

Background: Polyglutamine aggregates can be internalized by mammalian cells and gain access to the cytoplasmic compartment, but the properties of the aggregates and cell surface that mediate these processes are unknown.
Results: Introduction of net negative charge and disruption of fibrillar structure greatly reduced the capacity of polyglutamine aggregates to bind and be internalized by mammalian cells.
Conclusion: Aggregate uptake is influenced by the structure and net charge of aggregates and is mediated by two classes of binding sites on the cell surface.
Significance: Elucidating how protein aggregates are internalized by cells is important for understanding the pathogenesis of many neurodegenerative disorders.
Keywords: Amyloid, Huntington Disease, Polyglutamine, Prions, Protein Aggregation, Huntingtin
Abstract
The pathogenesis of most neurodegenerative diseases, including transmissible diseases like prion encephalopathy, inherited disorders like Huntington disease, and sporadic diseases like Alzheimer and Parkinson diseases, is intimately linked to the formation of fibrillar protein aggregates. It is becoming increasingly appreciated that prion-like intercellular transmission of protein aggregates can contribute to the stereotypical spread of disease pathology within the brain, but the mechanisms underlying the binding and uptake of protein aggregates by mammalian cells are largely uninvestigated. We have investigated the properties of polyglutamine (polyQ) aggregates that endow them with the ability to bind to mammalian cells in culture and the properties of the cell surface that facilitate such uptake. Binding and internalization of polyQ aggregates are common features of mammalian cells and depend upon both trypsin-sensitive and trypsin-resistant saturable sites on the cell surface, suggesting the involvement of cell surface proteins in this process. polyQ aggregate binding depends upon the presence of a fibrillar amyloid-like structure and does not depend upon electrostatic interaction of fibrils with the cell surface. Sequences in the huntingtin protein that flank the amyloid-forming polyQ tract also influence the extent to which aggregates are able to bind to cell surfaces.
Introduction
Aggregation of proteins into fibrillar organized megadalton assemblies underlies the pathogenesis of a large number of human diseases, including most neurodegenerative disorders and peripheral diseases such as type 2 diabetes and systemic amyloidoses (1–4). The ability of these non-native protein polymers to act as seeds that nucleate the incorporation of monomeric or oligomeric forms of proteins or peptides containing the same amino acid sequences (i.e. homotypic polymers) underlies the self-replication associated with prions (5) and is increasingly recognized as a potential mechanism for the stereotypical spread of neurodegenerative pathology within the brain (6). For example, migration of aggregated forms of the scrapie prion protein PrPSc from the intestine to the brain underlies its infectivity, and intercellular movement of PrPSc within the brain is required for prion replication and spread of spongiform pathology (7, 8). It is becoming increasingly appreciated that both extracellular (e.g. amyloid β) and intracellular (α-synuclein and tau) protein amyloids are able to move and possibly replicate within the brains of individuals with Alzheimer and Parkinson diseases, contributing thereby to the spread of pathology in a prion-like manner (6, 9–11). In order for such spread to be transmitted by aggregated forms of normally cytoplasmic proteins, the pathogenic proteins must be released from and taken up by cells. However, despite the rapidly accumulating evidence supporting a role for intercellular transmission of protein aggregates in pathological spread in animal and cell culture disease models, little is known about the unique properties of these pathogenic protein aggregates that endow them with the ability to interact with membranes or the properties of cell membranes that support binding to and internalization of protein aggregates.
Huntington disease belongs to a family of dominantly inherited neurodegenerative diseases that are caused by expansion of CAG tracts within protein-coding regions of genes (12). In Huntington disease, the mutant gene encodes a variant of the Htt (huntingtin) protein containing a homopolymeric tract of polyglutamine (polyQ)3 in excess of the pathogenic threshold of ∼35 glutamines (13). The N-terminal fragment of Htt containing the expanded polyQ tract (Httexon1Qn) is sufficient to cause neurological disease in primate, rodent, and invertebrate models of Huntington disease (14). Additionally, recombinant protein fragments (Httexon1Q>35) (15) or synthetic peptides containing >35 glutamines (16) spontaneously assemble into highly insoluble fibrillar β-sheet rich polymers resembling those that are deposited into intraneuronal inclusion bodies in Huntington disease. The kinetics of assembly of these polyQ-containing peptides or protein fragments in vitro into polymeric assemblies exhibit classical nucleation behavior in which the presence of a “seed” of preformed fibrils reduces the thermodynamic barrier required for polymerization of proteins or protein fragments containing polyQ tracts shorter than the pathogenic threshold (e.g. Httexon1Q<35) (17).
