

Background: Ada3 is a core component of HAT containing coactivator complexes.
Results: Germline deletion of Ada3 is embryonic lethal, and cell deletion leads to abnormal cell cycle progression.
Conclusion: Ada3 is a critical protein at organismic and cellular level.
Significance: This study describes a novel role of Ada3, a component of HAT complexes, as a critical regulator of cell survival.
Keywords: Adaptor Proteins, Cell Cycle, Chromatin Histone Modification, Mitosis, p53, Ada3 KO Mouse, G1/S, Histone Acetylation
Abstract
Ada3 protein is an essential component of histone acetyl transferase containing coactivator complexes conserved from yeast to human. We show here that germline deletion of Ada3 in mouse is embryonic lethal, and adenovirus-Cre mediated conditional deletion of Ada3 in Ada3FL/FL mouse embryonic fibroblasts leads to a severe proliferation defect which was rescued by ectopic expression of human Ada3. A delay in G1 to S phase of cell cycle was also seen that was due to accumulation of Cdk inhibitor p27 which was an indirect effect of c-myc gene transcription control by Ada3. We further showed that this defect could be partially reverted by knocking down p27. Additionally, drastic changes in global histone acetylation and changes in global gene expression were observed in microarray analyses upon loss of Ada3. Lastly, formation of abnormal nuclei, mitotic defects and delay in G2/M to G1 transition was seen in Ada3 deleted cells. Taken together, we provide evidence for a critical role of Ada3 in embryogenesis and cell cycle progression as an essential component of HAT complex.
Introduction
The eukaryotic cell cycle progression depends on proper coordination of DNA replication and duplication of chromosomes to daughter cells (1), a process precisely regulated by modification of chromatin that allows the accessibility to factors involved in transcription (2). Thus, proteins involved in modulating the structure of chromatin play an important role in cell cycle progression. The post-translational modification of core histones (H2A, H2B, H3, and H4) is an essential process for altering chromatin structure (3, 4). Histone acetyl transferases (HATs)6 and histone deacetylases are required to maintain steady state levels of acetylation (5). Several HAT enzymes, such as general control nonderepressible 5 (Gcn5), p300/CBP-associated factor (PCAF), p300, and CREB-binding protein (CBP), have been identified over the years (6, 7). Most of the HATs are part of large complexes such as the human TBP-free TAF complex (TFTC); the Spt3/Taf9/Gcn5 acetyltransferase complex (STAGA) (human homolog of yeast SAGA complex) and the Ada2a-containing (ATAC) complex that play a role in several important processes, such as cell cycle (8, 9). Additionally, previous studies from our laboratory and that of others have demonstrated the presence of p300 HAT in Ada3-containing protein complexes (10, 11). Given the combined presence of Ada3 with Gcn5 in a number of distinct HAT complexes, recent evidence for a role of Gcn5 in regulating DNA replication as well as mitosis (12–14) suggest that Ada3 may also play a role in cell cycle. Despite the range of established and potential cellular functions of Ada3 as part of multiple HAT complexes, the in vivo physiological role of mammalian Ada3 is not known.
We previously identified human Ada3 as a novel human papillomavirus 16 E6-binding protein (15). Human Ada3 is the homologue of the yeast Ada3, an essential component of the Ada transcriptional coactivator complex composed of Ada2, Ada3, and a HAT component Gcn5 (16). Genetic studies in yeast have demonstrated that Ada3 functions as a critical component of coactivator complexes that link transcriptional activators, bound to specific promoters, to histone acetylation and basal transcriptional machinery (17–19). We showed that Ada3 binds and stabilizes the tumor suppressor p53 protein and is required for p53 acetylation by p300 (20). Work from our laboratory has also shown that Ada3 is required for HAT recruitment to estrogen receptors and their transcription activation function (11). We and others have shown that Ada3 also associates with and regulates transcriptional activity of other nuclear hormone receptors, including retinoic acid receptor (21) and androgen receptor (22).
Here, we used conditional deletion of mouse Ada3 gene to explore the physiological importance of mammalian Ada3. We demonstrate that homozygous deletion of Ada3 is early embryonic lethal. Ada3 deletion in Ada3Flox/Flox (Ada3FL/FL) MEFs showed that Ada3 is required for efficient cell cycle progression through G1 to S transition as well as for proper mitosis. Detailed analyses in this system revealed an Ada3-c-Myc-Skp2-p27 axis that controls G1 to S phase progression and partly contributes to cell cycle delay upon Ada3 deletion. Additionally, loss of Ada3 showed dramatic decrease in acetylation of core histones that are known to play an important role in cell cycle. Loss of Ada3 also resulted in several changes in gene expression as observed by microarray analyses. Notably, many of the genes affected were involved in mitosis. Taken together, we present evidence for an essential role of mammalian Ada3 in embryonic development and cell cycle progression.
EXPERIMENTAL PROCEDURES
Generation of Ada3 Gene-targeted Mice, Isolation of Mouse Embryos and PCR Genotyping
Details concerning generation of conditional Ada3 knock-out construct and Ada3 knock-out mouse as well as PCR genotyping strategies are described in the supplemental data.
