

Background: The molecular mechanism of cognitive deficit in Huntington disease (HD) is poorly understood.
Results: Altered function of Ube3a could lead to increased levels of synaptic Arc and decreased expression of AMPA receptors and various synaptic proteins.
Conclusion: Dysfunction of Ube3a might be associated with abnormal synaptic function in HD mice.
Significance: Aberrant function of Ube3a might be linked with mental illness in HD.
Keywords: E3 Ubiquitin Ligase, Huntington Disease, Proteasome, Protein Aggregation, Synapses
Abstract
Huntington disease (HD) is a hereditary neurodegenerative disorder characterized by progressive cognitive, psychiatric, and motor symptoms. The disease is caused by abnormal expansion of CAG repeats in the gene encoding huntingtin, but how mutant huntingtin leads to early cognitive deficits in HD is poorly understood. Here, we demonstrate that the ubiquitin ligase Ube3a, which is implicated in synaptic plasticity and involved in the clearance of misfolded polyglutamine protein, is strongly recruited to the mutant huntingtin nuclear aggregates, resulting in significant loss of its functional pool in different regions of HD mouse brain. Interestingly, Arc, one of the substrates of Ube3a linked with synaptic plasticity, is also associated with nuclear aggregates, although its synaptic level is increased in the hippocampus and cortex of HD mouse brain. Different regions of HD mouse brain also exhibit decreased levels of AMPA receptors and various pre- and postsynaptic proteins, which could be due to the partial loss of function of Ube3a. Transient expression of mutant huntingtin in mouse primary cortical neurons further demonstrates recruitment of Ube3a into mutant huntingtin aggregates, increased accumulation of Arc, and decreased numbers of GluR1 puncta in the neuronal processes. Altogether, our results suggest that the loss of function of Ube3a might be associated with the synaptic abnormalities observed in HD.
Introduction
Huntington disease (HD)2 is an autosomal dominant progressive neurodegenerative disorder caused by a trinucleotide (CAG) repeat expansion in the gene encoding the protein huntingtin (1). The glutamine repeat length in normal individual varies from 6 to 36, whereas the disease is associated with >35 repeats, with longer repeats being observed with early onset of the disease. The disease is invariably fatal, and affected individuals show dementia, depression, and anxiety, which usually precede severe motor impairment. Neuropathologically, the disease is characterized by progressive degeneration of neurons in the striatum, certain layers of the cerebral cortex, and hippocampus as well as by generalized atrophy in the majority of brain regions (2–4).
One of the most common pathological features of HD and other related polyglutamine disorders is the accumulation of intracellular aggregates of the disease protein (3, 4). Cytoplasmic and nuclear aggregates of N-terminal fragments of mutant huntingtin have been observed in the brains of HD patients as well as in several animal models of HD. Studies in animal models and post-mortem HD brain samples proposed multiple mechanisms to explain the disease pathogenesis, including aberrant interactions and interference with gene transcription (5, 6), mitochondrial dysfunction and oxidative stress (7, 8), and malfunction of protein folding and the clearance system, etc. (9, 10). Interestingly, many of the altered signaling pathways are linked with the disruption of synaptic function (11, 12). In fact, several studies demonstrate cognitive disturbances in HD patients long before onset of classic motor symptoms like chorea (13–15). Similarly, transgenic mouse models of HD also show that the onset of symptoms is associated with synaptic and neuronal dysfunction and that neurodegeneration takes place at a much later stage (16–19). The HD model mice also display early deficits in spatial memory and defects in hippocampal and cortical plasticity (16, 18, 20, 21). The dysfunction in cortical neurons might contribute to both motor and cognitive dysfunction (19), but how expression of mutant huntingtin leads to early synaptic dysfunction is not yet clear.
Recently, we showed that Ube3a functions as a cell quality control ubiquitin ligase and is involved in the clearance of misfolded polyglutamine proteins (22–24). Ube3a is a HECT (homologous to E6-AP C terminus) domain family ubiquitin ligase, and the loss of function of maternally inherited Ube3a causes Angelman syndrome, a neurodevelopmental disorder with severe learning and memory impairment (25, 26). Recent studies have also indicated that Ube3a localizes in the synapse and plays a prominent role in experience-dependent cortical and neocortical plasticity (27–30). In this investigation, we studied the role of Ube3a in synaptic function using the R6/2 transgenic mouse model of HD. We found that Ube3a selectively redistributes to the nuclear aggregates of mutant huntingtin and that its soluble level is significantly reduced in different regions of HD mouse brain. The partial loss of function of Ube3a might lead to improper synaptic function, as evident from the altered levels of Arc, AMPA receptors, and some pre- and postsynaptic proteins.
EXPERIMENTAL PROCEDURES
Mice
Ovarian-transplanted hemizygous females carrying the HD exon 1 gene with ∼150 CAG repeats (strain B6CBA-Tg(HDexon1)62Gpb/3J) were purchased from The Jackson Laboratory and crossed with B6CBAF1/J males. Genotyping was carried out using PCR as described previously (31, 32). Heterozygous Ube3a mice were also obtained from The Jackson Laboratory (strain 129-Ube3atm1Alb/J) and maintained in the C57BL/6 background. Genotyping was carried out as described (33). Different crosses of mice were used to obtain maternally deficient mice as shown previously (33). Transgenic mice 6 and 12 weeks of old and age-matched controls were used for experimental purposes. All animal experiments were performed according to the protocol approved by the Institutional Animal Ethics Committee of the National Brain Research Centre. Animals had free access to a pelleted diet and water ad libitum. All efforts were made to minimize animal suffering. Animals were killed by cervical dislocation, brains were collected, and different parts were carefully dissected out and stored at −80 °C.