It was previously reported that mammalian cells can internalize large fibrillar assemblies formed from synthetic Q44 peptides (K2Q44K2) (17). Subsequent work from our laboratory demonstrated that these fibrils are able to interact with and nucleate specific homotypic aggregation of cytoplasmic fluorescent protein “reporters” containing Q25 tracts that are too short to form stable assemblies alone (18). These findings raise important questions as to the mechanism by which protein aggregates enter cells. In this study, we have investigated the properties of aggregates, including the role of net charge and fibrillar organization, and the properties of the cell surface required for internalization of polyQ aggregates.
EXPERIMENTAL PROCEDURES
Aggregate Preparation and Labeling
Synthetic polyQ peptides were assembled into fibrils as described previously (16, 17). Amorphous assemblies were obtained by solubilizing the synthetic peptides in 50% TFA and 50% hexafluoroisopropyl alcohol (4 mm), followed by a 40-fold dilution into distilled water at room temperature. K2Q44K2 and D2Q44D2 assembly was monitored turbidimetrically at 350 nm on a Hewlett-Packard 8453 diode array spectrophotometer using 1-cm path length cuvettes.
Recombinant His-tagged maltose-binding protein-tobacco etch virus-Httexon1Qn and maltose-binding protein-tobacco etch virus-Qn were purified from Escherichia coli with amylose resin and eluted with maltose. The maltose-binding protein fusion was cleaved with tobacco etch virus protease at a mass ratio of 5:1 for 1 h at 37 °C; bound to TALON resin (Clontech) at 4 °C in 20 mm Tris-HCl (pH 7.5), 200 mm NaCl, and 10 mm imidazole; and eluted in 20 mm Tris-HCl (pH 7.5), 200 mm NaCl, and 200 mm imidazole. The eluate containing the Httexon1Qn or Qn protein was filtered through a 0.2-μm filter, flash-frozen in liquid nitrogen, and stored at −80 °C. To assemble the fibrils, the recombinant proteins were rapidly thawed at 37 °C and incubated at 37 °C until steady-state fibrillization had been reached. For labeling, the fibrils were pelleted by centrifugation, washed with 50 mm HEPES (pH 7.5), labeled with Alexa Fluor dyes (Invitrogen) according to the manufacturer's protocol, and washed with 50 mm HEPES (pH 7.5).
Aggregate Acetylation
FITC-K2Q44K2 (1 mg) was dissolved in hexafluoroisopropyl alcohol. Following evaporation under a nitrogen stream in a 5-ml glass tube, the peptide was dissolved in 1 ml of 0.3 m HEPES, and the pH adjusted to 8.5 using NaOH. The glass tube was transferred to an ice bath, and 50 μl of acetic anhydride was added in 10-μl aliquots at 10-min intervals with constant stirring. The solution was then frozen in dry ice and lyophilized. The pellet was washed twice with ethanol, dried, and dissolved in 50% TFA and 50% hexafluoroisopropyl alcohol.
Fibrillar acetylated FITC-K2Q44K2 assemblies were generated as described above following evaporation of the solvents. The extent of acetylation was assessed by MALDI-TOF-MS measurements on a Bruker OmniFLEX-NT system operated in a reflectron mode using sinapinic acid as a matrix. Based on the mass increase due to acetylation (42 Da), on average, three lysines within K2Q44K2 were found acetylated.