Cell Culture Procedures and Viral Infections
Embryonic day 13.5 embryos were dissected from Ada3FL/+ intercrossed females, and MEFs were isolated and immortalized following the 3T3 protocol (23). MEFs were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Adenoviruses expressing EGFP-Cre or enhanced green fluorescent protein (EGFP) alone were purchased from the University of Iowa (Gene Transfer Vector Core). An adenovirus dose of 50–100 MOI diluted in 4 ml of serum-free medium was added to cells in 100-mm culture dishes (at about 30% confluence) and incubated for 1 h each at room temperature and at 37 °C followed by the addition of 7 ml of complete medium. After overnight incubation at 37 °C, medium was replaced with complete medium, and cells were carried further for various experiments. To generate retroviral FLAG-hAda3 vector, full-length FLAG-hAda3 (15) was cloned into pMSCVpuro vector (Clontech). Retroviruses were generated by transiently transfecting this retroviral construct into the Phoenix ecotropic packaging cell line using the calcium phosphate co-precipitation method. The retroviruses were transduced into Ada3FL/FL MEFs by three infections at 12-h intervals using supernatant from transfected Phoenix cells to generate Ada3FL/FL MEFs expressing FLAG-hAda3. Scrambled shRNA (5′-GGTTAAAACCTTACGATGT-3′) or p27 shRNA (5′-GTGGAATTTCGACTTTCAG-3′) was introduced into Ada3FL/FL MEFs by using three infections at 12-h intervals of the shRNA bearing pSUPER.retro.puro (Oligoengine) retrovirus containing supernatants from Phoenix cells. Retroviral infections were carried out in the presence of 8 μg/ml Polybrene (Sigma) and were followed by selection in 2 μg/ml puromycin for 48 h until complete loss of uninfected cells.
Proliferation Assay, Colony Formation Efficiency Assay, and Cell Cycle Analysis
To perform proliferation assays, 1 day after adenovirus infection, cells were plated at different numbers in 6-well plates in triplicates (5 × 104 (for counting on day 3), 2.5 × 104 (for counting on day 5), 1.25 × 104 (for counting on day 7), and 0.625 × 104 (for counting on day 9) and counted at the indicated time points. For colony formation assay, cells 3 days after adenovirus-infection were trypsinized and plated at 1000 cells per 100-mm culture dishes in triplicates and carried for 15 more days with medium change as required. At the end of incubation, colonies in dishes were fixed and stained with crystal violet solution (0.25% crystal violet in 25% methanol) and photographed. For cell cycle analysis, 2 days after plating and adenoviral infection of 2 × 105 cells in 100-mm culture dishes, cells were synchronized by replacing the complete medium with DMEM + 0.1% FCS and incubating for 72 h. Synchronized cells were stimulated with complete medium (DMEM + 10% FCS) for various time points and harvested and stained with propidium iodide (PI) for FACS analysis. For synchronization of cells at G2/M phase, 48 h after adenovirus infection, cells were switched to complete medium containing 125 ng/ml nocodazole for 18 h. Following synchronization, cells were washed three times with PBS and stimulated with complete medium for various time points and analyzed by FACS after PI staining.
Generation of Ada3 Monoclonal Antibody and Immunoblotting
Antibodies used in this study can be found in the supplemental data.
In Vitro Kinase Assay
In vitro kinase assay was performed using purified histone H1 (Roche Applied Science) or Rb (769) (Santa Cruz Biotechnology sc-4112) as a substrate. Adenovirus-infected MEFs were starved for 3 days and stimulated with serum. Cells were harvested in lysis buffer (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.5% Nonidet P-40, 0.1 mm Na4VO3, 1 mm NaF, and protease inhibitor mixture), and cyclin-dependent kinase (Cdk) complex was recovered by immunoprecipitation with 2 μg of either anti-Cdk4 (sc-56277)/Cdk6 (sc-53638) antibodies mixture or anti-Cdk2 (sc-6248) antibody (Santa Cruz Biotechnology). Cdk4/6 or Cdk2 complexes were captured with protein G-agarose for 1 h and washed with lysis buffer followed by one wash with kinase buffer (50 mm Tris-HCl (pH 7.5), 7.5 mm MgCl2, 1 mm dithiothreitol, 0.1 mm Na4VO3, and 1 mm NaF). Cdk2 complex was incubated with histone H1 (2 μg) or Rb (500 ng), whereas Cdk4/6 complex was incubated with only Rb (500 ng) in kinase buffer containing 10 mm β-glycerophosphate, 33 μm ATP, and 10 μCi of [γ-32P]ATP (10 mCi/ml, 6000 Ci/mmol) at room temperature for 20 min. The products were subjected to SDS-PAGE, transferred to polyvinylidene difluoride membranes (PVDF), and autoradiographed.
Analysis of the p27 Protein Turnover
Ada3FL/FL MEFs were plated in 100-mm dishes and infected with control or Cre adenoviruses. For analyzing p27 protein half-life in exponentially growing cells, 2 days after adenovirus infection, cells were treated with 50 μg/ml cycloheximide (Sigma) and harvested at the indicated time points. For analyzing p27 protein half-life in serum-starved cells, 2 days after adenovirus infection, cells were starved for 72 h in 0.1% serum-containing medium. Subsequently, 50 μg/ml cycloheximide was added to the medium, and cells were harvested at the indicated time points. Total cell extracts were prepared, and equivalent amounts were run on SDS-PAGE and analyzed by Western blotting. Densitometry analysis was carried out on scanned images using ImageJ software.
RNA Extraction and Quantitative Real-time PCR
TRIzol reagent (Invitrogen) was used to isolate total RNA from MEFs infected with control virus or Cre adenovirus. 2 μg of total RNA was used for reverse transcriptase reaction using SuperScriptTM II reverse transcriptase (Invitrogen). Real-time PCR quantification was performed in triplicates using SYBR Green PCR master mix (Applied Biosystems) and the primers listed in supplemental Table S3. Expression levels were normalized against β-actin mRNA levels, and the results were calculated by the ΔΔCt method.
