the modeled structure were obtained from the ConSurf server (29). Hydrophobicity values were obtained from the Kyte-Doolittle hydrophobicity scale (30).
Identification of Conserved Signatures in ZnT-2 Sequence
Indentifying conserved sequence profiles (signatures) possibly corresponding to elementary functional loops in ZnT-2 was performed using BLOSUM62 substitution matrix (31), which scores alignments between evolutionarily divergent protein sequences (non redundant sequences were obtained from Swissprot).
Graphic Imaging
Image of sequence analysis was produced using the Jalview package (32). Molecular graphic images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH Grant P41 RR001081) (33).
Cell Culture and Transient Transfection of HA-tagged ZnT-2
Mouse mammary epithelial HC11 cells were a gift from Dr. Jeffery Rosen (Houston, TX) and were used with permission of Dr. Bernd Groner (Institute for Biomedical Research, Frankfurt, Germany). HC11 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, gentamicin (50 μg/ml; Sigma), insulin (5 μg/ml; Sigma), and epidermal growth factor (10 ng/ml; Sigma) at 37 °C in a humidified atmosphere of 5% CO2. Human embryonic kidney HEK-293 cells and breast cancer MCF-7 cells were grown under monolayer conditions in RPMI 1640 medium (Invitrogen) containing 10% fetal calf serum, 2 mm glutamine, 100 μg/ml penicillin, and 100 units/ml streptomycin (Biological Industries, Beth-Haemek, Israel) in a humidified atmosphere of 5% CO2. Cells were seeded in 6-well plates (2.5 × 106 HC11cells/well) for cytoplasmic zinc pool assay or in 24-well plates (6 × 105 HC11 cells/well as well as 5 × 104 HEK-293 and MCF-7 cells/well) for zinc secretion, accumulation, and subcellular localization experiments or in 10-cm plates (2 × 106 HEK-293 and MCF-7 cells/plate) for Western blot analysis and cultured overnight until a confluence of ∼95% was achieved. Cells were transiently transfected with 1–1.6 μg of DNA (24-well plates), 4 μg of DNA (6-well plates), or 8 μg of DNA (10-cm plates) using transfection reagents (Lipofectamine 2000 for HC11 cells (Invitrogen) and JetPEI for HEK-293 and MCF-7 cells (Polyplus transfections)) according to the manufacturer's specifications for up to 24 h.
Immunoprecipitation of HA-ZnT-2
To explore ZnT-2 dimer formation, HC11 cells were transiently transfected with HA-tagged ZnT-2 cDNA. 24 h after transfection cells were scraped off in ice-cold PBS, sedimented by centrifugation, and lysed in radioimmunoprecipitation buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitors) for 30 min at 4 °C with rotation. Samples were centrifuged for 10 min at 15,000 × g, and the protein concentration in the lysates was determined by using the Bradford assay. Cell lysates were preincubated with anti-HA agarose (Pierce) overnight at 4 °C with rotation followed by 3 washes with radioimmunoprecipitation buffer. Then, proteins were eluted by boiling at 95 °C for 5 min in elution buffer, run on 10% SDS-PAGE gels under non-reducing conditions, and transferred to nitrocellulose membranes. ZnT-2 dimerization was assessed by immunoblotting using an anti-HA antibody (Roche Applied Science) and detected with horseradish peroxidase-conjugated secondary antibodies. Proteins were visualized by enhanced chemiluminescence (SuperSignal Femto, Pierce).
Subcellular Localization of HA-ZnT-2
Subcellular localization of HA-G87R ZnT-2 that was transiently transfected (transfection was performed as mentioned above) into HC11 cells was determined as follows. Cells plated onto glass coverslips were fixed with 4% paraformaldehyde in PBS for 10 min and permeabilized with 0.2% Triton X-100 in PBS for 5 min. Nonspecific binding was blocked for 1 h with 5% goat serum, 1% bovine serum albumin in PBS. For the detection of Golgi apparatus, cells were incubated for 1 h with rabbit anti-p58 (Golgi marker) antibody (1 μg/ml, Abcam), and after extensive washes with PBS, cells were incubated with anti-rabbit IgG antibody conjugated to Alexa 568 for 20 min. Cells were then rewashed with PBS, and nonspecific binding was blocked with 5% goat serum, 1% bovine serum albumin in PBS for 20 min. Thereafter, detection of HA-ZnT-2 was performed by incubation with mouse Alexa 488-conjugated anti-HA (1 μg/ml, Invitrogen) for 1 h, protected from light. Immunofluorescence imaging was performed using an Olympus FV1000 fluorescence microscope equipped with PlanApo 60× oil lens N.A. 1.42 and digital images were captured sequentially (FV10-ASW, Version 4.5; Olympus). Pearson's coefficient for the colocalization of G87R ZnT-2 and p58 was 0.83, indicating a positive correlation (see statistical analysis); no co-localization for the WT ZnT-2 and p58 was observed. Subcellular localization of HA- G87R ZnT-2 in MCF-7 and HEK-293 transfectants was determined similarly with some modification as previously described (34). For detection of HA-ZnT-2, mouse anti-HA monoclonal antibody (generously provided by Prof. Ami Aronheim, Rappaport School of Medicine, Technion, Israel) was used as a primary antibody (1:50 dilution), and FITC-conjugated anti-mouse antibody (1:100; Jackson ImmunoResearch Laboratories, West Grove, PA) was used as a secondary antibody. For calnexin (an endoplasmic reticulum (ER) marker) detection in MCF-7 and HEK-293 cells, rabbit anti-calnexin (Abcam, 5 μg/ml) was used as a primary antibody, and rhodamine-conjugated anti rabbit antibody (1:100; Jackson ImmunoResearch) was used as a secondary antibody. DAPI (1 μg/ml) was used for the staining of nuclear DNA. Immunofluorescence imaging was performed using a Zeiss Axiovert 200 Cell Observer inverted microscope. To establish the effect of G87R ZnT-2 transfection on the subcellular localization of the WT ZnT-2, MCF-7 cells were co-transfected with specific combinations of HA-ZnT-2 or HA-reduced folate carrier (RFC) (35) (0.5 μg) along with untagged ZnT-2 (0.5 μg), thereby enabling the following of specific tagged protein. HA-ZnT-2 and HA-RFC were detected using mouse anti-HA monoclonal antibody as a primary antibody and FITC-conjugated anti mouse antibody as a secondary antibody. Nuclei were stained using DAPI (1 μg/ml). In each independent experiment 300 cells were counted for secretory vesicles or perinuclear localization.
Vesicular Zinc Accumulation Assay
To determine whether G87R ZnT-2 affects labile zinc accumulation, HC11 cells were plated in antibiotic-free growth medium in 24-well plates and cultured until 90–95% confluence was achieved. Cells were then transfected with plasmids containing HA-WT ZnT-2, HA-G87R ZnT-2, or with an empty vector (mock) for 24 h, and FluoZin-3 fluorometry assay was performed as previously described (36).
65Zn Secretion Assay
To explore the zinc secretion ability of G87R ZnT-2, HC11 cells were transfected with HA-WT ZnT-2, G87R ZnT-2, and empty vector. Transfected cells were loaded with serum-free medium containing 0.1 μm ZnSO4 and 65Zn (0.1 μCi; Oakridge National Laboratory, Oakridge, TN) for 3 h at 37 °C, after which cells were washed briefly with cold PBS containing 1 mm EDTA to remove exofacial-bound zinc. Cells were then cultured in serum-free medium for up to 120 min and the amount of 65Zn secreted to the culture medium was quantified using a gamma counter. To determine whether co-expression of the G87R allele results in a dominant negative effect, hence impairing WT ZnT-2 function, cells were co-transfected with equal amounts of plasmids as described under “Results” (0.8 μg of each 1× plasmid), and 65Zn secretion was determined.