Cell Culture
HeLa, HEK, Neuro-2a, U2OS, CHO, mouse embryonic fibroblast, and COS-7 cells were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin. AtT-20α5 cells (a kind gift from J. Schwarzbauer, Princeton University) (3) were grown in DMEM/nutrient mixture F-12 and HEPES (Invitrogen) supplemented with 10% FBS, 10% horse serum, 4 mm l-glutamine, and 0.25 mg/ml G418. Jurkat cells were grown in RPMI 1640 medium supplemented with 10% FBS and penicillin/streptomycin.
Electron Microscopy
Transmission electron micrographs were acquired as described previously (18). Briefly, peptide or protein aggregates were placed on carbon/Formvar-coated copper grids (Electron Microscopy Sciences) and stained with 1% uranyl acetate in distilled water for 1 min. The grids were analyzed at the Cell Sciences Imaging Facility at Stanford University using a JEOL 1230 electron microscope at an excitation voltage of 80 kV.
Flow Cytometry
Adherent cells were detached with trypsin or EDTA as indicated and resuspended in PBS at room temperature unless indicated otherwise. Fluorescently labeled aggregates were added to cells and analyzed at the indicated time points using a FACSCalibur flow cytometer (BD Biosciences). The cell population was gated by forward and side scatter, and at least 10,000 cells per condition were recorded. The geometric mean of the gated population was determined using FlowJo software (TreeStar Inc).
Binding experiments were done after incubation of cells with fibrils for 1–10 min on ice, which represented equilibrium binding conditions. For experiments comparing Httexon1Q44 and Q44, a longer time course was used at 37 °C to observe the reduced binding of Httexon1Q44 fibrils.
Competition Experiments
Recombinant polyQ fibrils labeled with Alexa Fluor 488 were diluted in PBS to 2 μm in a microcentrifuge tube on ice. Unlabeled polyQ fibrils were diluted to 20, 50, and 100 μm in PBS in a microcentrifuge tube on ice. ∼2 × 105 COS-7 cells were pelleted by centrifugation after lifting either EDTA (10 mm in PBS) or trypsin (0.5 mg/ml in PBS) and washing with PBS. Labeled and unlabeled fibrils were mixed immediately before addition to cells, and cells plus fibrils were incubated on ice for ∼5 min, which represented equilibrium binding conditions.
Nucleation Experiments
Nucleation experiments were performed essentially as described (18). In summary, HEK cells stably expressing mCherry fluorescent protein (chFP)-Httexon1Q25 were grown on lysine-coated glass coverslips and treated with synthetic FITC-labeled polyQ or Alexa Fluor 488-labeled recombinant Httexon1Q44 and Q44 fibrils or amorphous K2Q44K2 assemblies at 37 °C in DMEM/FBS. At the indicated times, the coverslips were removed and washed with PBS, followed by fixation with 4% paraformaldehyde (Electron Microscopy Sciences or Sigma).
HeLa cells stably expressing chFP-Httexon1Q25 were grown in Lab-Tek 4-well chambered slides (Nunc) at 37 °C in DMEM/FBS. At the indicated times, the cells were washed with PBS and fixed with 4% paraformaldehyde. Cells were permeabilized with 0.1% Triton X-100 and blocked for 1 h at room temperature with 1% BSA in PBS. Subsequently, they were immunostained with Living Colors anti-DsRed polyclonal antibody (Clontech) at a dilution of 1:400 in blocking buffer and with anti-rabbit IgG conjugated to Alexa Fluor 594 (Invitrogen) at a dilution of 1:400. Cells were imaged on a Zeiss Axiovert 200M microscope, and images were processed with NIH ImageJ software.