Materials
The truncated N-terminal huntingtin (tNhtt) expression constructs fused to enhanced GFP (EGFP; pIND-tNhtt-16Q-EGFP and pIND-tNhtt-150Q-EGFP) and the generation of the stable cell lines of these constructs (HD16Q and HD150Q) have been described previously (34). All cell culture reagents were obtained from Sigma. Ponasterone A, LipofectamineTM LTX transfection reagent, Zeocin, and G418 were purchased from Invitrogen. Rabbit polyclonal anti-GAPDH, anti-Ube3a, anti-Arc, and β-tubulin antibodies and goat polyclonal anti-huntingtin antibody were from Santa Cruz Biotechnology. Mouse monoclonal anti-Ube3a antibody was purchased from BD Biosciences. Rabbit polyclonal anti-ubiquitin antibody was purchased from Dako, anti-PSD95 antibody was from Cell Signaling, anti-synapsin-1 antibody was from Stressgen, and anti-glutamate receptor (GluR) 1 and anti-GluR2 antibodies were from Chemicon. Rhodamine-conjugated goat anti-rabbit IgG, FITC-conjugated goat anti-mouse IgG, and alkaline phosphatase-conjugated anti-mouse and anti-rabbit IgG and an ABC kit were purchased from Vector Laboratories.
Cell Culture and Immunofluorescence Staining
The stable Neuro-2a cell lines HD16Q and HD150Q were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum and antibiotics (0.4 mg/ml Zeocin and 0.4 mg/ml G418). Cells were plated onto 2-well chamber slides and induced with 1 μm ponasterone A for 48 h and then subjected to immunofluorescence staining as described (35). Briefly, cells were washed twice with PBS, fixed with 4% paraformaldehyde in PBS for 20 min, permeabilized with 0.3% Triton X-100 in PBS for 5 min, washed extensively, and blocked with 5% nonfat dry milk in TBS/Tween for 1 h. The cells were then incubated with anti-Arc antibody (1:500 dilution) overnight at 4 °C. After three washings with TBS/Tween, cells were incubated with rhodamine-conjugated secondary antibody (1:1000 dilution) for 2 h, washed several times, and mounted. Samples were visualized using a Zeiss ApoTome system, and digital images were arranged using Adobe Photoshop.
Immunoblot Analysis
HD16Q and HD150Q cells were plated onto 6-well plates and induced with ponasterone A for different time periods. Cells were then washed with cold PBS, scraped, pelleted by centrifugation, lysed with SDS sample buffer, and processed for immunoblot analysis. Different areas of brain were homogenized in radioimmune precipitation assay buffer (10 mm Tris (pH 7.4), 150 mm NaCl, 10 mm EDTA, 2.5 mm EGTA, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 10 mm NaF, 5 mm Na4P2O7, 0.1 mm Na2VO5, and Complete protease inhibitor mixture); the lysates were briefly sonicated and centrifuged for 10 min at 15,000 × g at 4 °C; and the supernatants were used for immunoblotting as described previously (36). Anti-Ube3a, anti-Arc, anti-PSD95, anti-synaptophysin, and anti-GAPDH antibodies were used at 1:1000 dilution; anti-synapsin-1 antibody was used at 1:10,000 dilution; and anti-β-tubulin antibody was used at 1:5000 dilution.
Semiquantitative and Quantitative Real-time RT-PCR Analysis
Total RNA was extracted using TRIzol reagent, and semiquantitative RT-PCR was carried out with a RT-PCR kit (Takara Bio). Quantitative real-time PCR for Ube3a was carried out using iQ SYBR Green Super Mix (Bio-Rad) after cDNA synthesis from total RNA. Real-time PCR was performed using an ABI Prism 7500 system, and results were analyzed using the sequence detection software (Applied Biosystems). All reactions were normalized with 18 S rRNA as an internal control. The primer sequences for Ube3a were as follows. Ube3aF, 5′-AACTGAGGGCTGTGGAAATG-3′; and Ube3aR, 5′-TCCGAAAGCTCAGAACCAGT-3′. PCR conditions for Ube3a and β-actin were the same: an initial denaturation step at 94 °C for 4 min; 25 (for Ube3a) or 20 (for β-actin) cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 45 s; and a final extension step at 72 °C for 5 min.
Immunohistochemical Staining
The transgenic mice and their age-matched controls were anesthetized and then perfused with PBS containing 4% paraformaldehyde. Brain samples were collected and processed for cryosectioning (20-μm thickness). The sections were then subjected to immunohistochemical staining for Ube3a, Arc, ubiquitin, GluR1, GluR2, synapsin-1, and PSD95 as described previously (34). Staining was carried out using the ABC kit. Anti-PSD95, anti-GluR1, anti-GluR2, anti-synapsin-1, and anti-Arc antibodies were used at 1:500 dilution, and anti-Ube3a antibody was used at 1:100 dilution.
Preparation of Synaptosomes
Total brains collected from 12-week-old wild-type or R6/2 mice were homogenized in 5 ml of 0.32 m sucrose. After removal of debris by centrifugation at 1000 × g, the supernatant was layered onto 4 ml of 1.2 m sucrose and centrifuged at 160,000 × g for 15 min. A thin cloudy layer of synaptosomes was formed at the interface between the 1.2 and 0.32 m sucrose layers. Diluted synaptosomes were layered onto 4 ml of 0.8 m sucrose and centrifuged at 160,000 × g for 15 min to obtain synaptosomal pellets. Pellets were resuspended in Nonidet P-40 lysis buffer, sonicated, centrifuged, and subjected to co-immunoprecipitation as described previously (22).