Cytoplasmic Zinc Pool Assay
To assess changes in the cytoplasmic zinc pools, we used a bioluminescent reporter assay. The pGL3-luciferase reporter vector containing 4× metal-responsive element (MRE) was kindly provided by Dr. Colin Duckett (University of Michigan Medical School, Ann Arbor, MI) and used as previously described (36). Briefly, HC11 cells were co-transfected with either the empty pGL3 vector (0.8 μg) or the 4× MRE pGL3 luciferase reporter (0.8 μg) and pRL-TK renilla vector (0.05 μg) in addition to pcDNA3.1-WT ZnT-2 or pcDNA3.1-G87R ZnT-2 (0.8 μg), and the assay was performed as described previously (36).
Western Blot Analysis of WT and G87R ZnT-2
To examine the protein expression level of WT and G87R ZnT-2 as well as the effect of G87R ZnT-2 expression on the expression of WT ZnT-2, HEK-293 and MCF-7 cells were co-transfected with equal amounts of two specific plasmids as described under “Results” (4 μg of each construct). HA-ZnT-2 expression was determined by Western blot analysis as previously described (35) and detected with mouse anti-HA monoclonal antibody (generously provided by Prof. Ami Aronheim, Rappaport School of Medicine, Technion, Israel) (1:100 dilution) overnight at 4 °C. Blots were then rinsed 3 times in TBS buffer for 10 min each at room temperature and detected with horseradish peroxidase-conjugated goat anti-mouse IgG (1:10,000 dilution; Jackson ImmunoResearch) for 1 h at room temperature. After three 10-min washes in TBS at room temperature, enhanced chemiluminescence detection was performed according to the manufacturer's instructions (Biological Industries). Similarly, equal loading of samples was confirmed using rabbit anti α-subunit of Na+/K+ ATPase polyclonal antibody (KETTY at 1:3000 dilution) and detected with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:15,000 dilution; Jackson ImmunoResearch).
Statistical Analysis
Results are presented as the means ± S.D. Statistical comparisons were performed using Student's t test (Prism Graph Pad, Berkeley, CA), and a significant difference was demonstrated when p < 0.05. To quantify the subcellular localization of G87R-HA in specific subcellular compartments, statistical analysis of the correlation of the intensity values of green and red pixels in a dual-channel image was performed using correlation coefficient (Pearson's coefficient). The value can range from +1 to −1, with +1 illustrating a positive correlation, −1 illustrating a negative correlation, and zero revealing a lack of correlation (37).
RESULTS
Identification of a Heterozygous G87R ZnT-2 Mutation in Two Distinct Families
2.5- and 4-month-old females (subjects 1 and 2, respectively) were born to non-consanguineous parents of Ashkenazi Jewish descent (subjects 3 and 4 and subjects 5 and 6, respectively) and had been exclusively breast-fed (Fig. 1). Infant 1 was born at 36 weeks after decreased fetal movements and sonographic findings of pericardial effusion and ascites. She displayed zinc deficiency symptoms including dermatitis eruption over the face and perineal regions that appeared 2.2 months after birth. A cutaneous examination showed extensive, erosive, crusted erythematous plaques that were located on the face and over the perineum extending on to the thighs and gluteal region (data not shown).
FIGURE 1.

Identification of a heterozygous ZnT-2 mutation in two distinct families afflicted with TNZD. Pedigree of two different Ashkenazi Jewish families (family members are denoted by Arabic numbers) reveals two infants diagnosed with severe TNZD confirmed by serum zinc concentration analysis (denoted by solid circles and termed 1 and 2). The infants were exclusively breast-fed by their mothers who were diagnosed with low milk zinc concentration (denoted by hatched circles and numbered as 3 and 5). Based on family history, the brother of infant 1 was exclusively breast-fed and displayed mild zinc deficiency symptoms (denoted by solid circle and termed 7), whereas the sisters of infant 2 were healthy despite the fact that they were exclusively breast-fed (denoted by empty circles and termed 8 and 9).
The pregnancy and birth of infant 2 were normal. However, 2 months after birth she displayed severe dermatitis accompanied with seborrhea-like rash and secondary infection around the mouth, head, and back. Partial alopecia of the eyebrows, eyelashes, and temple area was also noticed (data not shown). Clinical examination of the mothers (subjects 3 and 5, respectively) of infants 1 and 2 revealed low milk zinc concentration (0.35 and 0.17 mg/liter, respectively; normal range, 1–3 mg/liter (38)) that resulted in low serum zinc concentration of their exclusively breast-fed infants (45 and 13 μg/dl, respectively; normal range, 70–120 μg/dl (39)). Clinical history of the family of infant 1 revealed that her brother (subject 7) had been exclusively breast-fed as well and displayed mild dermatitis that appeared 2 months after birth and resolved after 4 months of zinc supplementation. Infant 2 had two healthy sisters (subjects 8 and 9) that were exclusively breast-fed as infants but did not exhibit any zinc deficiency symptoms (Fig. 1). These symptoms and clinical manifestations were consistent with reported cases of TNZD that developed in infants who were breast-fed zinc-deficient milk (13–15). Hence, both infants were treated with zinc supplementation as follows. Infant 1 received oral zinc acetate (40 mg/day) with rapid improvement of skin lesions within days and complete resolution after 3 weeks. Consistently, infant 2 was treated with zinc acetate (3 mg/kg per day) and had a dramatic improvement after 30 days (data not shown).
Zip-4 (SLC39A4) gene sequencing revealed no mutations in genomic DNA from infants 1 and 2, thereby excluding the possibility of AE. Thus, ZnT-2 sequencing was performed on genomic DNA of affected mothers (subjects 3 and 5) based on our previous study showing that ZnT-2 plays a role in zinc secretion into milk and that an H54R mutation in ZnT-2 is associated with TNZD (16). Both mothers were found to carry a heterozygous missense mutation in exon 2 that substituted a G nucleotide at position 259 to A in the coding region of ZnT-2, thereby resulting in a glycine to arginine substitution at amino acid 87 (G87R) (data not shown). To establish a clear-cut association between the G87R mutation and TNZD as well as to rule out the possibility that G87R is a common polymorphism, DNA from 103 random healthy Ashkenazi Jewish women was examined by restriction enzyme assay, and none was found to harbor this G87R mutation (data not shown).