Chromatin Immunoprecipitation Experiments
Approximately 0.7 million Ada3FL/FL MEFs were plated in 100-mm dishes and infected with control or Cre adenoviruses. Forty-eight hours after infection, cells were synchronized with DMEM + 0.1% FCS for 72 h and then stimulated with complete medium (DMEM + 10% FCS) for 0–60 min as indicated for each experiment in Fig. 8C. ChIP experiment was performed by using the ChIP-IT Express kit from Active Motif. PCR amplification was performed using primers for the c-myc enhancer (forward, 5′-CTAGAACCAATGCACAGAGC-3′; reverse, 5′-CTCCCAGGACAAACCCAAGC-3′) and for the Skp2 promoter (forward, 5′-GCCATCGAGACCCCGGAGAT-3′; reverse, 5′-TGAGTCCCTTCCAGACGCTGT-3′). Control PCR was performed using primers for the c-myc distal site (forward, 5′-ACACACCTTGAATCCCGT-3′; reverse, 5′-CCCAGCTAGAATGAAGAAG-3′) and the Skp2 distal site (forward, 5′-GTGCTAGCTGCTTACCTTTGT-3′; reverse, 5′-GATAAGGATGCACTCTGGGGC-3′). PCR products were analyzed on 2% agarose/Tris-acetate-EDTA gels with ethidium bromide stain. PCR of the input DNA prior to immunoprecipitation was used as a control.
FIGURE 8.

Abnormal cell division and delayed G2/M to G1 transition in Ada3-deleted cells. A, images of Ada3FL/FL cells after 5 days of infection with Cre adenovirus showing abnormal (fragmented, lobulated, or multi) nuclei. B, quantification of abnormal nuclei from cells infected with control (Ctrl) or Cre adenovirus; 5 days after infection, cells were fixed and stained with Giemsa stain and scored for abnormal nuclei (at least 100 cells from each group were counted). Error bars show mean ± S.E. from three independent experiments. C, control- and Cre adenovirus-infected MEFs were treated for 20 h with nocodazole and were harvested at the indicated time points after release, stained with PI, and subjected to FACS analysis. D, graph showing the percentage of cells entering G1 phase after release from nocodazole treatment at various time points from experiments as in C. Error bars are mean ± S.E. from three independent experiments (*, p = 0.034; **, p = 0.0038 and 0.007 for 4 and 8 h, respectively, by two tailed Student's t test).
Generation of Recombinant Baculoviruses and Ada3-His Expression Using Bac-to-Bac® Expression System
Ada3 baculoviral construct information and recombinant protein purification are detailed in the supplemental data.
HAT Assay
Protocol used for in vitro HAT assay can be found in the supplemental data.
Microarray Analyses
Protocol for microarray analyses is described in the supplemental data. The microarray data from this publication have been submitted to the GEO database and have been assigned the following Series record: GSE37542.
RESULTS
Deletion of Ada3 Leads to Early Embryonic Lethality in Mice
The targeting construct generated using the recombineering technique (supplemental Fig. S1A; see supplemental Materials and Methods) was electroporated into an ES cell line derived from the 129/Ola strain of mice. Screening of resultant neomycin-resistant colonies yielded three correctly targeted clones (supplemental Fig. S1B). One positive clone was microinjected into blastocysts. The resulting chimeras transmitted the targeted allele to their progeny as verified by PCR. The neomycin cassette flanked by Frt recombination sites was removed by crossing the Ada3-targeted mice to FlpE recombinase transgenic mice (B6.Cg-Tg (ACTFLPe) 9205Dym/J; stock number 005703). Homozygous Ada3FL/FL mice were viable and fertile and exhibited no gross abnormalities when compared with Ada3FL/+ or Ada3+/+ controls. To achieve Ada3 deletion, heterozygous Ada3-targeted mice (Ada3FL/+ mice) were bred with transgenic mice expressing the adenovirus EIIa promoter-driven Cre (B6.FVB-Tg (EIIa-Cre) C5379Lmgd/J). EIIa directs Cre expression in a wide range of tissues including germ cells. Heterozygous Ada3-targeted, Cre transgene-positive mice were crossed to C57BL/6J (wild-type) mice to generate heterozygous Ada3-deleted, Cre transgene-negative (Ada3+/−) mice. Heterozygous Ada3+/− mice of a mixed 129/Sv × C57BL/6 background were viable and fertile, and their median life span of more than 18 months was comparable with that of their control littermates (data not shown). Heterozygous Ada3+/− mice were intercrossed to obtain homozygous Ada3-null mice. No Ada3−/− mice were observed among 224 live born pups screened (Table 1). The ratio of wild type to heterozygous offspring was 1:2, indicating that the loss of one Ada3 allele does not lead to haploinsufficiency in mice.
TABLE 1.