Filter Trap Assay
Different brain regions were homogenized in homogenization buffer (50 mm Tris (pH 7.4), 150 mm NaCl, 1% Triton X-100, 1 mm PMSF, and one Complete protease inhibitor tablet), briefly sonicated, and centrifuged at 13,000 × g for 15 min. Precipitated pellets were treated with 2% SDS at room temperature for 5 min and allowed to filter through cellulose acetate membranes. Membranes were then processed for immunoblotting with anti-Ube3a, anti-Arc, and anti-huntingtin antibodies. Nitrocellulose membrane was placed at the bottom of the cellulose acetate membrane, which was also immunoblotted with the abovementioned antibodies to check equal loading of the samples.
Primary Cortical Culture
Postnatal day 1 mice were anesthetized by hypothermia on ice. The cortex was isolated and dissociated in 1× ice-cold Hanks' balanced salt solution containing 0.25% trypsin, 0.3% glucose, and 1.2 units/ml DNase at 30 °C for 7 min. After centrifugation, the cell pellet was washed and resuspended in serum-containing DMEM. Cells were plated onto chamber slides coated with poly-d-lysine. After 24 h, the medium was changed, and cells were grown in Neurobasal medium supplemented with 2 mm glutamine, 0.3% glucose, and 2% B-27 supplement. One-half of the medium was then replaced with supplemented Neurobasal medium every 2 days. After 4 days of incubation, cortical neurons were transfected with the pIND-tNhtt-150Q-EGFP plasmid using Lipofectamine LTX transfection reagent and processed for immunofluorescence staining using antibodies against Ube3a, Arc, and GluR1. In another experiment, cortical neuronal cultures isolated from wild-type and maternal Ube3a-deficient mice were immunostained with anti-Ube3a, anti-Arc, and anti-GluR1 antibodies.
Statistical Analysis
Statistical analysis was performed using SigmaStat software. Values are expressed as means ± S.D. Intergroup comparisons were performed by Student's two-tailed t test. p < 0.05 was considered statistical significant.
RESULTS
Recruitment of Ube3a into Nuclear Aggregates of Mutant Huntingtin in R6/2 Transgenic Mouse Brain
We reported previously that the ubiquitin ligase Ube3a selectively promotes proteasomal degradation of cellular misfolded proteins, including expanded polyglutamine proteins and mutant α-synuclein (22–24). We have also shown that Ube3a strongly co-localizes with the misfolded mutant disease proteins (22). We further attempted to study the possible role of Ube3a in the clearance of mutant huntingtin and HD pathogenesis using the R6/2 mouse model. First, we analyzed the localization of Ube3a in huntingtin aggregates in different regions of R6/2 mouse brain. In wild-type mice, Ube3a was widely expressed in the cortex, hippocampus, striatum, and cerebellum and was localized in the nucleus and neuronal processes (Fig. 1). However, in R6/2 mice, Ube3a was strongly recruited to the mutant huntingtin nuclear aggregates present in cortical, hippocampal, and striatal neurons as well as in cerebellar Purkinje cells (Fig. 1A). Double immunofluorescence staining studies using anti-ubiquitin and anti-Ube3a antibodies indicated that >80–90% of the ubiquitin-positive nuclear aggregates were also positive for Ube3a (Fig. 1B). Because Ube3a was recruited to the nuclear aggregates in HD mouse brain, we next compared its soluble pool in wild-type and transgenic mouse brains by immunoblot analysis. As shown in Fig. 2 (A and B), the soluble levels of Ube3a were significantly reduced in the cortex, striatum, and cerebellum of R6/2 mouse brain compared with age-matched wild-type mouse brain. The mRNA level of Ube3a was unaltered in different brain regions of wild-type and R6/2 mice (Fig. 2, C and D). The numbers of NeuN-stained cells were similar in wild-type and R6/2 mouse brains, indicating the absence of neuronal loss in R6/2 mice at 12 weeks of age (data not shown). These results indicate that the decrease in the protein levels of Ube3a in R6/2 transgenic mice could be due the redistribution of Ube3a into aggregates. The filter trap assay also demonstrated the presence of Ube3a in insoluble fractions in R6/2 mouse brain (Fig. 2E). Ube3a was barely detectable in insoluble fractions from wild-type mouse brain. Interestingly, we also noticed that Arc, one of the recently identified substrates of Ube3a, also appeared in insoluble fractions in the filter trap assay (Fig. 2E).
FIGURE 1.

Recruitment of Ube3a into huntingtin nuclear aggregates. A, representative immunohistochemical staining of Ube3a in different brain regions of wild-type and R6/2 mice. Brain sections (20-μm thickness) collected from 10–12-week-old mice were used for staining. B, double immunofluorescence staining of Ube3a and ubiquitin in the hippocampal region of R6/2 mouse brain. Scale bars = 10 μm. Arrows indicate the localization of Ube3a in nuclear aggregates.
FIGURE 2.

Protein level of Ube3a is significantly reduced in different brain regions of R6/2 mice. A, immunoblot analysis of the levels of Ube3a in the cortices, striatums, and cerebellums of wild-type and R6/2 mice (10–12 weeks of age). B, the band intensities of Ube3a collected from five different mice in each group were quantitated using NIH ImageJ analysis software and normalized against β-tubulin. *, p < 0.01 in comparison with wild-type mice. C and D, brain samples (cortex and cerebellum) were collected from wild-type and R6/2 mice and processed for semiquantitative RT-PCR (C) and quantitative real-time RT-PCR (D). R6/2 mouse cortex and cerebellum did not exhibit any significant decrease in Ube3a mRNA levels compared with wild-type mouse cortex and cerebellum (p > 0.2). Values are means ± S.D. (n = 4). E, the total brain lysates collected from wild-type and R6/2 mice were subjected to filter trap assay on cellulose acetate membrane and blotted with anti-huntingtin (Htt), anti-Ube3a, and anti-Arc antibodies. Nitrocellulose membrane was placed at the bottom of the cellulose acetate membrane in the filter trap assay and probed with the abovementioned antibodies to monitor the equal loading of the samples. The negative control (NC) was developed without primary antibody.