Gly-87 Conservation, Predicted Transmembrane Localization, and Three-dimensional Modeling
Multiple blast alignments revealed that Gly-87 is a conserved residue among closely related zinc transporters including ZnT-3 and ZnT-4. Moreover, Gly-87 was found to be located in a highly conserved region encompassing amino acids 75–106 (Fig. 2A). According to multiple bioinformatics tools (OCTOPUS, HMMTOP, PHOBIUS, MEMSAT3, and TMHMM), ZnT-2 is predicted to contain six transmembrane helices (TM) with Gly-87 localizing in TM1 (data not shown). Amino acid helical wheel prediction in TM1 (residues 74–96) indicated that Gly-87 resides between two hydrophobic residues (data not shown). To characterize the impact of the G87R mutation on ZnT-2 structure and function, we first generated a three-dimensional model. Using the profile-profile HHpred engine to identify the closest template with a known three-dimensional structure, the Escherichia coli zinc transporter YiiP (Protein Data Bank id 3h90, chain A) emerged as the closest homologue of ZnT-2. YiiP is a homodimeric transporter that mediates Zn2+/H+ exchange across the inner membrane of E. coli (40). Both E. coli YiiP and mammalian ZnT-2 belong to the conserved family of zinc transporters yet share only 21% amino acid identity. Despite the relatively low level of amino acid identity, the following experimental line of evidence supports the suitability of the template. (a) Known structural and functional similarity was found between both transporters (40, 41). (b) The monomer and dimer models created by Modeler achieved scores of 0.0277 and 0.0369 by ConQuass, respectively, indicating a reasonable structural quality. (c) The crystal structure of YiiP was successfully used to generate a predicted three-dimensional model of close homologues of ZnT-2 including ZnT-3 and ZnT-8 (42, 43). (d) Nine additional models of the ZnT-2 monomer were generated using Modeler based on the same alignment with YiiP. All 10 models displayed a very similar orientation of the six TMs, whereas loop conformations were more variable (Fig. 2B). (e) According to this predicted ZnT-2 model, the most conserved residues of ZnT-2 form a putative permeation pathway of zinc atoms (that were imported from the YiiP crystal structure) (40) (Fig. 2C) that is compatible with the zinc permeation pathway in YiiP (data not shown). According to all 10 models, one could predict that Gly-87 is very likely to face the hydrophobic membrane core of the lipid bilayer (Fig. 2B). Consistently, hydrophobicity analysis of this model shows that Gly-87 maps to an exclusive hydrophobic domain (Fig. 2D). Moreover, based on the high evolutionary conservation (data not shown) along with the putative homodimer model interface (formed by superimposing the one modeled monomer onto one interacting YiiP monomer), ZnT-2 is likely to form a homodimer that is similar to multiple zinc transporters that have been shown to form homodimeric or heterodimeric structures including ZnT-3, ZnT-5, ZnT-6, and ZnT-8 (42, 44, 45).
FIGURE 2.

Gly-87 conservation and structural model of ZnT-2. A, shown is a section of the multiple amino acid alignment of ZnT-2 (residues 75–106 according to the human ZnT-2 sequence) and close homologues that belong to the ZnT transporter family. The human ZnT-2 appears in the first line. Protein names are presented according to the UniProt data base, and alignment was colored according to the Taylor scale. Note the remarkable degree of conservation at this segment and the Gly-87, which is conserved in ZnT transporters including ZnT-3 and ZnT-4. B, 10 predicted ZnT-2 models generated based on the monomeric crystal structure of E. coli YiiP demonstrate that Gly-87 (colored in red) points toward the hydrophobic membrane lipid core. C, according to this predicted model of ZnT-2 (the model is on the right, and YiiP template is on the left), α-carbons of most conserved residues (colored in purple and ranked 9 according to ConSurf scale) form a permeation pathway of divalent zinc cations (colored in green). Gly-87 is colored in yellow. D, hydrophobicity calculations (the scale ranges between highly hydrophobic residues (colored in red) and highly hydrophilic residues (colored in blue)) of predicted ZnT-2 model (the model is on the right) consistently indicate that Gly-87 (colored in yellow) maps to a hydrophobic region.
ZnT-2 Forms Homodimers
To explore the likely possibility that ZnT-2, like its sibling transporters, also forms homodimers, HC11 cells were transfected with HA-ZnT-2 or empty vector, and immunoprecipitation was performed using anti-HA antibodies (Fig. 3). SDS-PAGE performed under non-reducing conditions followed by immunoblotting using an anti-HA antibody revealed a band with a molecular mass of ∼43 kDa that is compatible with the size of the ZnT-2 monomer (16) and a ∼85-kDa band, consistent with the molecular mass of a ZnT-2 dimer. These results are concordant with the bioinformatics analysis predicting that ZnT-2 forms homodimers.
FIGURE 3.
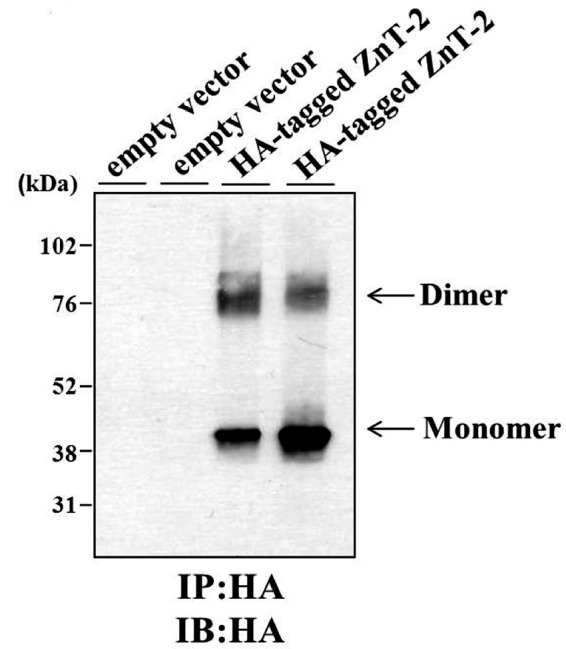
Immunoprecipitation (IP) of HA-tagged ZnT-2. Proteins were purified from HC11 cells that were transiently transfected with empty vector or HA-tagged ZnT-2. Immunoprecipitation was performed using anti-HA antibody, and the immunoprecipitation products were run on 10% SDS-PAGE gels under non-reducing conditions. Immunoblotting (IB) was performed using anti-HA antibody. Enhanced chemiluminescence of immunoprecipitation products purified from HA-tagged ZnT-2 transfectants revealed an ∼43-kDa band representing ZnT-2 monomer and an ∼85-kDa band that represents putative ZnT-2 dimer. The results are displayed in a representative photograph from two independent experiments, and independent duplicates are shown for each treatment.