Genotype analysis of embryos from heterozygous intercrosses
| Stage | Total no. of embryos | No. (%) of embryos |
|||
|---|---|---|---|---|---|
| WT | Heterozygous | KO | Resorbed | ||
| Live born | 224 | 75 (33) | 149 (66) | 0 | 0 |
| E12.5 | 14 | 3 (21) | 5 (36) | 0 | 6 (43) |
| E 9.5 | 15 | 8 (53) | 2 (13) | 0 | 5 (33) |
| E 8.5 | 44 | 12 (27) | 27 (61) | 0 | 5 (11) |
| E 3.5 | 15 | 4 (27) | 7 (47) | 4 (27) | 0 |
To assess the specific period of developmental failure in the Ada3 knock-out mice, embryos derived from Ada3+/− intercrosses were genotyped at different stages of gestation using a duplex PCR method (supplemental Fig. S1, C and D). Because no homozygous mutant embryos were recovered beyond embryonic day 8.5 (E8.5; Table 1), blastocysts were isolated at 3.5 days postcoitum and genotyped directly by PCR (supplemental Fig. S1E). When compared with blastocysts of Ada3+/+ and Ada3+/− genotypes, Ada3−/− blastocysts that attached to culture dishes showed severe growth retardation of the trophoblast layer, and the inner cell mass was absent (supplemental Fig. S1F). PCR analysis revealed that ∼25% of blastocysts analyzed were null for Ada3 (Table 1). These results demonstrate that Ada3 plays a critical role in early embryogenesis in mice. The failure of Ada3−/− embryos to remain viable beyond E3.5 suggests a potential role of Ada3 in cell proliferation because extensive cellular proliferation occurs during this early stage of embryogenesis (see later sections).
Ada3 Is Ubiquitously Expressed in Adult Mouse Tissues
Embryonic lethality of Ada3−/− mice suggested a potential role of Ada3 in growth and development of many tissues. To examine whether Ada3 is expressed in adult tissues, we analyzed the relative levels of Ada3 protein expression in a range of adult mouse tissues. For this purpose, lysates from various tissues of 8-week-old wild-type mice were subjected to immunoblotting using an anti-Ada3 monoclonal antibody generated in our laboratory (see supplemental Materials and Methods). As seen in supplemental Fig. S2, Ada3 is ubiquitously expressed in all the tissues with higher levels seen in the mammary gland, lung, and thymus. These results suggest potentially ubiquitous functional roles of Ada3 and are consistent with embryonic lethal phenotype of its germline deletion.
Conditional Ada3 Deletion in MEFs Leads to Proliferation Arrest
Given the embryonic lethality as a result of Ada3 deletion, we resorted to a cellular model of conditional Ada3 deletion to investigate its roles at the cellular level. For this purpose, we generated Ada3FL/FL mice by interbreeding Ada3FL/+ mice and established MEFs from these mice. Conditional Ada3 deletion was obtained by infecting Ada3FL/FL MEFs with an adenovirus expressing the Cre recombinase (adeno-Cre), with adeno-GFP serving as a control. To assess the effects of Ada3 on cell proliferation, equal numbers of control- and adeno-Cre-infected MEFs were plated a day after adenoviral infection, and cells were counted at the indicated time points up to 9 days. Notably, Ada3-deleted MEFs exhibited a significantly slower rate of proliferation when compared with control MEFs (Fig. 1A, left). To confirm that the defect in cell proliferation was specifically due to depletion of Ada3, we generated Ada3FL/FL/hAda3 MEFs by retrovirally introducing human Ada3 (hAda3) with an N-terminal FLAG tag into Ada3FL/FL MEFs. These transfectants were verified to be expressing the exogenous FLAG-tagged Ada3 protein (Fig. 1B). Similar to Ada3FL/FL MEFs, adeno-Cre infection of these cells led to deletion of endogenous Ada3 and loss of its protein product (Fig. 1B). Notably, however, Cre-mediated deletion of Ada3 in Ada3FL/FL/hAda3 MEFs had a minimal effect on the proliferation of MEFs, whereas similar treatment of Ada3FL/FL MEFs led to reduction in the rate of proliferation; thus, the proliferative defect induced by deletion of mouse Ada3 in MEFs was rescued by exogenous hAda3 (Fig. 1A, right). Colony formation efficiency assay, as an independent method to measure the extent of cell proliferation, further confirmed the proliferative defect of Ada3-deleted MEFs that could be rescued by reconstitution with exogenous hAda3 (Fig. 1, C and D).
FIGURE 1.

Ablation of Ada3 causes proliferation defect in MEFs. A, growth curves of Ada3FL/FL (left) and Ada3FL/FL/fhAda3 (right) MEFs after control adenovirus (Ctrl)or Cre adenovirus (Cre) infection. Data are mean ± S.E. from three independent experiments performed in triplicates. B, Ada3 protein levels at different time points after Cre adenovirus infection. Note that reconstituted control cells express both mouse (mAda3; lower band) and human (FLAG hAda3; upper band) proteins, whereas only hAda3 is seen in Cre adenovirus-infected cells. C, colony formation assay. Crystal violet staining of the indicated cells infected with control virus or Cre adenovirus grown for 10 days is shown. D, Western blotting of lysates from C showing exogenous and endogenous Ada3.
Ada3 Is Required for Cell Cycle Progression through G1 to S Phase
We reasoned that the proliferation defect upon Ada3 deletion in MEFs could reflect a role of Ada3 in cell cycle progression. To directly examine whether Ada3 plays a role in cell cycle progression, Ada3FL/FL MEFs were infected with control and Cre adenoviruses, arrested in G0/G1 by serum deprivation for 72 h, and then synchronously released into cell cycle by serum stimulation. FACS-based cell cycle analysis of propidium iodide-stained cells showed significant delay in G1 to S progression in Ada3-deleted MEFs when compared with control MEFs (Fig. 2A). Of note, the relative distribution of S phase in Ada3-null MEFs after 20 h of serum stimulation was about half (31.6 ± 2.33 S.E. %) of the control virus-infected MEFs (56.05 ± 4.71 S.E. %) (Fig. 2B). These results demonstrate that conditional deletion of Ada3 leads to delay in G1 to S progression in MEFs, indicating an essential role of Ada3 in efficient G1/S progression.