Altered Function of Arc in R6/2 Mouse Brain
The data presented in Figs. 1 and 2 indicate that Ube3a function might be compromised in R6/2 transgenic mouse brain. In addition to possible quality control ligase function, Ube3a also plays an important role in regulating synaptic plasticity. A recent report demonstrated that Ube3a modulates synaptic function by regulating the levels of Arc, a synaptic protein involved in AMPA receptor internalization and implicated in synaptic plasticity (27). Because Arc is a substrate of Ube3a and appears in insoluble factions along with Ube3a in R6/2 mouse brain, we checked for possible alterations of the levels of Arc in R6/2 mouse brain. Immunohistochemical staining of Arc indicated that this protein was localized in the nucleus as well as in neuronal processes in wild-type mouse brain. In R6/2 mouse brain, Arc was strongly localized with the nuclear aggregates (Fig. 3A). Interestingly, the neuritic Arc levels in R6/2 mouse brain were clearly increased compared with those in wild-type mouse brain (Fig. 3A). However, the total level of Arc in R6/2 mouse brain was not significantly altered compared with that in the wild-type control (Fig. 3B). We next analyzed the levels of Arc and Ube3a in synaptosomal fractions isolated from wild-type and R6/2 mouse brains. As shown in Fig. 4, the synaptosomal level of Arc was significantly increased in R6/2 mouse brain compared with that in the wild-type control. The synaptosomal fraction of R6/2 mouse brain also showed decreased levels of Ube3a and GluR1 compared with the control. Increased accumulation of ubiquitinated Arc was also detected in the synaptosomal fraction of R6/2 mouse brain (Fig. 4C). These data indicate that the synaptic Arc might be increased in R6/2 mouse brain, although extrasynaptic Arc associated with nuclear aggregates. We further checked the expression and localization of Arc in the cellular model of HD, where exon 1 of wild-type huntingtin and that of mutant huntingtin were stably and inducibly expressed as GFP fusions in mouse Neuro-2a cells. These cell lines were named HD16Q and HD150Q. The cell lines were induced with 1 μm ponasterone A to express the wild-type and mutant proteins and then subjected to immunofluorescence staining using anti-Arc antibody. As expected, Arc was co-localized with mutant huntingtin aggregates in HD150Q cells (Fig. 5A). In HD16Q cells, Arc was diffusely stained both in the cytoplasm and nucleus (Fig. 5A). Interestingly, the level of Arc was significantly increased in HD150Q cells compared with HD16Q cells (Fig. 5B). The uninduced HD150Q cells also showed increased levels of Arc compared with the uninduced HD16Q cells, which could be due to the basal expression of the 150Q protein without induction. The increased level of Arc in HD150Q cells could be due to proteasomal dysfunction as well as reduced levels of Ube3a, which we have reported previously (22, 34).
FIGURE 3.

R6/2 mouse brain exhibits altered levels and localization of Arc. A, representative immunohistochemical staining of Arc in the cortices, hippocampuses, and cerebellums of wild-type and R6/2 mouse brains (12 weeks old). Scale bar = 10 μm. Arrows indicate the localization of Arc in nuclear aggregates, and arrowheads point to neuritic Arc (increased levels in R6/2 brain). B, immunoblot analysis of Arc in different brain regions of wild-type and R6/2 mice (6 and 12 weeks old).
FIGURE 4.
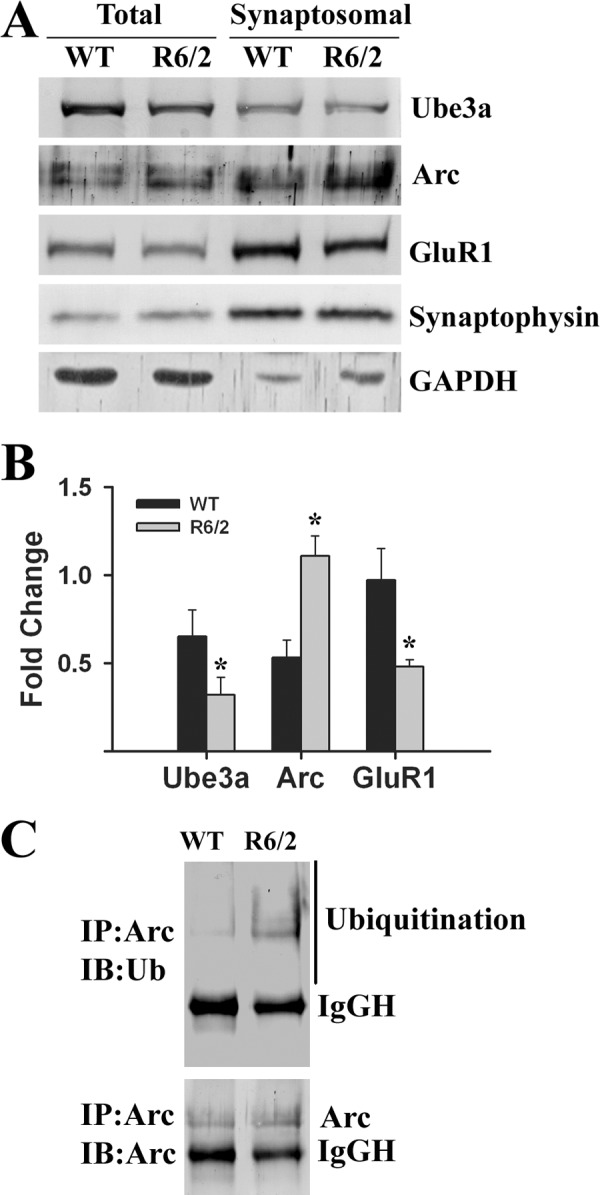
Analysis of levels of Ube3a, Arc, and GluR1 in synaptosomal fractions. A, representative immunoblot of Ube3a, Arc, GluR1, and synaptophysin in the synaptosomal preparations obtained from wild-type and R6/2 mouse (12 weeks old) total brain. B, quantification of the band intensities of Arc, Ube3a and GluR1 shown in A. *, p < 0.01 in comparison with wild-type mice (n = 3). C, co-immunoprecipitation (IP) of Arc in the synaptosomal preparation. Blots were probed with anti-Arc and anti-ubiquitin antibodies. IB, immunoblot; IgGH, IgG heavy chain.