RESULTS
Cell Surface Properties That Contribute to Fibril Binding
To assess whether the ability to bind fibrillar protein aggregates is a general property of mammalian cells, we incubated nine different cell lines with fibrils prepared from FITC-labeled synthetic K2Q44K2 peptides (Fig. 1A), generated as described previously (18). Each cell line bound a characteristic amount of the fibrils, indicating that the capacity to interact with polyQ fibrils is a general property of mammalian cells, although non-adherent cells (AtT-20 and Jurkat) bound distinctly less. The reduced binding capacity of these non-adherent cells was not directly due to a deficiency in the adhesion protein fibronectin (Fn), as Fn−/− mouse embryonic fibroblasts (19) bound similar amounts of K2Q44K2 fibrils compared with Fn+/− mouse embryonic fibroblasts (supplemental Fig. S1, A and E), and binding of aggregates to AtT-20 cells, which are intrinsically deficient in Fn (20, 21), was not enhanced by plating the cells on Fn-coated culture dishes (supplemental Fig. S1, B and E). The reduced aggregate-binding capacity of these non-adherent cells was also not due to the absence of the Fn receptor, as overexpression of either α5 integrin (supplemental Fig. S1, C and F) or β1 integrin (supplemental Fig. S1D) did not enhance the capacity of integrin-deficient cells to bind K2Q44K2 fibrils. Thus, the differing capacity of mammalian cells to bind fibrillar cationic polyQ aggregates is unrelated to interactions between the extracellular matrix protein Fn and its cognate receptor.
FIGURE 1.

Binding of synthetic polyQ fibrils to cells. A, binding of K2Q44K2 fibrils (1 μm) to mammalian cells. B, binding of K2Q44K2 fibrils (1 μm) to HEK cells at 25 and 4 °C. Error bars indicate S.E. AFU, arbitrary fluorescence units.
At room temperature, binding of K2Q44K2 fibrils to HEK cells was rapid, with near maximal binding observed at 1 min. At 4 °C, binding was slightly slower but reached comparable levels by 2.5 min (Fig. 1B). All subsequent experiments were performed at room temperature.
K2Q44K2 fibril binding was strongly reduced by treatment of cells with trypsin (Fig. 2A), suggesting that cell surface proteins contribute to aggregate-binding sites. In addition to our observations in HEK cells, we also observed a strong reduction in binding with trypsin treatment in COS-7 and HeLa cells (data not shown). Fibril binding was also reduced by inclusion of 1% BSA, either when added to cells together with the fibrils (Fig. 2A) or by pretreatment of cells (data not shown), indicating that low affinity nonspecific sites on the cell surface contribute to aggregate binding. Increasing the concentration of BSA beyond 5% did not further reduce binding of K2Q44K2 fibrils (data not shown). The effect of BSA was not additive with that of trypsin (Fig. 2A), suggesting that the proteinaceous sites removed by trypsin are unlikely to constitute a unique population of high affinity fibril-binding sites. Trypsin-sensitive binding of fibrils appeared to saturate at K2Q44K2 concentrations above 50 μm, whereas total binding exhibited apparent saturation at concentrations above 100 μm (Fig. 2B), supporting the conclusion that aggregate binding to HEK cells is dominated by proteinaceous sites. Unlabeled fibrils competed for binding to trypsin-sensitive sites with an apparent IC50 of ∼30 μm, representing an unlabeled/labeled ratio of ∼15:1 (Fig. 2C). Together, these data indicate that cell surface proteins contribute significantly to binding of K2Q44K2 fibrillar aggregates to cells.
FIGURE 2.

Effect of BSA and trypsin on polyQ fibril binding. A, binding of K2Q44K2 fibrils (1 μm) to HEK cells after treatment of cells with 1% BSA or trypsin. B, saturable binding of K2Q44K2 fibrils to HEK cells lifted with EDTA or trypsin. C, competition for binding of Q44 fibrils to COS-7 cells lifted with EDTA or trypsin. Error bars indicate S.E. for A and C, whereas B is the result of a single experiment (n = 1). AFU, arbitrary fluorescence units.