Identification of a Conserved Motif Predicted to Be Involved in ZnT-2 Homodimerization
We identified a conserved (F/Y)G(W/Y/F)XRXE signature (FGWQRAE in ZnT-2 encompassing amino acids 134–141) that resides at the homodimer interface region (Fig. 4A) in which glycine (Gly-135), arginine (Arg-138), and glutamate (E140) are highly conserved. We, therefore, theorized that this motif may be important for protein-protein interactions due to the presence of aromatic and charged side chains (46). To examine the possible role of this conserved motif in ZnT-2 homodimerization, we substituted the most conserved core residues in this motif i.e. Phe-134, Gly-135, Arg-138, and Glu-140 to alanine using site-directed mutagenesis as previously reported in studies aimed at identification of residues necessary for dimerization (46). All four mutations (i.e. F134A, G135A, R138A, and E140A) were introduced into the same construct harboring an HA-tagged ZnT-2. After transient transfection into MCF-7 cells, which are essentially devoid of WT ZnT-2, immunofluorescence microscopy revealed that HA-tagged WT ZnT-2 as well as the HA-tagged mutant harboring F134A/G135A/R138A/E140A mutations were properly targeted to cytoplasmic secretory vesicles (Fig. 4B). Although equal amounts of protein were loaded, Western blot analysis under non-reducing conditions revealed that the level of the F134A/G135A/R138A/E140A mutant protein was ∼3-fold higher than the protein level of HA-tagged WT ZnT-2 in transiently transfected MCF-7 cells (Fig. 4C). Immunoprecipitation assay in which MCF-7 cells were transiently transfected with HA-tagged WT ZnT-2, HA-tagged F134A/G135A/R138A/E140A ZnT-2 or an empty vector revealed that the abundance of the homodimer form of HA-tagged mutant F134A/G135A/R138A/E140A ZnT-2 was ∼3-fold higher than its monomeric form. Conversely, the abundance of the dimer form of the HA-tagged WT ZnT-2 was ∼2-fold lower than the level of the monomeric form (Fig. 4D). Hence, these results suggest that substitution of the most conserved core residues within the (F/Y)G(W/Y/F)XRXE signature to alanine results in a markedly enhanced homodimerization and increased stability of the monomer possibly due to a stabilization mediated through homodimer formation. Furthermore, the observation that the (F/Y)G(W/Y/F)XRXE signature, which was predicted to map to the dimer interface, is likely involved in homodimerization further supports the validity of the structure model of ZnT-2. In the majority of published papers alanine substitution of native residues that are required for dimerization resulted in interference with homodimerization. In contrast, in some reports it was found that substitution of these residues to alanine resulted in enhanced dimerization as found in this study. For example, the substitution to alanine of His-234 or Ser-148, which localized to the established interface of the outer membrane phospholipase A (OMPLA), resulted in an enhanced dimerization when compared with wild-type OMPLA (47). Moreover, substitution of the second arginine within the RKTR motif in one of the protomers of the serine/threonine kinase RAF to alanine also led to a strongly increased homo- and heterodimer formation (48). Likewise, the substitution of Gly-79 and Gly-83 in the conserved dimerization motif GXXXG to alanine in glycophorin A also resulted in an enhanced homodimerization. The substitution of residues at the dimer interface to alanine contributes to dimerization, as alanine has a small side chain that can facilitate a closer approach of the transmembrane helices to maintain stronger van der Waals interactions (49). However, additional research is necessary to determine the definitive involvement of certain residues in the (F/Y)G(W/Y/F)XRXE signature in homodimerization.
FIGURE 4.

Identification of conserved motif presumably involved in ZnT-2 dimerization. A, profile analysis revealed a conserved signature simplified as (F/Y)G(W/Y/F)XRXE (appears in ZnT-2 sequence as FGWQRAE). Signature residues (colored according to ConSurf scale), which are presumably important for protein-protein interactions, are found to map to the predicted ZnT-2 interface. Gly-87 is colored in yellow. B, localization of HA-tagged WT ZnT-2 (ii) or HA-tagged F134A/G135A/R138A/E140A ZnT-2 (iii) in MCF-7 cells was examined using inverted cell observer microscopy. Empty vector transfection (i) was used as negative control, and HA-tagged G87R ZnT-2 transfection (iv) was used as the control for the localization of ZnT-2 to the ER compartment. Immunofluorescent detection of ZnT-2 was performed using a mouse anti-HA antibody (green fluorescence; ii, iii, iv). Nuclei were stained with DAPI. Merged images illustrate vesicular localization of WT ZnT-2 as well as F134A/G135A/R138A/E140A ZnT-2 mutant in MCF-7 cells (ii and iii, respectively), whereas HA-tagged G87R ZnT-2 showed perinuclear localization in MCF-7 cells (iv). A magnification of ×63 under immersion oil was used. C, MCF-7 cells were transiently transfected with equal amounts of empty vector or HA-tagged WT ZnT-2 or 4Mut ZnT-2 (i.e. F134A/G135A/R138A/E140A ZnT-2). Equal amounts of lysate were loaded, and Western blot analysis was performed under non-reducing conditions. HA-tagged ZnT-2 was detected with an anti-HA antibody and displayed a molecular mass of 43 kDa. D, proteins were isolated from MCF-7 cells that were transiently transfected with an empty vector or HA-tagged WT ZnT-2 or HA-tagged 4Mut construct (i.e. F134A-G135A-R138A-140A ZnT-2). Immunoprecipitation (IP) was performed using an anti-HA antibody, and the immunoprecipitation products were resolved on 10% SDS-PAGE gels under non-reducing conditions. Immunoblotting (IB) was performed using the anti-HA antibody. Enhanced chemiluminescence of immunoprecipitation products isolated from HA-tagged ZnT-2 transfectants revealed an ∼43-kDa band representing the ZnT-2 monomer and an ∼85-kDa band representing the ZnT-2 dimer.
Subcellular Localization of HA-WT ZnT-2 and G87R ZnT-2
HA-WT ZnT-2 and G87R ZnT-2 were transfected into HC11, MCF-7, and HEK-293 cells (Fig. 5). WT ZnT-2 was found to properly localize to intracellular secretory vesicles in HC11 and MCF-7 cells (Fig. 5, A–C, and G–I, respectively). In contrast, mutant G87R ZnT-2 was largely retained at the Golgi apparatus in HC11 cells as revealed by co-localization with the Golgi marker p58 (Fig. 5, D–F) and showed punctuated perinuclear localization in MCF-7 cells that did not perfectly overlap with the localization of the ER chaperone calnexin that was dispersed throughout the cell (Fig. 5, J–L). However, some vesicular localization was also detected with G87R ZnT-2 in both cell lines. In addition, WT ZnT-2 was detected at the cell periphery in HEK-293 cells (Fig. 5, M–O), whereas G87R ZnT-2 accumulated in the ER as indicated by the co-localization with calnexin (Fig. 5, P–R).
FIGURE 5.

Subcellular localization of WT- and G87R ZnT-2. Localization of HA-tagged WT ZnT-2 (A–C) and HA-tagged G87R ZnT-2 (D–F) in HC11 transfectants was examined using confocal microscopy. Alexa 488-conjugated anti-HA antibody (green fluorescence; A and D) was used for the detection of ZnT-2, and anti-P-58 antibody (red fluorescence; B and E) was used to follow Golgi apparatus. Merged images illustrate vesicular localization of WT ZnT-2 (C) or co-localization with Golgi apparatus of G87R ZnT-2 (F). A magnification of ×100 under immersion oil was used. Localization of HA-tagged WT (G–I and M–O) and G87R ZnT-2 (J–L and P–R) in MCF-7 and HEK-293 cells was examined using inverted cell observer microscopy. Detection of ZnT-2 was performed using a mouse anti-HA antibody (green fluorescence; G, J, M, and P), and detection of ER was accomplished using rabbit anti-calnexin antibody (red fluorescence; H, K, N, and Q). Nuclei were stained with DAPI. Merged images illustrate vesicular localization of WT ZnT-2 in MCF-7 cells (I) and plasma membrane localization in HEK-293 cells (O), whereas HA-tagged G87R ZnT-2 showed perinuclear localization in MCF-7 cells (L) and co-localization with ER in HEK-293 cells (R). A magnification of ×63 under immersion oil was used.