FIGURE 5.

A, localization of Arc with mutant huntingtin aggregates in HD150Q cells. HD16Q and HD150Q cells were plated onto 2-well tissue culture chamber slides and induced with 1 μm ponasterone A for 48 h. Cells were then processed for immunofluorescence staining using anti-Arc antibody. Rhodamine-conjugated secondary antibody was used to detect Arc. Arrows indicate the localization of Arc in the huntingtin aggregates. Scale bar = 20 μm. B, Arc levels are increased in HD150Q cells. HD16Q and HD150Q cells were left uninduced or induced with ponasterone A for 48 h, and the cell lysates were made and subjected to immunoblot analysis using antibodies against Arc, Ube3a, GAPDH, and GFP (to detect 16Q and 150Q proteins).
Down-regulation of AMPA Receptors and Synaptic Proteins
Arc is known to be involved in the internalization and down-regulation of the AMPA subtype of glutamate receptors (37, 38). Because the levels of synaptic Arc are increased along with decreases in GluR1 in R6/2 transgenic mouse brain, we further analyzed the expression levels of GluR1 and GluR2 (classes of AMPA receptors). We observed a significant decrease in the expression levels of GluR1 and GluR2 in R6/2 mouse cortex and hippocampus (Fig. 6). Previous reports also demonstrated the decreased ligand binding of AMPA receptors and reduced AMPA currents in R6/2 mouse brain. All of these data indicate that AMPA receptor function is altered in R6/2 mouse brain. We also compared the expression and localization of various pre- and postsynaptic proteins in wild-type and transgenic mouse brains because maternal Ube3a-deficient mice demonstrate decreased dendritic spines and synaptic proteins (30, 39). Fig. 7 shows the significant decrease in the expression of synapsin (presynaptic protein) and PSD95 (postsynaptic protein) in the cortex, hippocampus, and cerebellum of R6/2 mouse brain. However, the level of another synaptic protein, synaptophysin, was unaffected in R6/2 mouse brain, indicating alteration of specific types of synaptic proteins in R6/2 mouse brain.
FIGURE 6.

Expression of GluR1 and GluR2 is decreased in R6/2 mouse brain. A and B, representative immunohistochemical staining of GluR1 and GluR2 in the cortices and hippocampuses of wild-type and R6/2 mice. Scale bars = 10 μm. C, immunoblot analysis of GluR1 and GluR2 in the cortex and hippocampal (Hippo) regions of wild-type and R6/2 mouse brains (10–12 weeks old). D and E, GluR1 and GluR2 blots obtained from four different mice in each group were quantitated using NIH ImageJ analysis software and normalized against β-tubulin. *, p < 0.05 in comparison with wild-type mice.
FIGURE 7.

Decreased levels of synapsin-1 and PSD95 in R6/2 mouse brain. A, immunoblot analysis of PSD95, synapsin-1, synaptophysin, and β-tubulin in cortices, striatums, and cerebellums of wild-type and R6/2 mice. B and C, the band intensities of PSD95 and synapsin-1, respectively, collected from four different mice in each group were quantitated using NIH ImageJ analysis software and normalized against β-tubulin. *, p < 0.01 in comparison with wild-type mice. D, representative immunohistochemical staining of PSD95 and synapsin-1 in the hippocampuses of wild-type and R6/2 mice. Scale bar = 10 μm.
The functional relationship among Ube3a, Arc, and GluR in mutant huntingtin-expressing cells was further studied in mouse primary cortical neuronal culture upon transient transfection with the mutant huntingtin plasmid. As shown in Fig. 8A, mutant huntingtin-expressing cells exhibited significantly reduced numbers of neuronal processes. Ube3a was localized with mutant huntingtin aggregates, and its staining was significantly reduced in the cell soma and neuronal processes. Arc also co-localized with mutant huntingtin aggregates, and its neuritic level was increased. In untransfected cortical neurons, GluR1 puncta were clearly visible in the neuronal processes, but were drastically reduced in mutant huntingtin-expressing cells. We performed immunofluorescence staining of Arc and GluR1 in cultured primary cortical neurons isolated from wild-type and maternal Ube3a-deficient mouse brains. Maternal Ube3a-deficient mice show a complete absence of Ube3a expression in neurons because of paternal-specific imprinting (33). We found that primary cortical neurons isolated from maternal Ube3a-deficient mice showed clear increases in Arc immunostaining and decreases in GluR1 puncta compared with wild-type cortical neurons (Fig. 8B). Similar findings have been reported previously (27). Altogether, our data suggest that the partial loss of function of Ube3a might be associated with abnormal synaptic function in HD transgenic mouse brain.
FIGURE 8.