Effect of Net Charge on polyQ Fibril Binding and Internalization
To assess the effect of electrostatic interactions on fibril binding, we prepared aggregates from synthetic Q44 peptides flanked by strongly cationic (K2Q44K2), neutral (acetylated lysine; Ac-K2Q44K2), or anionic (aspartate; D2Q44D2) amino acids. Examination of these fibrils by negative stain transmission electron microscopy did not reveal significant differences in either the dimensions or morphology of the fibrils (Fig. 3A). The neutral Ac-K2Q44K2 fibrils bound to HEK cells to the same extent as the cationic K2Q44K2 fibrils (Fig. 3B), suggesting that electrostatic attraction does not contribute to a significant extent to fibril binding. However, binding of negatively charged D2Q44D2 fibrils was significantly reduced compared with that of positive or neutral fibrils in both HEK cells (Fig. 3B) and COS-7 cells (data not shown). D2Q44D2 fibril binding was reduced by ∼80% by BSA, similar to the behavior of K2Q44K2 fibrils (Fig. 3C). However, no significant effect on D2Q44D2 fibril binding was observed upon treatment of cells with trypsin (Fig. 3C). These data suggest that, although electrostatic interactions do not contribute to cell surface binding of polyQ fibrils, binding can be disrupted by repulsion between negatively charged aggregates and the net negative charge of the cell surface.
FIGURE 3.

Effect of charge on polyQ fibril binding. A, transmission electron micrographs of charged and neutral synthetic Q44 fibrils. Scale bars = 0.5 μm. B, binding of charged and neutral Q44 fibrils(1 μm) to HEK cells. C, binding of negatively charged D2Q44D2 fibrils (1 μm) to HEK cells treated with 1% BSA or trypsin. Cell-associated fluorescence was normalized to the 0% BSA/EDTA sample. Error bars indicate S.E. AFU, arbitrary fluorescence units.
To determine the extent to which fibril charge affects cytoplasmic entry, we treated cells expressing soluble mCherry-labeled Htt exon 1 with 25 glutamines (chFP-Httexon1Q25) with neutral or negatively charged FITC-labeled Q44 fibrils (Fig. 4). Puncta containing both green and red fluorophores were observed as early as 1 h following aggregate addition. All three (i.e. with positive, neutral, and negative net charges) types of fibrils were competent to nucleate the cytoplasmic chFP-Httexon1Q25 reporter (Fig. 4A), and consistent with the binding behavior, the neutral and positively charged fibrils induced formation of puncta with similar time courses, whereas the negatively fibrils nucleated cytoplasmic aggregation more slowly, ultimately reaching similar levels by 17 h (Fig. 4B). These data suggest that fibrillar species are able to enter cells and nucleate cytoplasmic aggregation irrespective of charge.
FIGURE 4.
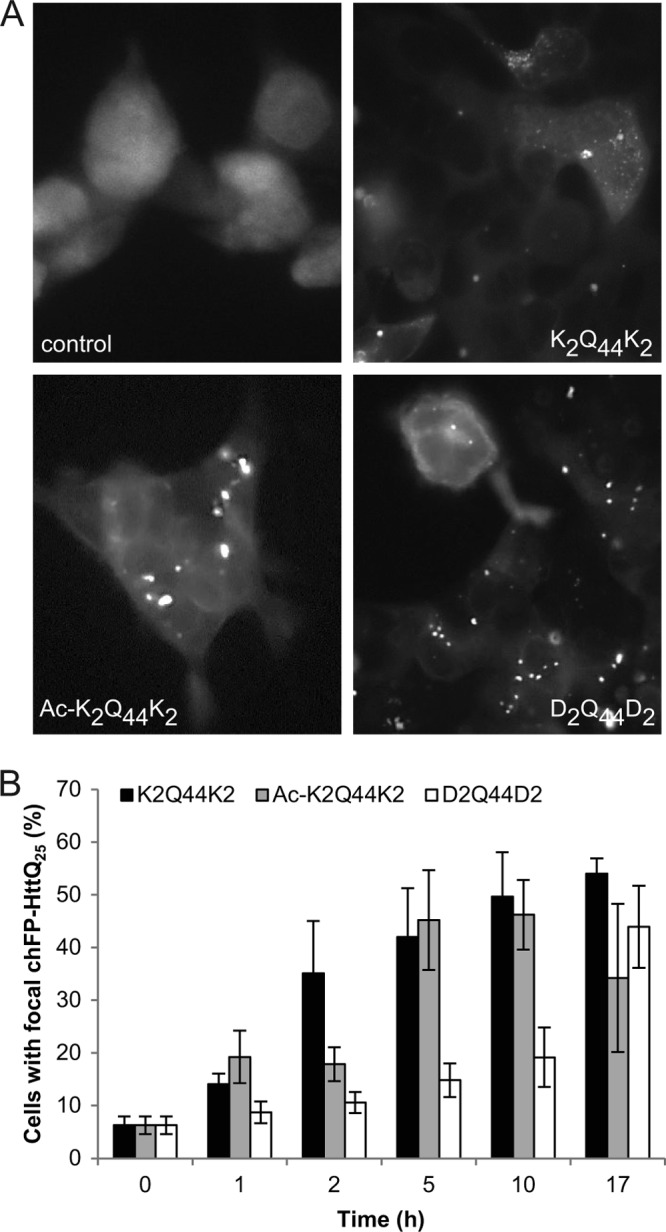
Effect of charge on polyQ fibril internalization. A, fluorescence micrographs of HEK cells expressing chFP-Httexon1Q25 and treated with charged and neutral synthetic Q44 fibrils for 16 h at 37 °C. B, quantification of HEK cells with focal chFP-Httexon1Q25 after treatment with synthetic Q44 fibrils for the indicated times. Error bars indicate S.E.