Loss of Zinc Transport Function of G87R ZnT-2
To assess the impact of the G87R mutation on its zinc transport activity, several functional assays were performed in HC11 transfectants. We first explored the ZnT-2-dependent accumulation of labile zinc using FluoZin-3 fluorometry (36) (Fig. 6A); whereas overexpression of WT ZnT-2 resulted in ∼1.4-fold higher FluoZin-3 fluorescence compared with mock-transfected cells (p < 0.01), there was no effect of ectopic expression of mutant G87R ZnT-2 on FluoZin-3 fluorescence. We further explored the capacity of G87R ZnT-2 to facilitate zinc secretion by measuring the secretion of 65Zn from transfectant cells (Fig. 6B). Cells overexpressing WT ZnT-2 displayed an ∼1.5-fold greater zinc secretion (p < 0.01) compared with mock-transfected cells or cells expressing G87R ZnT-2; this result was consistent with impaired accumulation of labile zinc pools. We finally assessed the consequences of the decreased labile zinc accumulation and zinc secretion of G87R ZnT-2 transfectants on the cytoplasmic zinc pool by co-transfection of HC11 cells with a 4× MRE pGL3-luciferase reporter plasmid along with G87R ZnT-2, WT ZnT-2, or an empty vector; in this assay a larger cytoplasmic zinc pool is reflected by high chemiluminescence activity, which stems from increased zinc binding to the MRE (Fig. 6C). WT ZnT-2 transfectants, which displayed a significantly higher labile zinc accumulation and zinc secretion compared with mock and G87R ZnT-2 transfectants, consistently showed 2.6-fold lower cytoplasmic zinc pool (p < 0.01). In contrast, G87R ZnT-2 transfectants, which exhibited similar labile zinc accumulation and zinc secretion as compared with mock transfectants, consistently displayed comparable cytoplasmic zinc pool, thereby establishing the loss of G87R ZnT-2-dependent zinc transport.
FIGURE 6.

Establishing loss of function of G87R ZnT-2. A, labile zinc accumulation was quantified after loading of HC11 cells that were transfected with empty vector, WT ZnT-2, or G87R ZnT-2 with FluoZin-3 AM for 1 h. Values represent labile zinc accumulation (mean fluorescence in arbitrary units/μg of protein ± S.D.; n = 8 from three independent experiments). B, zinc secretion of mock, G87R ZnT-2, and WT ZnT-2 HC11 transfectants was measured over 120 min after incubation with 0.1 μm ZnSO4 and 0.1 μCi of 65Zn for 3 h. Values represent zinc secreted from cells (mean pmol/μg of protein ± S.D.; n = 3 from three independent experiments. C, cytoplasmic zinc pool was assessed 24 h after transfection of HC11 cells with 4× MRE pGL3-luciferase reporter plasmid in addition to empty vector, G87R ZnT-2, or WT ZnT-2. After transfection, cells were treated with 1 μm ZnSO4 for 24 h to activate the promoter. Chemiluminescence values were determined using a luminometer (mean relative light units (RLU) determined by the ratio of firefly:renilla luciferase activity ± S.D.; n = 3 from three independent experiments). The asterisks indicate that the loss of function of G87R ZnT-2 was statistically significant as determined by Student's t test (p < 0.01).
Dominant Negative Effect of G87R ZnT-2 over WT ZnT-2
Given the apparent dominant inheritance of the heterozygous G87R mutation, we undertook experiments to determine whether or not G87R ZnT-2 inflicts a dominant negative effect on the WT ZnT-2. We first explored the effect of G87R ZnT-2 on the subcellular localization of WT ZnT-2 in MCF-7 cells. To visualize and quantify the impact of the mutant G87R ZnT-2 on the subcellular localization of WT ZnT-2, co-transfection of MCF-7 cells with equal amounts of expression plasmids harboring HA-ZnT-2 or HA-RFC (50) along with untagged ZnT-2 was performed. ZnT-2 was detected using anti-HA antibody, thus solely following HA-tagged proteins (Fig. 7). A representative immunofluorescence image was captured for each co-transfection (Fig. 7A), and cells were counted for vesicular/plasma membrane localization or perinuclear localization of HA-tagged protein (Fig. 7B). Consistent with the results described above, HA-WT ZnT-2 showed vesicular localization in the majority of transfectant cells (∼91%) when co-transfected with an empty vector or WT ZnT-2 (Fig. 7, A, i-ii, and B). In contrast, any co-transfection with HA-G87R ZnT-2 resulted in perinuclear localization of this protein in almost all cells (∼95%) (Fig. 7, A, iii-v, and B). Importantly, co-transfection of HA-WT ZnT-2 along with G87R ZnT-2 resulted in mislocalization of WT-ZnT-2 and its retention in a perinuclear compartment in the majority of cells (85%) (Fig. 7Avi and B), suggesting a dominant negative effect of mutant G87R ZnT-2 on the subcellular localization of WT ZnT-2 (p < 0.01). In contrast, the subcellular localization of HA-RFC, a folate transporter known to form homooligomers (50), was not altered by co-transfection with either G87R ZnT-2 (Fig. 7Avii), WT ZnT-2 (Fig. 7Aviii), or empty vector (Fig. 7Aix), as nearly all cells (∼97%) showed cell membrane localization and some vesicular localization (Fig. 7B). These results suggest that the putative physical interaction between WT ZnT-2 and mutant G87R ZnT-2 is specific.
FIGURE 7.

Dominant negative effect of G87R ZnT-2 over the subcellular localization of WT ZnT-2. A, MCF-7 cells were transiently co-transfected with equal amounts of DNA (0.5 μg of each construct) of the following plasmids: HA-tagged WT ZnT-2 and empty vector (i), HA-tagged WT ZnT-2 and untagged WT ZnT-2 (ii), HA-tagged G87R ZnT-2 and empty vector (iii), HA-tagged G87R ZnT-2 and untagged G87R ZnT-2 (iv), HA-tagged G87R ZnT-2 and untagged WT ZnT-2 (v), HA-tagged WT ZnT-2 and untagged G87R ZnT-2 (vi), HA-tagged RFC and empty vector (vii), HA-tagged RFC and untagged WT ZnT-2 (viii), HA-tagged RFC and untagged G87R ZnT-2 (ix). HA-tagged proteins were detected using mouse anti-HA antibody (green fluorescence), and nuclei were stained with DAPI (blue fluorescence). The common localization of the HA-tagged protein in co-transfected cell population is illustrated in representative photographs. B, 300 co-transfected cells were analyzed in each of three independent experiments for vesicular or perinuclear localization of HA-tagged protein. Values represent the percent of cells showing vesicular or perinuclear localization of HA-tagged protein out of 300 cells evaluated (mean ± S.D.; n = 1 from three independent experiments). Asterisks indicates that the retention of HA-tagged ZnT-2 in perinuclear compartments due to co-transfection with G87R ZnT-2 is statistically significant as determined by Student's t test (p < 0.01).
We next explored the impact of G87R ZnT-2 on the protein level of WT ZnT-2 using Western blot analysis; the protein level of HA-G87R ZnT-2 was ∼10-fold lower than that of HA-WT ZnT-2 when each one of them was co-transfected with its untagged form (Fig. 8, A and B). The markedly diminished G87R ZnT-2 protein level is possibly a result of the decreased stability and/or enhanced degradation of this misfolded mutant. Moreover, the protein level of WT HA-ZnT-2 was ∼3-fold lower after its co-transfection with mutant G87R ZnT-2 when compared with HA-WT ZnT-2 protein level when co-transfected with its untagged form (Fig. 8, A and B). This dominant negative effect may be explained by the likely destabilization of WT ZnT-2 upon homodimerization with the unstable G87R ZnT-2. Interestingly, the protein levels of HA-WT ZnT-2 were higher when co-transfected with WT ZnT-2, possibly due to homodimerization with the more stable WT ZnT-2 (Fig. 8, A and B). The equal levels of the α-subunit of Na+/K+ ATPase (99 kDa) observed in all treatments confirmed the equal loading of protein samples.