Immunofluorescence staining of Ube3a, Arc, and GluR1 in primary cortical neuronal culture. A, primary cortical neurons (after 4 days of culture) were transiently transfected with tNhtt-150Q-EGFP constructs for 36 h and then subjected to immunofluorescence staining using antibodies against Ube3a, Arc, and GluR1. Rhodamine-conjugated secondary antibody was used to detect Ube3a, Arc, and GluR1. Nuclei were counterstained with DAPI. Arrows indicate the localization of Arc in the mutant huntingtin aggregates; arrowheads points to GluR1 puncta. Scale bar = 10 μm. The tNhtt-150Q-transfected cells showed significantly reduced numbers of neuronal processes compared with the control cells: control, 15.25 ± 3.6; and tNhtt-150Q, 4.64 ± 1.52 (p < 0.01). Neuronal branching of untransfected and transfected cells was calculated using NeuronStudio (Beta) software. At least nine cultured neurons in each group were used for analysis. B, double immunofluorescence staining of Arc/Ube3a and GluR1/Ube3a in primary cortical neuronal cultures (5 days) obtained from wild-type (m+/p+) and maternal Ube3a-deficient (m−/p+) mouse brains. Rhodamine-conjugated secondary antibody was used to detect Ube3a (mouse-specific), whereas FITC-conjugated secondary antibody was used to label Arc and GluR1 (rabbit-specific). Nuclei were counterstained with DAPI in overlaid images. Scale bars = 10 μm.
DISCUSSION
The data presented in this study indicate that a defective function of the ubiquitin ligase Ube3a not only can increase the aggregate burden but can also lead to cognitive and motor deficits observed in a transgenic mouse model of HD. We reported previously that Ube3a promotes proteasomal degradation of misfolded mutant huntingtin and that overexpression of Ube3a suppresses mutant huntingtin aggregation and mutant huntingtin-induced cellular toxicity (22). Here, we have demonstrated the strong recruitment of Ube3a into the mutant huntingtin nuclear aggregates in the brains of HD transgenic mice, and as a consequence, the soluble pool of Ube3a decreases. The recruitment of Ube3a into the huntingtin aggregates could be an adaptive response of the cell to handle misfolded and aggregated huntingtin, and association of Ube3a with the aggregates over time could result in its partial loss of function. The inefficient clearance of mutant huntingtin or its aggregates would worsen the situation over time (10, 34).
Apart from its quality control ligase activity, Ube3a also plays an important role in synaptic function and experience-dependent synaptic plasticity (27–29). Interestingly, loss of function of maternally inherited Ube3a causes Angelman syndrome, which is characterized by severe cognitive and motor deficits (25, 26). Maternal Ube3a-deficient mice also exhibit deficits in learning and memory as well as motor function (33, 40). These mice also exhibit a significantly reduced number of dendritic spines and defects in hippocampal long-term potentiation (30, 33). Ube3a is widely expressed in the cerebral cortex, hippocampus, striatum, and cerebellar Purkinje cells and is predominantly localized in the nucleus and synapses (30, 41). Because the HD transgenic mice also display deficits in learning and memory and defects in hippocampal and cortical plasticity (13, 16, 18–20), it is conceivable that the widespread loss of function of Ube3a in HD transgenic mouse brain could contribute to cognitive as well as motor dysfunction.
Recently, Ube3a has been shown to play a crucial role in regulating synaptic plasticity through the ubiquitination and subsequent proteasomal degradation of Arc, a synaptic protein that promotes endocytosis of AMPA subtypes of glutamate receptors (27). Overexpression of Arc in primary cultured hippocampal neurons not only down-regulates the surface expression of AMPA receptors through increased endocytosis but also significantly decreases the total AMPA receptor protein levels (37). Ube3a knock-out mice demonstrate few synaptically expressed AMPA receptors (27). We have noticed increased levels of synaptic Arc in the cortical and hippocampal neurons of HD transgenic mice compared with age-matched controls, which could be due to decreased levels of Ube3a. The proteasomal dysfunction at the synaptic level reported previously (42) could also lead to increased levels of synaptic Arc. In fact, our observations of increased accumulation of ubiquitinated Arc in the synaptosomal fraction could be due to inhibition of proteasomal function at the synapses. Our findings also indicate that Arc could be ubiquitinated not only by Ube3a but also by other ubiquitin ligase. Presently, we do not know the implication of the association of extrasynaptic Arc with the huntingtin aggregates. HD mouse brain shows a significant decrease in ligand binding to AMPA receptors compared with wild-type mouse brain, and the cortical pyramidal neurons of R6/2 mice exhibit decreased AMPA receptor-mediated current (43–45). Furthermore, ampakine treatment slows the disease progression in R6/2 mice (46, 47). These findings, along with our data, indicate that there is a significant loss of the AMPA subtypes of glutamate receptors in R6/2 mouse cortical or hippocampal neurons, which could be due to the increased levels of Arc. Thus, partial loss of function of Ube3a in R6/2 mouse brain could lead to down-regulation of AMPA receptors, leading to the abnormalities in synaptic function and synaptic plasticity. Abnormal synaptic function could also result in decreased levels of dendritic spines and alteration of synaptic proteins. In addition to abnormalities in AMPA receptors, ample evidence also suggests disrupted NMDA receptor function in various HD mouse models (13). In conclusion, our study provides evidence that there is a significant decrease in the levels of Ube3a in different brain regions of HD transgenic mice and that Ube3a dysfunction might be associated with increased aggregate burden and abnormal synaptic function.
Acknowledgments
We thank Dr. V. Rema (National Brain Research Centre) for providing anti-GluR1 and anti-GluR2 antibodies and Dr. N. Dhingra for providing anti-synapsin-1 antibody. We also thank Ankit Sharma for technical assistance.
This work was supported in part by a core grant from the Department of Biotechnology to the National Brain Research Centre, Government of India.