FIGURE 2.

Ada3 disruption delays G1 to S transition in MEFs. A, control (Ctrl)- or Cre- infected Ada3FL/FL MEFs were serum-starved for 72 h and then released from synchrony as described under “Experimental Procedures” and processed for PI staining followed by FACS analysis. Cells in different phases of the cell cycle are shown from a representative experiment. B, graph derived from three independent experiments performed as in A, showing the proportion of cells entering into S phase at the indicated times after serum restimulation. Error bars are mean ± S.E. from three independent experiments (**, p = 0.0096, two-tailed Student's t test).
Elevated p27Kip1 Levels and Impaired Rb Phosphorylation upon Conditional Ada3 Deletion
Given the delay in G1/S progression imposed by induced Ada3 deficiency, we examined the status of key proteins known to control the G1/S transition. A well established and critical event during G1 to S progression is the phosphorylation of Rb by Cdk complexes (particularly complexes containing Cyclins D, E, or A), such as Cdk4/6 and Cdk2 (24, 25); phosphorylation of Rb leads to its release from Rb/E2F complexes, relieves E2Fs from repression, and facilitates the expression of E2F-responsive genes important for S phase progression (24, 25). Furthermore, degradation of Cdk inhibitors, such as p27, is required for progression of cells from G1 to S phase (26, 27). Therefore, we carried out Western blotting of cell lysates obtained from control versus conditional Ada3-deleted MEFs released into synchronous cell cycle progression to assess the levels of proteins relevant to the G1 to S phase transition. Notably, although minimal to no changes were observed in the levels of Cdk2, Cdk4, Cdk6, p16, p21, cyclin E, and cyclin D, a significant increase in p27 levels, a delay in the cell cycle-associated increase in cyclin A levels, and a lower level of Rb phosphorylation were observed in MEFs upon Ada3 deletion when compared with control cells (Fig. 3A).
FIGURE 3.

Effect of Ada3 depletion on expression of cell cycle regulator proteins and Cdk2 kinase activity. A, Ada3FL/FL MEFs infected with control (Ctrl) and Cre adenoviruses serum-starved for 72 h, released from synchrony as described under “Experimental Procedures,” and processed for immunoblot analysis of the indicated cell cycle proteins. hyper, hyperphosphorylated; hypo, hypophosphorylated. B, anti-Cdk2 or anti-Cdk4/6 immunoprecipitations performed using 300-μg extracts of Ada3FL/FL MEFs infected with control or Cre adenovirus were subjected to in vitro kinase assay using histone H1 or Rb as a substrate. WB, Western blot; IP, immunoprecipitation.
In view of increased levels of p27 without a significant change in the levels of Cdk proteins in cells with Ada3 deletion, we assessed the level of Cdk2 kinase activity using an in vitro kinase assay on immunoprecipitates from cells. Although the Cdk4/6 kinase activity was comparable between control- and adeno-Cre-infected MEFs (Fig. 3B), the level of Cdk2 kinase activity was substantially reduced in Cre-infected MEFs when compared with control MEFs (Fig. 3B). These results suggest the potential reduction of Cdk2 kinase activity in the Ada3-deleted cells as a result of an increase in the levels of p27, accounting for defective Rb phosphorylation.
Accumulation of p27 upon Ada3 Deletion Is due to Increased Stability of p27
As accumulation of p27 levels upon Ada3 deletion appeared to be functionally important, we examined whether this accumulation was at the transcriptional or post-transcriptional level. Real-time PCR analysis showed that serum stimulation resulted in a marked reduction in the levels of Cdkn1b mRNA in both the control-infected and the Cre-infected cells (Fig. 4A); furthermore, the levels of Cdkn1b mRNA at various time points after serum addition remained comparable between the two cell populations, reinforcing the idea that the increase in p27 protein levels in Ada3-deleted cells was likely to be at a post-transcriptional level. As alterations in protein stability are a prominent mechanism to control Cdk inhibitor levels (28), we compared the half-life of p27 protein in WT versus Ada3-deleted MEFs using two distinct experimental formats; the first one utilized exponentially growing cultures, whereas the second one utilized cells first arrested in G1 by serum deprivation for 72 h followed by synchronous release into cell cycle by serum addition. In each case, Ada3FL/FL MEFs infected with control or Cre adenoviruses were treated with cycloheximide to block new protein synthesis, and p27 levels in cell lysates following cycloheximide treatment were quantified using immunoblotting at various time points. Previous work has shown that p27 half-life in exponentially growing MEFs is about 3 h and increases to about 8 h in serum-starved cells (29). We found the p27 half-life in cells infected with control adenovirus was consistent with published results, i.e. approximately 2 h and 40 min in exponentially growing MEFs, whereas in growth-arrested cells, half-life was approximately 3 h and 30 min (Fig. 4, B–E). Notably, in both experimental formats, we observed a substantial increase in p27 protein half-life upon Cre-dependent Ada3 deletion, with approximate half-lives of 4 h and 10 min and 6 h in exponentially growing versus synchronous culture formats, respectively. These results strongly support our conclusion that accumulation of p27 protein upon Ada3 deletion is due to its increased stability.