To assess the contribution of the protein context of fibrillar polyQ to its ability to bind and enter cells, we generated fibrils from bacterially expressed recombinant Htt exon 1 (Httexon1Q44). Although these fibrils were similar in overall shape (Fig. 5A) and length (supplemental Fig. S2) to recombinant Q44 aggregates, they were greatly reduced in the ability to interact with cells (Fig. 5B), although the small amounts that bound did so with similar kinetics to aggregates prepared from pure Q44 (Fig. 5C). Some morphological differences were observed between these fibrils and fibrils obtained from synthetic polyQ peptides. This is likely due to the effect of sonication, which was performed prior to transmission electron microscopy of recombinant (but not synthetic) polyQ fibrils. Indeed, when synthetic polyQ fibrils were sonicated, smaller fibrils were observed that more closely resembled recombinant polyQ and Httexon1Q44 fibrils. Both Httexon1Q44 and Q44 fibrils were internalized into HeLa cells and nucleated cytoplasmic chFP-Httexon1Q25; however, consistent with the binding data, the amount of internalization was drastically reduced with Httexon1Q44 fibrils (Fig. 5, D and E). These data demonstrate that recognition of the polyQ structure on amyloid fibrils by the cell surface can be strongly influenced by flanking sequences that distinguish Htt exon 1 from pure polyQ.
FIGURE 5.

Effect of flanking protein sequences on polyQ fibril binding and internalization. A, transmission electron micrographs of recombinant Httexon1Q44 and Q44 fibrils. B, comparison of binding of Httexon1Q44 and Q44 fibrils to COS-7 cells after 24 h of treatment at 37 °C. C, time-dependent binding of both Httexon1Q44 and Q44 fibrils to COS-7 cells. D, quantification of HeLa cells with focal chFP-Httexon1Q25 after treatment with Httexon1Q44 and Q44 fibrils for the indicated times. E, fluorescence micrographs of HeLa cells expressing chFP-Httexon1Q25 and treated with Httexon1Q44 and Q44 fibrils for 24 h. Error bars indicate S.E. AFU, arbitrary fluorescence units.
To determine whether the fibrillar state is a requisite for binding of polyQ aggregates, we generated non-fibrillar (amorphous) K2Q44K2 aggregates (Fig. 6A). CD spectroscopy showed that fibrillar assemblies had high β-sheet content, whereas amorphous aggregates were composed primarily of α-helix and random coil (supplemental Fig. S3). COS-7 cells bound amorphous K2Q44K2 aggregates to a far lesser extent than their fibrillar counterparts (Fig. 6B), suggesting that the organized β-sheet-rich assemblies are essential for the binding of polyQ aggregates. Furthermore, consistent with the reduced binding of amorphous K2Q44K2 aggregates to COS-7 cells, reduced internalization and nucleation of cytoplasmic chFP-Httexon1Q25 in HEK reporter cells were observed (Fig. 6C), suggesting that fibrillar β-sheet-rich assemblies nucleate soluble Httexon1Q25 more efficiently.