- HD
- Huntington disease
- tNhtt
- truncated N-terminal huntingtin
- EGFP
- enhanced GFP
- GluR
- glutamate receptor.
REFERENCES
- 1. Huntington Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington disease chromosomes. Cell 72, 971–983 [DOI] [PubMed] [Google Scholar]
- 2. Gatchel J. R., Zoghbi H. Y. (2005) Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 6, 743–755 [DOI] [PubMed] [Google Scholar]
- 3. Landles C., Bates G. P. (2004) Huntingtin and the molecular pathogenesis of Huntington disease. Fourth in molecular medicine review series. EMBO Rep. 5, 958–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Orr H. T., Zoghbi H. Y. (2007) Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621 [DOI] [PubMed] [Google Scholar]
- 5. Cha J. H. (2000) Transcriptional dysregulation in Huntington disease. Trends Neurosci. 23, 387–392 [DOI] [PubMed] [Google Scholar]
- 6. Cha J. H. (2007) Transcriptional signatures in Huntington disease. Prog. Neurobiol. 83, 228–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Browne S. E., Ferrante R. J., Beal M. F. (1999) Oxidative stress in Huntington disease. Brain Pathol. 9, 147–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quintanilla R. A., Johnson G. V. (2009) Role of mitochondrial dysfunction in the pathogenesis of Huntington disease. Brain Res. Bull. 80, 242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jana N. R., Nukina N. (2003) Recent advances in understanding the pathogenesis of polyglutamine diseases: involvement of molecular chaperones and ubiquitin-proteasome pathway. J. Chem. Neuroanat. 26, 95–101 [DOI] [PubMed] [Google Scholar]
- 10. Rubinsztein D. C. (2006) The roles of intracellular protein degradation pathways in neurodegeneration. Nature 443, 780–786 [DOI] [PubMed] [Google Scholar]
- 11. Luthi-Carter R., Strand A., Peters N. L., Solano S. M., Hollingsworth Z. R., Menon A. S., Frey A. S., Spektor B. S., Penney E. B., Schilling G., Ross C. A., Borchelt D. R., Tapscott S. J., Young A. B., Cha J. H., Olson J. M. (2000) Decreased expression of striatal signaling genes in a mouse model of Huntington disease. Hum. Mol. Genet. 9, 1259–1271 [DOI] [PubMed] [Google Scholar]
- 12. Luthi-Carter R., Apostol B. L., Dunah A. W., DeJohn M. M., Farrell L. A., Bates G. P., Young A. B., Standaert D. G., Thompson L. M., Cha J. H. (2003) Complex alteration of NMDA receptors in transgenic Huntington disease mouse brain: analysis of mRNA and protein expression, plasma membrane association, interacting proteins, and phosphorylation. Neurobiol. Dis. 14, 624–636 [DOI] [PubMed] [Google Scholar]
- 13. Milnerwood A. J., Raymond L. A. (2010) Early synaptic pathophysiology in neurodegeneration: insights from Huntington disease. Trends Neurosci. 33, 513–523 [DOI] [PubMed] [Google Scholar]
- 14. Foroud T., Siemers E., Kleindorfer D., Bill D. J., Hodes M. E., Norton J. A., Conneally P. M., Christian J. C. (1995) Cognitive scores in carriers of Huntington disease gene compared to non-carriers. Ann. Neurol. 37, 657–664 [DOI] [PubMed] [Google Scholar]
- 15. Peavy G. M., Jacobson M. W., Goldstein J. L., Hamilton J. M., Kane A., Gamst A. C., Lessig S. L., Lee J. C., Corey-Bloom J. (2010) Cognitive and functional decline in Huntington disease: dementia criteria revisited. Mov. Disord. 25, 1163–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Milnerwood A. J., Cummings D. M., Dallérac G. M., Brown J. Y., Vatsavayai S. C., Hirst M. C., Rezaie P., Murphy K. P. (2006) Early development of aberrant synaptic plasticity in a mouse model of Huntington disease. Hum. Mol. Genet. 15, 1690–1703 [DOI] [PubMed] [Google Scholar]
- 17. Milnerwood A. J., Gladding C. M., Pouladi M. A., Kaufman A. M., Hines R. M., Boyd J. D., Ko R. W., Vasuta O. C., Graham R. K., Hayden M. R., Murphy T. H., Raymond L. A. (2010) Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington disease mice. Neuron 65, 178–190 [DOI] [PubMed] [Google Scholar]
- 18. Van Raamsdonk J. M., Pearson J., Slow E. J., Hossain S. M., Leavitt B. R., Hayden M. R. (2005) Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington disease. J. Neurosci. 25, 4169–4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Milnerwood A. J., Raymond L. A. (2007) Corticostriatal synaptic function in mouse models of Huntington disease: early effects of huntingtin repeat length and protein load. J. Physiol. 585, 817–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Usdin M. T., Shelbourne P. F., Myers R. M., Madison D. V. (1999) Impaired synaptic plasticity in mice carrying the Huntington disease mutation. Hum. Mol. Genet. 8, 839–846 [DOI] [PubMed] [Google Scholar]
- 21. Murphy K. P., Carter R. J., Lione L. A., Mangiarini L., Mahal A., Bates G. P., Dunnett S. B., Morton A. J. (2000) Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington disease mutation. J. Neurosci. 20, 5115–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mishra A., Dikshit P., Purkayastha S., Sharma J., Nukina N., Jana N. R. (2008) E6-AP promotes misfolded polyglutamine proteins for proteasomal degradation and suppresses polyglutamine protein aggregation and toxicity. J. Biol. Chem. 283, 7648–7656 [DOI] [PubMed] [Google Scholar]