FIGURE 8.

Dominant negative effect of G87R ZnT-2 on the protein expression levels and function of WT ZnT-2. A, HEK-293 and MCF-7 cells were co-transfected with equal amounts (4 μg of each construct) of HA-tagged ZnT-2 together with empty vector or untagged ZnT-2 plasmids. Representative immunoblots from two independent experiments (50 μg of protein/lane) are shown, and HA-tagged ZnT-2 was detected with an anti-HA antibody and displayed a molecular mass of 43 kDa. Detection of the α-subunit of Na+/K+ ATPase was used to confirm equal protein loading. B, the table describes a comparison between HA-tagged ZnT-2 protein level derived from co-transfection with specific untagged ZnT-2 as shown in the representative immunoblot (mean ± 30%S.D.; n = 1 from two independent experiments). C, HC11 cells were transiently transfected with empty pcDNA3.1 plasmid (1.6 μg; 2× empty vector), WT ZnT-2 (1.6 μg; 2× HA-tagged WT ZnT-2), or G87R ZnT-2 (1.6 μg; 2× HA-tagged G87R ZnT-2), or co-transfected with a combination of empty vector and WT ZnT-2, empty vector and G87R ZnT-2, and WT ZnT-2 together with G87R ZnT-2 (0.8 μg; 1× plasmid). 1.2 × 106 Cells were preincubated in serum-free medium containing 0.1 μm ZnSO4 and 0.1 μCi of 65Zn for 3 h. Medium was then replaced, and 65Zn secretion was measured over 120 min. Values represent 65Zn secreted into medium (mean counts/min ± S.D., n = 4, from three independent experiments). Asterisks indicate that the loss of function of WT ZnT-2 upon co-transfection with G87R ZnT-2 was statistically significant as determined by Student's t test (p < 0.05).
Based on the apparent dominant negative effect of G87R ZnT-2 on WT ZnT-2, we explored the impact of G87R ZnT-2 on the zinc transport capacity of WT ZnT-2 (Fig. 8C). HC11 cells co-transfected with equal amounts of expression plasmids harboring WT ZnT-2 and G87R ZnT-2 had significantly decreased zinc secretion (p < 0.01) similar to that obtained with cells transfected with G87R ZnT-2 or the empty vector. This result constitutes the first functional evidence for the dominant negative effect of the mutant G87R ZnT-2 on the zinc transport function of WT ZnT-2.
DISCUSSION
Although multiple cases of TNZD have been reported (13–15), a single genetic basis for this specific disorder has only been recently identified (16). We have previously identified a heterozygous mutation (H54R) in ZnT2/SLC30A2 in two related women in a family that resulted in reduced zinc secretion into milk. Hence, the infants of these mothers harboring the H54R mutation developed TNZD (16). Herein we identified a second heterozygous mutation in ZnT-2 (G87R) in two unrelated Israeli mothers of Ashkenazi Jewish descent. This mutation also resulted in reduced zinc transport in cultured cells and consequently reduced the zinc concentration in mothers milk. As a result, their exclusively breast-fed infants exhibited TNZD. Consistent with our previous report (16), no mutations were found in Zip-4, which plays a key role in intestinal zinc absorption (8, 9). Hence, our current findings provide further evidence that loss-of-function mutations in ZnT-2 result in TNZD, thereby further establishing the critical role that ZnT-2 plays in zinc secretion into the milk.
Several lines of evidence support the conclusion that the G87R mutation in ZnT-2 resulted in loss of zinc transport activity. We found that whereas WT ZnT-2 was properly localized to secretory vesicles, the mutant G87R ZnT-2 transiently transfected into two different mammary epithelial cell lines displayed mislocalization to the Golgi apparatus in HC11 cells and to the perinuclear compartment in MCF-7 cells. Importantly, only a minimal fraction of G87R ZnT-2 was associated with secretory vesicles. Hence, the mislocalization of mutant G87R ZnT-2 strongly suggested that normal zinc transport function would be compromised. Indeed, functional zinc transport assays including labile zinc accumulation, 65Zn secretion, and cytoplasmic zinc pool determination in HC11 transfectants demonstrated that G87R ZnT-2 was impaired in its ability to accumulate or secrete labile zinc. The retention of G87R ZnT-2 in a perinuclear compartment and the consequent marked impairment in zinc transport can possibly be explained by the position of Gly-87 in ZnT-2 as predicted from the putative three-dimensional structure that was based on the crystal structure of the bacterial YiiP homologue (40). In this respect, Gly-87, which is predicted to map to putative TM1, resides in a hydrophobic domain and points toward the hydrophobic core of the lipid bilayer. Substitution of a small uncharged glycine residue by a bulky, positively charged arginine, is likely to disrupt the proper folding of ZnT-2 and, hence, alter its normal subcellular localization. Consequently, transfectants failed to express similar amounts of WT ZnT-2 and G87R ZnT-2 possibly due to decreased protein stability and enhanced degradation of the misfolded mutant G87R ZnT-2. Hence, these results highly support the conclusion that the capacity of G87R ZnT-2 to secrete zinc into milk was impaired.
Determination of the pattern of inheritance and penetrance of inactivating mutations in ZnT-2 that result in TNZD is difficult as this phenotype goes unrecognized unless women exclusively nurse their infants (16). In a previous study we showed that two related women who were heterozygous for the H54R ZnT-2 mutation produced zinc-deficient milk, and their infants consequently developed TNZD. However, the inheritance of the mutation in this family was not fully understood for the reasons delineated above (16). In the United States and Israel, only 13 and 11% of infants (51), respectively, are exclusively nursed for 6 months, which further complicates the understanding of this disorder. In this study, two unrelated women who were heterozygous for the G87R ZnT-2 mutation also produced zinc-deficient milk, and their infants developed TNZD, thus supporting the notion that TNZD is inherited in a dominant manner. Moreover, an independent study of TNZD (52) also supported the possibility of the dominant inheritance as in one affected family three of four related sisters had low breast milk zinc concentration, and their infants developed TNZD. In this study, however, the brother of infant 1, who was exclusively breast-fed by the same mother, developed only moderate zinc deficiency symptoms. Moreover, the two older sisters of infant 2 who were exclusively breast-fed did not develop any TNZD symptoms. Unfortunately, zinc concentration in mother's milk as well as in infants' serum was not determined in these cases. The lack of these clinical data leaves the pattern of inheritance of ZnT-2 mutations complicated and yet unclear. However, this information suggests that mutations in ZnT-2 may have low penetrance, which can be modified by dietary factors such as consumption of infant formula, zinc-containing foods, etc.
Several lines of evidences suggest that the possible dominant inheritance of G87R ZnT-2 mutation is a result of its dominant negative effect over the WT ZnT-2. (a) Immunofluorescence microscopy revealed that expression of the mutant G87R ZnT-2 allele has a dominant negative effect on the subcellular mislocalization of the WT ZnT-2. Specifically, 85% of cells co-transfected with equal amounts of HA-WT ZnT-2 and G87R ZnT-2 showed perinuclear retention of the WT ZnT-2. Moreover, co-transfection of HA-tagged G87R ZnT-2 and WT ZnT-2 preserved the perinuclear retention of G87R ZnT-2. (b) Western blot analysis showed that co-transfection of WT ZnT-2 with G87R ZnT-2 resulted in an ∼3-fold reduction in ZnT-2 protein levels. (c) Co-transfection of WT ZnT-2 and G87R ZnT-2 resulted in a loss of zinc transport function of the WT ZnT-2. This result constitutes the first direct evidence for the dominant negative effect that G87R ZnT-2 inflicts on the zinc transport function of the WT ZnT-2.