FIGURE 4.

Deletion of Ada3 does not affect p27 transcription but extends p27 protein half-life. A, unaltered p27 mRNA levels after Ada3 deletion. Real-time RT-PCR analysis of p27 mRNA levels from cells as treated in Fig. 2 was performed. Signals were normalized to β-actin levels and plotted relative to the level of p27 mRNA in starved control (Ctrl) cells. Error bars show mean ± S.E. from three independent experiments. B–E, Ada3 deletion in MEFs extends p27 half-life. B, 48 h after adenovirus infection, MEFs were treated with 50 μg/ml cycloheximide and harvested at the indicated time points, and p27 and β-actin protein levels were analyzed by immunoblotting. C, the intensity of p27 bands was quantified by densitometry, normalized to β-actin using ImageJ software, and plotted against the time of cycloheximide treatment. Each decrease of 1 unit of log 2 is equivalent to one half-life. The lines were generated by linear regression formula. D, after 48 h of adenovirus infection, MEFs were starved using 0.1% serum-containing medium for 72 h and subsequently treated with 50 μg/ml cycloheximide and harvested at the indicated time points. Cell lysates were analyzed by Western blotting using antibodies against p27 and β-actin. E, graph made from experiment in D by using the same procedure as in C.
Depletion of p27 from Conditionally Deleted Ada3 MEFs Causes a Partial Rescue of G1/S Progression Defects
Reduced activity of the p27 target Cdk2 in Ada3-deleted MEFs strongly suggested a role for p27 in defective cell cycle progression in these cells. To directly establish whether this is the case, we generated stable p27 knockdown Ada3FL/FL MEFs (Ada3FL/FL/p27shRNA) by infecting Ada3FL/FL MEFs with a retrovirus expressing a p27-specific shRNA followed by selection in puromycin, which resulted in a significant knockdown of p27 expression in these cells (Fig. 5A). Next, we infected the Ada3FL/FL/p27shRNA MEFs with control or Cre adenovirus and analyzed these for cell cycle progression using serum deprivation followed by serum stimulation, as above (Fig. 5B). Notably, a partial but clear rescue of the G1/S delay was observed in p27 shRNA-expressing cells, as seen by a much larger percentage of cells entering the S phase (41.4 ± 3.5 S.E. % in p27shRNA expressing conditionally deleted Ada3 MEFs versus 31.6 ± 2.33 S.E. % in Ada3-deleted MEFs at 20 h; compare Fig. 5C with Fig. 2B). Importantly, the levels of cyclin A, which is known to be expressed during G1/S transition and to peak in the S phase, as well as hyperphosphorylation of Rb, were essentially fully rescued by p27 shRNA knockdown (Fig. 5D; compare with Fig. 3A). Taken together, these results clearly demonstrate an important role of Ada3-dependent control of p27 levels in promoting cell cycle progression.
FIGURE 5.

p27 depletion partially rescues G1 to S transition defects seen in Ada3-null MEFs. A, Ada3FL/FL MEFs were infected with retrovirus-expressing scrambled or p27 shRNA followed by selection for 2 days in puromycin and analyzed by immunoblotting using p27 and β-actin antibodies. B, PI staining and FACS analysis of Ada3FL/FL MEFs expressing p27 shRNA that were infected with either control (Ctrl) or Cre adenoviruses and synchronized as in Fig. 2. C, graph derived from three experiments as in B showing the proportion of cells entering into S phase at the indicated times after serum restimulation. Error bars indicate mean ± S.E. from three independent experiments. D, immunoblotting of protein samples from B showing rescue of hyperphosphorylated (hyper) Rb and cyclin A levels. hypo, hypophosphorylated.
Deletion of Ada3 Leads to Reduced Protein and mRNA Levels of Skp2 and c-Myc
Given the causal link established above between p27 accumulation and G1/S cell cycle delay upon Ada3 deletion, we wished to examine the molecular mechanism by which loss of Ada3 promotes p27 stability. Published studies have established a major role of Skp2-containing E3 ubiquitin ligases in regulating p27 protein turnover during cell cycle progression (30). As Skp2 is a transcriptional target of c-Myc (31) and Ada3-containing STAGA complex has been shown to increase myc mRNA transcription (32, 33), the possibility of an Ada3-c-Myc-Skp2-p27 regulatory pathway appeared to be a plausible mechanism for our findings. To explore this hypothesis, we first examined the effects of Ada3 deletion on the levels of Skp2 mRNA (real-time PCR) and protein (immunoblotting). For this purpose, Ada3FL/FL cells infected with control or Cre adenovirus were serum-deprived and released into synchronous cell cycle progression by adding serum followed by analyses of Skp2 mRNA and protein at various time points. Notably, Skp2 mRNA and protein levels were substantially lower at each comparable time point in adeno-Cre-infected versus control MEFs (Fig. 6, A and B). These results indicate that Ada3 deletion indeed leads to reduction in Skp2 levels and that this effect is likely due to reduced Skp2 gene transcription.
FIGURE 6.