FIGURE 6.

Effect of structure on polyQ fibril binding and internalization. A, transmission electron micrographs of fibrillar and amorphous K2Q44K2 aggregates. B, binding of fibrillar and amorphous K2Q44K2 aggregates to COS-7 cells (p = 6 × 10−5). C, quantification of HEK cells with focal chFP-Httexon1Q25 after treatment with fibrillar and amorphous K2Q44K2 aggregates for 16 h (p = 0.002). Error bars indicate S.E.
DISCUSSION
It is becoming increasingly evident that many neurodegenerative diseases, including Alzheimer and Parkinson diseases, spread through the brain by a non-cell autonomous process that appears to share key features with prion infectivity (6, 9–11). Prions are composed of fibrillar amyloid aggregates rich in β-sheet structure that are able to act as seeds or templates to grow by monomer addition (5, 7). Intracellular protein aggregates, such as those composed of α-synuclein, tau, and Htt, are linked to diseases not traditionally associated with prions yet nonetheless share similar biophysical properties that underlie prion infectivity. This raises the possibility that their spread within the brain could account for some of the non-cell autonomous properties associated with disease pathogenesis. For this mechanism to contribute to disease progression, aggregate seeds that are released from cells must be able to enter neighboring or synaptically connected cells and gain access to the cytoplasm, where they can nucleate the aggregation of endogenous proteins that share a common aggregation-prone sequence. In this study, we have begun to define the properties by which mammalian cells are able to internalize fibrillar polyQ protein aggregates. Our data establish that binding of polyQ aggregates is a property common to all mammalian cell types examined and is mediated primarily by saturable proteinaceous sites on the cell surface.
The finding that aggregate binding can be strongly diminished by pretreatment of the cell surface with BSA suggests that a substantial fraction of aggregate binding is mediated by nonspecific sites on the cell surface. That the effect of BSA is not additive with that of trypsin suggests that the sites blocked by BSA are proteinaceous and are likely to include the trypsin-sensitive sites that are saturable and can be out-competed by unlabeled fibrils. Our data indicate that binding is not dominated by electrostatic attraction but can be diminished by charge repulsion between negatively charged aggregates and the negatively charged cell surface.
The dramatic difference in binding observed between fibrillar and amorphous polyQ aggregates suggests that the primary surface of interaction of aggregates with the cell surface is mediated in some way by the regular repeating structure of the polyQ amyloid. Furthermore, sequences that flank the polyQ amyloid structure, such as the N and C termini of Htt exon 1, can dramatically interfere with the binding of the polyQ amyloid. The polyQ tract in the context of Htt exon 1 is flanked by a moderately hydrophobic 17-amino acid N-terminal sequence that adopts an α-helical conformation in fibrils (22) and a C-terminal proline-rich domain of indeterminate structure. The N-terminal segment strongly influences the kinetics of polyQ aggregation (22, 23) but not the β-sheet-rich amyloid structure of the neighboring polyQ region (22). Further studies will be required to determine the extent to which the reduced binding capacity observed for the Htt construct is due to the steric or screening effect of the N- or C-terminal extensions.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant NS042842 (to R. R. K.) and Predoctoral Training Grant GM007276 (to R. S. T.). This work was also supported by Agence Nationale de la Recherche Grant ANR-09-MNPS-013-01 and a Coup d'Élan à la Recherche Française award from the Bettencourt Schueller Foundation (to R. M.), the Human Frontiers Science Program (to R. R. K. and R. M.), and a postdoctoral fellowship from the Hereditary Disease Foundation (to J. E. L.).

This article contains supplemental “Experimental Procedures,” Figs. S1–S3, and additional references.
- polyQ
- polyglutamine
- chFP
- mCherry fluorescent protein
- Fn
- fibronectin.