This study suggests that the dominant negative effect of the G87R mutation can be explained by the formation of ZnT-2 homodimers as inferred from the following line of evidence. First, the zinc transporters ZnT-3, ZnT-4, and ZnT-8 which are close homologues of ZnT-2, form homodimers or heterodimers (42, 44, 45). Second, there is high evolutionary conservation along the putative interface of the homodimeric ZnT-2 model, which was created based on the crystal structure of the E. coli homodimeric zinc transporter YiiP. Moreover, immunoprecipitation of ZnT-2 illustrated that ZnT-2 forms dimers with a molecular mass that is twice that of the ZnT-2 monomer, thus strongly supporting the formation of ZnT-2 homodimers that enhance stability as revealed by Western blot analysis. Finally, we identified a conserved (F/Y)G(W/Y/F)XRXE signature that resides at the interface of the predicted homodimer. Immunoprecipitation experiments suggest that this motif is involved in ZnT-2 homodimerization, as substitution of its most conserved core residues to alanine resulted in markedly enhanced homodimerization and monomer stabilization.
Although the G87R mutation displayed a dominant negative effect and resulted in the loss of function of WT ZnT-2, the H54R mutation did not exhibit a dominant negative effect (16). Interestingly, however, comparable zinc concentrations were found in milk produced by women heterozygous for G87R mutation and by mothers heterozygous for the H54R mutation (16) (0.26 and 0.23 mg of zinc/liter in women heterozygous for the H54R mutation as well as 0.35 and 0.17 mg/liter in women heterozygous for the G87R mutation). This apparent discrepancy is not fully understood and can possibly be explained by the fact that the zinc transport activity achieved by a single WT ZnT-2 allele is not sufficient for producing milk with normal zinc concentration (i.e. haploinsufficiency) along with the difference in the functionally redundant activities of other zinc transporters such as ZnT-4 or ZnT-1 (18, 53) that may compensate, to some extent, for the loss of ZnT-2 function. Hence, further studies are warranted to shed light on the molecular mechanisms underlying the dominant inheritance of certain ZnT-2 mutations and the dominant negative effect observed with certain ZnT-2 mutations.
In summary, we identified a second heterozygous mutation in ZnT-2 (G87R) that resulted in zinc-deficient milk in two unrelated women with consequent TNZD in their exclusively breast-fed infants. This finding further establishes causative associations between inactivating mutations in ZnT-2 and this specific disorder and significantly expands our understanding of the role of genetic alterations leading to TNZD in exclusively breast-fed infants. Moreover, these results provide the first evidence for a mutation inflicting a dominant negative effect mediated through homodimer formation, thus shedding light on the molecular mechanism underlying the frequent dominant inheritance in TNZD. Moreover, these results imply that not only ZnT-2 is involved in zinc transport to milk but that mutations in ZnT-2 may not be the only defect in mothers of infants with TNZD, hence, expanding the knowledge that genetic variation in zinc transporters that result in TNZD is important for optimal infant growth and development.
Supplementary Material

This article contains supplemental Tables S1 and S2.
- AE
- acrodermatitis enteropathica
- ER
- endoplasmic reticulum
- TNZD
- transient neonatal zinc deficiency
- RFC
- reduced folate carrier
- MRE
- metal-responsive element
- TM
- transmembrane helice.
REFERENCES
- 1. Vallee B. L., Falchuk K. H. (1993) The biochemical basis of zinc physiology. Physiol. Rev. 73, 79–118 [DOI] [PubMed] [Google Scholar]
- 2. MacDonald R. S. (2000) The role of zinc in growth and cell proliferation. J. Nutr. 130, 1500S–1508S [DOI] [PubMed] [Google Scholar]
- 3. Dorea J. G. (2000) Zinc in human milk. Nutr. Res. 20, 1645–1688 [Google Scholar]
- 4. Bhatnagar S., Taneja S. (2001) Zinc and cognitive development. Br. J. Nutr. 85, S139–S145 [DOI] [PubMed] [Google Scholar]
- 5. Dórea J. G. (2002) Zinc deficiency in nursing infants. J. Am. Coll. Nutr. 21, 84–87 [DOI] [PubMed] [Google Scholar]
- 6. Wang K., Zhou B., Kuo Y. M., Zemansky J., Gitschier J. (2002) A novel member of a zinc transporter family is defective in acrodermatitis enteropathica. Am. J. Hum. Genet. 71, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Küry S., Dréno B., Bézieau S., Giraudet S., Kharfi M., Kamoun R., Moisan J. P. (2002) Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat. Genet. 31, 239–240 [DOI] [PubMed] [Google Scholar]
- 8. Aggett P. J. (1983) Acrodermatitis enteropathica. J. Inherit. Metab. Dis. 6, 39–43 [DOI] [PubMed] [Google Scholar]
- 9. Moynahan E. J. (1974) Acrodermatitis enteropathica. A lethal inherited human zinc deficiency disorder. Lancet 2, 399–400 [DOI] [PubMed] [Google Scholar]
- 10. Huang L., Gitschier J. (1997) A novel gene involved in zinc transport is deficient in the lethal milk mouse. Nat. Genet. 17, 292–297 [DOI] [PubMed] [Google Scholar]
- 11. Lee D. Y., Shay N. F., Cousins R. J. (1992) Altered zinc metabolism occurs in murine lethal milk syndrome. J. Nutr. 122, 2233–2238 [DOI] [PubMed] [Google Scholar]
- 12. Piletz J. E., Ganschow R. E. (1978) Zinc deficiency in murine milk underlies expression of the lethal milk (lm) mutation. Science 199, 181–183 [DOI] [PubMed] [Google Scholar]
- 13. Aggett P. J., Atherton D. J., More J., Davey J., Delves H. T., Harries J. T. (1980) Symptomatic zinc deficiency in a breast-fed preterm infant. Arch. Dis. Child. 55, 547–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parker P. H., Helinek G. L., Meneely R. L., Stroop S., Ghishan F. K., Greene H. L. (1982) Zinc deficiency in a premature infant fed exclusively human milk. Am. J. Dis. Child. 136, 77–78 [DOI] [PubMed] [Google Scholar]
- 15. Zimmerman A. W., Hambidge K. M., Lepow M. L., Greenberg R. D., Stover M. L., Casey C. E. (1982) Acrodermatitis in breast-fed premature infants. Evidence for a defect of mammary zinc secretion. Pediatrics 69, 176–183 [PubMed] [Google Scholar]
- 16. Chowanadisai W., Lönnerdal B., Kelleher S. L. (2006) Identification of a mutation in SLC30A2 (ZnT-2) in women with low milk zinc concentration that results in transient neonatal zinc deficiency. J. Biol. Chem. 281, 39699–39707 [DOI] [PubMed] [Google Scholar]
- 17. Kelleher S. L., Lönnerdal B. (2005) Zip3 plays a major role in zinc uptake into mammary epithelial cells and is regulated by prolactin. Am. J. Physiol. Cell Physiol. 288, C1042–C1047 [DOI] [PubMed] [Google Scholar]
- 18. Kelleher S. L., Lönnerdal B. (2003) Zinc transporter levels and localization change throughout lactation in rat mammary gland and are regulated by zinc in mammary cells. J. Nutr. 133, 3378–3385 [DOI] [PubMed] [Google Scholar]