Deletion of Ada3 from MEFs leads to reduced mRNA and protein levels of Skp2 and c-Myc. A, analysis of Skp2 mRNA levels by real-time RT-PCR from cells as treated in Fig. 2. Signals were normalized to β-actin levels and plotted relative to the level of Skp2 mRNA in starved control cells. Error bars represent mean ± S.E. from three independent experiments (*, p = 0.015, 0.036, 0.043, and 0.032 for 16, 20, 24, and 28 h, respectively by two-tailed Student's t test). B, immunoblots showing Skp2 protein levels in cells treated as in A. C, analysis of c-Myc mRNA levels by real-time RT-PCR from cells as treated in Fig. 5. Signals were normalized to β-actin levels and plotted as in A. Error bars show mean ± S.E. from three independent experiments. D, immunoblots showing c-Myc protein levels in cells treated as in C (*, p = 0.023 and 0.027 for 1 and 3 h, respectively; **, p = 0.008 by two-tailed Student's t test). E, occupancy of Ada3 on the c-myc enhancer. Chromatin fragments from control (Ctrl) and Cre Ada3FL/FL MEFs cells were immunoprecipitated with anti-Ada3 antibody. Chromatin fragments were prepared from Asynchronous (Asyn.) cells as well as from cells synchronized with 0.1% serum containing DMEM for 72 h (lane 0) and stimulated with serum with indicated time points. The immunoprecipitated DNA was analyzed by PCR, using c-Myc enhancer-specific primers. Primers amplifying a region that is 5 kb upstream of the c-Myc enhancer were used as a negative control. RNA Pol II, RNA polymerase II.
Next, we asked whether Ada3 deletion alters c-Myc mRNA levels and whether Ada3 directly binds to c-myc promoter. Indeed, analysis of control versus Ada3-deleted MEFs stimulated with serum to undergo cell cycle progression demonstrated that c-Myc mRNA as well as protein levels were significantly lower at each time point examined upon deletion of Ada3 from cells (Fig. 6, C and D). Consistent with this, we observed lower occupancy of mouse Skp2 promoter by c-Myc upon deletion of Ada3, which supports our results (supplemental Fig. S3). Finally, to establish that Ada3 indeed participates in the enhancement of myc gene transcription, we carried out ChIP analysis to assess whether Ada3 is recruited to c-myc enhancer during cell cycle progression. Indeed, a rapid recruitment of Ada3, as well as RNA polymerase II (used as positive control), to c-myc enhancer at −1.4 kb relative to transcription start site (but not to a distal site at −5 kb) was seen upon serum stimulation of MEFs (Fig. 6E). As expected, we did not detect any signals after immunoprecipitation with anti-Ada3 antibody in cells infected with adeno-Cre. These results therefore support the existence of a novel cell cycle-associated, Ada3-regulated signaling pathway that promotes G1/S cell cycle progression by regulating p27 stability through Myc-dependent control of Skp2 expression.
Ada3 Deletion Leads to Decreased Histone Acetylation
As we observed a partial rescue of G1/S transition in Ada3-deleted MEFs after knockdown of p27, we speculated that Ada3 deletion-induced cell cycle arrest may involve other pathways as well. Given the known literature on Ada3 as part of HAT complexes (8, 9), we examined whether Ada3 is involved in controlling global histone acetylation. Therefore, we assessed the effect of Ada3 deletion on lysine acetylation of various core histones. We expressed Cre recombinase in Ada3FL/FL MEFs and harvested protein samples from asynchronous cultures after 3 days of infection. Western blotting using antibodies against important acetylated lysine residues of all four core histones (H2A-K5, H2B-K5, H3-K9, H3-K56, and H4-K8) showed a significant reduction in acetylation at all these sites in Ada3-deficient MEFs when compared with control MEFs (Fig. 7A), indicating that Ada3 is essential in maintaining global histone acetylation.
FIGURE 7.

Ada3 deletion abrogates histone acetylation by destabilizing various HATs. A–C, Western blotting analysis of lysates from asynchronous (A and C) or serum-restimulated (B) Ada3FL/FL or Ada3FL/FL/fhAda3 MEFs infected with control (Ctrl) or Cre adenoviruses using the indicated antibodies. D and E, Ada3 enhances p300 HAT activity. In vitro HAT assay using purified recombinant human Ada3 and core histones (D) or histone H3 alone (E) along with their respective Ponceau blots to indicate equal loading is shown.
We further examined the effect of Ada3 deletion on acetylation of core histones after synchronizing cells in G1 phase and subsequent release. There was a dramatic down-regulation of H3-K9 acetylation and a slight decrease in acetylation of H2B-K5 in Ada3-deleted MEFs when compared with control-MEFs, whereas this defect was rescued in Ada3FL/FL MEFs reconstituted with exogenous human FLAG-Ada3 (Fig. 7B), suggesting that the defect in histone acetylation seen in Ada3-deleted MEFs was a consequence of Ada3 deletion. Histone acetylation has been shown to be important for deposition of histones during replication-coupled nucleosome assembly as well as for chromatin maturation following DNA replication (34, 35). Thus, the partial rescue in G1 to S transition observed upon knockdown of p27 in Ada3-deficient cells could be attributed to massive histone acetylation defects, which would create difficulties for cells to undergo DNA replication and thus delay transition through S phase.
Recombinant Ada3 Stabilizes HAT Enzymes and Enhances Their Activity
Ada3 protein has been identified as an important component of protein complexes containing HAT enzymes. Therefore, we subjected samples harvested after 3 days of Ada3 deletion to immunoblotting with two important HATs such as p300 and PCAF. Indeed, deletion of Ada3 caused drastic down-regulation of p300 and PCAF in MEFs (Fig. 7C). Notably, Ada3 deletion had no effect on the mRNA levels of p300 and PCAF (data not shown). Thus, the defects in histone acetylation seen in Ada3-null MEFs could be attributed to the effect of Ada3 deletion on stability of important HATs in cells.