REFERENCES
- 1. Carrell R. W., Lomas D. A. (1997) Conformational disease. Lancet 350, 134–138 [DOI] [PubMed] [Google Scholar]
- 2. Koo E. H., Lansbury P. T., Jr., Kelly J. W. (1999) Amyloid diseases: abnormal protein aggregation in neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 96, 9989–9990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ross C. A., Poirier M. A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17 [DOI] [PubMed] [Google Scholar]
- 4. Stefani M., Dobson C. M. (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases, and biological evolution. J. Mol. Med. 81, 678–699 [DOI] [PubMed] [Google Scholar]
- 5. Caughey B., Lansbury P. T. (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298 [DOI] [PubMed] [Google Scholar]
- 6. Brundin P., Melki R., Kopito R. (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prusiner S. B. (1994) Biology and genetics of prion diseases. Annu. Rev. Microbiol. 48, 655–686 [DOI] [PubMed] [Google Scholar]
- 8. Aguzzi A., Calella A. M. (2009) Prions: protein aggregation and infectious diseases. Physiol. Rev. 89, 1105–1152 [DOI] [PubMed] [Google Scholar]
- 9. Jucker M., Walker L. C. (2011) Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 70, 532–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aguzzi A., Rajendran L. (2009) The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64, 783–790 [DOI] [PubMed] [Google Scholar]
- 11. Soto C., Estrada L., Castilla J. (2006) Amyloids, prions, and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci. 31, 150–155 [DOI] [PubMed] [Google Scholar]
- 12. Paulson H. L., Bonini N. M., Roth K. A. (2000) Polyglutamine disease and neuronal cell death. Proc. Natl. Acad. Sci. U.S.A. 97, 12957–12958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Finkbeiner S. (2011) Huntington disease. Cold Spring Harb. Perspect. Biol. 3, a007476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rubinsztein D. C. (2002) Lessons from animal models of Huntington disease. Trends Genet. 18, 202–209 [DOI] [PubMed] [Google Scholar]
- 15. Scherzinger E., Sittler A., Schweiger K., Heiser V., Lurz R., Hasenbank R., Bates G. P., Lehrach H., Wanker E. E. (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington disease pathology. Proc. Natl. Acad. Sci. U.S.A. 96, 4604–4609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen S., Wetzel R. (2001) Solubilization and disaggregation of polyglutamine peptides. Protein Sci. 10, 887–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang W., Dunlap J. R., Andrews R. B., Wetzel R. (2002) Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 11, 2905–2917 [DOI] [PubMed] [Google Scholar]
- 18. Ren P. H., Lauckner J. E., Kachirskaia I., Heuser J. E., Melki R., Kopito R. R. (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 11, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saavedra A., Giralt A., Rué L., Xifró X., Xu J., Ortega Z., Lucas J. J., Lombroso P. J., Alberch J., Pérez-Navarro E. (2011) Striatal-enriched protein-tyrosine phosphatase expression and activity in Huntington disease: a STEP in the resistance to excitotoxicity. J. Neurosci. 31, 8150–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ortega Z., Díaz-Hernández M., Maynard C. J., Hernández F., Dantuma N. P., Lucas J. J. (2010) Acute polyglutamine expression in inducible mouse model unravels ubiquitin/proteasome system impairment and permanent recovery attributable to aggregate formation. J. Neurosci. 30, 3675–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sorolla M. A., Rodríguez-Colman M. J., Tamarit J., Ortega Z., Lucas J. J., Ferrer I., Ros J., Cabiscol E. (2010) Protein oxidation in Huntington disease affects energy production and vitamin B6 metabolism. Free Radic. Biol. Med. 49, 612–621 [DOI] [PubMed] [Google Scholar]
- 22. Sivanandam V. N., Jayaraman M., Hoop C. L., Kodali R., Wetzel R., van der Wel P. C. (2011) The aggregation-enhancing huntingtin N terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 133, 4558–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tam S., Spiess C., Auyeung W., Joachimiak L., Chen B., Poirier M. A., Frydman J. (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.