- 19. Michalczyk A., Varigos G., Catto-Smith A., Blomeley R. C., Ackland M. L. (2003) Analysis of zinc transporter, hZnT4 (Slc30A4), gene expression in a mammary gland disorder leading to reduced zinc secretion into milk. Hum. Genet. 113, 202–210 [DOI] [PubMed] [Google Scholar]
- 20. Erway L. C., Grider A., Jr. (1984) Zinc metabolism in lethal-milk mice. Otolith, lactation, and aging effects. J. Hered. 75, 480–484 [DOI] [PubMed] [Google Scholar]
- 21. Bairoch A., Apweiler R., Wu C. H., Barker W. C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Martin M. J., Natale D. A., O'Donovan C., Redaschi N., Yeh L. S. (2005) The Universal Protein Resource (UniProt). Nucleic Acids Res. 33, D154–D159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 23. Biegert A., Mayer C., Remmert M., Söding J., Lupas A. N. (2006) The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 34, W335–W339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Katoh K., Misawa K., Kuma K., Miyata T. (2002) MAFFT. A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Söding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, 244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Punta M., Forrest L. R., Bigelow H., Kernytsky A., Liu J., Rost B. (2007) Membrane protein prediction methods. Methods 41, 460–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2007) Comparative protein structure modeling with MODELLER. Curr. Protoc. Protein Sci. Chapter 2, Unit 2.9 [DOI] [PubMed] [Google Scholar]
- 28. Kalman M., Ben-Tal N. (2010) Quality assessment of protein model structures using evolutionary conservation. Bioinformatics 26, 1299–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., Ben-Tal N. (2005) ConSurf 2005. The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 31. Henikoff S., Henikoff J. G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89, 10915–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clamp M., Cuff J., Searle S. M., Barton G. J. (2004) The Jalview Java alignment editor. Bioinformatics 20, 426–427 [DOI] [PubMed] [Google Scholar]
- 33. Pettersen E. F., Goddard T. D., Huang C. C. (2004) UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 34. Ifergan I., Shafran A., Jansen G., Hooijberg J. H., Scheffer G. L., Assaraf Y. G. (2004) Folate deprivation results in the loss of breast cancer resistance protein (ABCG2) expression. A role for BCRP in cellular folate homeostasis. J. Biol. Chem. 279, 25527–25534 [DOI] [PubMed] [Google Scholar]
- 35. Rothem L., Berman B., Stark M., Jansen G., Assaraf Y. G. (2005) The reduced folate carrier gene is a novel selectable marker for recombinant protein overexpression. Mol. Pharmacol. 68, 616–624 [DOI] [PubMed] [Google Scholar]
- 36. Seo Y. A., Kelleher S. L. (2010) Functional analysis of two single nucleotide polymorphisms in SLC30A2 (ZnT2): implications for mammary gland function and breast disease in women. Physiol. Genomics 42, 219–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bolte S., Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 38. Krebs N. F., Reidinger C. J., Hartley S., Robertson A. D., Hambidge K. M. (1995) Zinc supplementation during lactation. Effects on maternal status and milk zinc concentrations. Am. J. Clin. Nutr. 61, 1030–1036 [DOI] [PubMed] [Google Scholar]
- 39. Kasperek K., Feinendegen L. E., Lombeck I., Bremer H. J. (1977) Serum zinc concentration during childhood. Eur. J. Pediatr. 126, 199–202 [DOI] [PubMed] [Google Scholar]
- 40. Lu M., Chai J., Fu D. (2009) Structural basis for autoregulation of the zinc transporter YiiP. Nat. Struct. Mol. Biol. 16, 1063–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schushan M., Ben-Tal N. (2010) in Introduction to Protein Structure Prediction: Methods and Algorithms (Rangwala H., Karypis G., eds) pp. 369–401, John Wiley & Sons, Inc., Hoboken, NJ [Google Scholar]
- 42. Salazar G., Falcon-Perez J. M., Harrison R., Faundez V. (2009) SLC30A3 (ZnT3) oligomerization by dityrosine bonds regulates its subcellular localization and metal transport capacity. PLoS ONE 4, e5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weijers R. N. (2010) Three-dimensional structure of β-cell-specific zinc transporter, ZnT-8, predicted from the type 2 diabetes-associated gene variant SLC30A8 R325W. Diabetol. Metab. Syndr. 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fukunaka A., Suzuki T., Kurokawa Y., Yamazaki T., Fujiwara N., Ishihara K., Migaki H., Okumura K., Masuda S., Yamaguchi-Iwai Y., Nagao M., Kambe T. (2009) Demonstration and characterization of the heterodimerization of ZnT5 and ZnT6 in the early secretory pathway. J. Biol. Chem. 284, 30798–30806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murgia C., Devirgiliis C., Mancini E., Donadel G., Zalewski P., Perozzi G. (2009) Diabetes-linked zinc transporter ZnT8 is a homodimeric protein expressed by distinct rodent endocrine cell types in the pancreas and other glands. Nutr. Metab. Cardiovasc. Dis. 19, 431–439 [DOI] [PubMed] [Google Scholar]
- 46. Moreira I. S., Fernandes P. A., Ramos M. J. (2007) Hot spots. A review of the protein-protein interface determinant amino acid residues. Proteins 68, 803–812 [DOI] [PubMed] [Google Scholar]
- 47. Ebie Tan A., Fleming K. G. (2008) Outer membrane phospholipase a dimer stability does not correlate to occluded surface area. Biochemistry 47, 12095–12103 [DOI] [PubMed] [Google Scholar]
- 48. Baljuls A., Mahr R., Schwarzenau I., Müller T., Polzien L., Hekman M., Rapp U. R. (2011) Single substitution within the RKTR motif impairs kinase activity but promotes dimerization of RAF kinase. J. Biol. Chem. 286, 16491–16503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Doura A. K., Kobus F. J., Dubrovsky L., Hibbard E., Fleming K. G. (2004) Sequence context modulates the stability of a GXXXG-mediated transmembrane helix-helix dimer. J. Mol. Biol. 341, 991–998 [DOI] [PubMed] [Google Scholar]
- 50. Hou Z., Matherly L. H. (2009) Oligomeric structure of the human reduced folate carrier. Identification of homo-oligomers and dominant-negative effects on carrier expression and function. J. Biol. Chem. 284, 3285–3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Berger-Achituv S., Shohat T., Garty B. Z. (2005) Breast-feeding patterns in central Israel. Isr. Med. Assoc. J. 7, 515–519 [PubMed] [Google Scholar]
- 52. Matsumoto G., Stojanovic A., Holmberg C. I., Kim S., Morimoto R. I. (2005) Structural properties and neuronal toxicity of amyotrophic lateral sclerosis-associated copper/zinc superoxide dismutase 1 aggregates. J. Cell Biol. 171, 75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Michalczyk A. A., Allen J., Blomeley R. C., Ackland M. L. (2002) Constitutive expression of hZnT4 zinc transporter in human breast epithelial cells. Biochem. J. 364, 105–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.