- 23. Mishra A., Godavarthi S. K., Maheshwari M., Goswami A., Jana N. R. (2009) The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 284, 10537–10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mulherkar S. A., Sharma J., Jana N. R. (2009) The ubiquitin ligase E6-AP promotes degradation of α-synuclein. J. Neurochem. 110, 1955–1964 [DOI] [PubMed] [Google Scholar]
- 25. Williams C. A. (2005) Neurological aspects of the Angelman syndrome. Brain Dev. 27, 88–94 [DOI] [PubMed] [Google Scholar]
- 26. Kishino T., Lalande M., Wagstaff J. (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73 [DOI] [PubMed] [Google Scholar]
- 27. Greer P. L., Hanayama R., Bloodgood B. L., Mardinly A. R., Lipton D. M., Flavell S. W., Kim T. K., Griffith E. C., Waldon Z., Maehr R., Ploegh H. L., Chowdhury S., Worley P. F., Steen J., Greenberg M. E. (2010) The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating Arc. Cell 140, 704–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sato M., Stryker M. P. (2010) Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. U.S.A. 107, 5611–5616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yashiro K., Riday T. T., Condon K. H., Roberts A. C., Bernardo D. R., Prakash R., Weinberg R. J., Ehlers M. D., Philpot B. D. (2009) Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 12, 777–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dindot S. V., Antalffy B. A., Bhattacharjee M. B., Beaudet A. L. (2008) The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17, 111–118 [DOI] [PubMed] [Google Scholar]
- 31. Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S. W., Bates G. P. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 [DOI] [PubMed] [Google Scholar]
- 32. Kotliarova S., Jana N. R., Sakamoto N., Kurosawa M., Miyazaki H., Nekooki M., Doi H., Machida Y., Wong H. K., Suzuki T., Uchikawa C., Kotliarov Y., Uchida K., Nagao Y., Nagaoka U., Tamaoka A., Oyanagi K., Oyama F., Nukina N. (2005) Decreased expression of hypothalamic neuropeptides in Huntington disease transgenic mice with expanded polyglutamine-EGFP fluorescent aggregates. J. Neurochem. 93, 641–653 [DOI] [PubMed] [Google Scholar]
- 33. Jiang Y. H., Armstrong D., Albrecht U., Atkins C. M., Noebels J. L., Eichele G., Sweatt J. D., Beaudet A. L. (1998) Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811 [DOI] [PubMed] [Google Scholar]
- 34. Jana N. R., Zemskov E. A., Wang G., Nukina N. (2001) Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 10, 1049–1059 [DOI] [PubMed] [Google Scholar]
- 35. Rao S. N., Sharma J., Maity R., Jana N. R. (2010) Co-chaperone CHIP stabilizes aggregate-prone malin, a ubiquitin ligase mutated in Lafora disease. J. Biol. Chem. 285, 1404–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jana N. R., Dikshit P., Goswami A., Kotliarova S., Murata S., Tanaka K., Nukina N. (2005) Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 280, 11635–11640 [DOI] [PubMed] [Google Scholar]
- 37. Chowdhury S., Shepherd J. D., Okuno H., Lyford G., Petralia R. S., Plath N., Kuhl D., Huganir R. L., Worley P. F. (2006) Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tzingounis A. V., Nicoll R. A. (2006) Arc/Arg3.1: linking gene expression to synaptic plasticity and memory. Neuron 52, 403–407 [DOI] [PubMed] [Google Scholar]
- 39. Mulherkar S. A., Jana N. R. (2010) Loss of dopaminergic neurons and resulting behavioral deficits in mouse model of Angelman syndrome. Neurobiol. Dis. 40, 586–592 [DOI] [PubMed] [Google Scholar]
- 40. Jiang Y., Lev-Lehman E., Bressler J., Tsai T. F., Beaudet A. L. (1999) Genetics of Angelman syndrome. Am. J. Hum. Genet. 65, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gustin R. M., Bichell T. J., Bubser M., Daily J., Filonova I., Mrelashvili D., Deutch A. Y., Colbran R. J., Weeber E. J., Haas K. F. (2010) Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol. Dis. 39, 283–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang J., Wang C. E., Orr A., Tydlacka S., Li S. H., Li X. J. (2008) Impaired ubiquitin-proteasome system activity in the synapses of Huntington disease mice. J. Cell Biol. 180, 1177–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cha J. H., Kosinski C. M., Kerner J. A., Alsdorf S. A., Mangiarini L., Davies S. W., Penney J. B., Bates G. P., Young A. B. (1998) Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc. Natl. Acad. Sci. U.S.A. 95, 6480–6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. André V. M., Cepeda C., Venegas A., Gomez Y., Levine M. S. (2006) Altered cortical glutamate receptor function in the R6/2 model of Huntington disease. J. Neurophysiol. 95, 2108–2119 [DOI] [PubMed] [Google Scholar]
- 45. Mandal M., Wei J., Zhong P., Cheng J., Duffney L. J., Liu W., Yuen E. Y., Twelvetrees A. E., Li S., Li X. J., Kittler J. T., Yan Z. (2011) Impaired α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor trafficking and function by mutant huntingtin. J. Biol. Chem. 286, 33719–33728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Simmons D. A., Rex C. S., Palmer L., Pandyarajan V., Fedulov V., Gall C. M., Lynch G. (2009) Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington disease knock-in mice. Proc. Natl. Acad. Sci., U.S.A. 106, 4906–4911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Simmons D. A., Mehta R. A., Lauterborn J. C., Gall C. M., Lynch G. (2011) Brief ampakine treatments slow the progression of Huntington disease phenotypes in R6/2 mice. Neurobiol. Dis. 41, 436–444 [DOI] [PMC free article] [PubMed] [Google Scholar]