In addition to the role of Ada3 in stability of HAT enzymes, we explored whether Ada3 catalyzes the activity of HAT enzymes. Although Ada3 is shown to be important in maintaining stability of HAT complexes, it has not been demonstrated whether Ada3 directly modulates the activity of known HAT enzymes such as p300. Thus, we expressed and purified baculoviral hAda3 and used it in an in vitro assay in which HAT activity of p300 histone acetyl transferase enzyme on histone substrates was measured. As seen in Fig. 7D, increasing amounts of Ada3 resulted in increased acetylation of histone H1 and histone H3 by p300, suggesting that Ada3 plays an important role in enhancing the HAT activity of p300. To further explore the role of Ada3 in histone acetylation, we used only histone H3 as a substrate and observed an Ada3 dose-dependent increase in acetylation of histone H3 by p300 (Fig. 7E). Thus, Ada3 manifests its effect on histone acetylation by maintaining the integrity of various HAT complexes and by enhancing the catalytic activity of HATs.
Deletion of Ada3 Leads to Global Gene Expression Changes
Given the links between Ada3 and transcriptional activation, we used control and Ada3-deleted cells to perform microarray analyses. As expected, the expression of multiple genes was altered; 539 genes were down-regulated and 928 genes were up-regulated ≥ 1.5-fold upon Ada3 deletion (supplemental Table S1). Validation of some of the deregulated genes from microarray by real-time PCR showed good co-relation with the microarray data (supplemental Fig. S4). Ingenuity pathway analyses showed that most of the genes affected were involved in controlling cell growth, proliferation, and cell death (supplemental Table S2, top biological functions affected; cell growth and proliferation (386 genes) and cell death (359 genes)). The top network affected was the RNA post-transcriptional modification and cellular assembly and organization network, whereas the cell cycle, endocrine system development and function, and cancer network was the third most affected network (supplemental Fig. S5). Notably, c-myc and Skp2 genes that we described above were down-regulated 1.4- and 1.43-fold, respectively. This is lower than what we observed by real-time PCR and could be attributed to the fact that microarray data were performed on asynchronous populations, whereas the real-time PCR data were performed on synchronous cells (Fig. 6, A and C). Interestingly, many of the genes present in cell growth and proliferation set were those involved in controlling cell division as well as some involved in DNA replication (Table 2).
TABLE 2.
List of deregulated genes involved in cell division and DNA replication
Genes down-regulated at least 1.5-fold upon loss of Ada3 as obtained from microarray analyses. The genes were classified based upon gene ontology biological processes.
| Gene symbol | Gene title | -Fold down-regulated |
|---|---|---|
| Genes involved in cell division | ||
| Kifc1 | Kinesin family member C1, similar to Kifc1 protein | 2.0 |
| Nfkbil1 | Nuclear factor of [kappa[ light polypeptide gene enhancer in B-cells inhibitor-like 1 | 2.0 |
| Fbxo5 | F-box protein 5 | 1.8 |
| Cenpf | Centromere protein F | 1.8 |
| Cdc6 | Cell division cycle 6 homolog (Saccharomyces cerevisiae) | 1.7 |
| Kntc1 | Kinetochore-associated 1 | 1.7 |
| Baz1b | Bromodomain adjacent to zinc finger domain, 1B | 1.6 |
| Mlf1ip | Myeloid leukemia factor 1 interacting protein | 1.6 |
| Myh10 | Myosin, heavy polypeptide 10, non-muscle | 1.6 |
| Kif11 | Kinesin family member 11 | 1.6 |
| Ccna2 | Cyclin A2 | 1.6 |
| Smc2 | Structural maintenance of chromosomes 2 | 1.6 |
| Plk1 | Polo-like kinase 1 (Drosophila) | 1.5 |
| Bub1b | Budding uninhibited by benzimidazoles 1 homolog, β (S. cerevisiae) | 1.5 |
| Aspm | asp (abnormal spindle)-like, microcephaly-associated (Drosophila) | 1.5 |
| Anln | Anillin, actin-binding protein | 1.5 |
| Zwilch | Zwilch, kinetochore-associated, homolog (Drosophila) | 1.5 |
| Mki67 | Antigen identified by monoclonal antibody Ki 67 | 1.5 |
| Mad2l1 | MAD2 mitotic arrest deficient-like 1 (yeast) | 1.5 |
| Smc4 | Structural maintenance of chromosomes 4 | 1.5 |
| Cdca8 | Cell division cycle-associated 8 | 1.5 |
| Kif20b | Kinesin family member 20B | 1.5 |
| Hells | Helicase, lymphoid-specific | 1.5 |
| Ccnb1 | Cyclin B1 | 1.5 |
| Cdca3 | Cell division cycle-associated 3 | 1.5 |
| Nuf2 | NUF2, NDC80 kinetochore complex component, homolog (S. cerevisiae) | 1.5 |
| Ndc80 | NDC80 homolog, kinetochore complex component (S. cerevisiae) | 1.5 |
| Birc5 | Baculoviral IAP repeat-containing 5 | 1.5 |
| Bub1 | Budding uninhibited by benzimidazoles 1 homolog (S. cerevisiae) | 1.5 |
| Suv39h2 | Suppressor of variegation 3–9 homolog 2 (Drosophila) | 1.5 |
| Aurkb | Aurora kinase B | 1.5 |
| Wee1 | WEE 1 homolog 1 (Schizosaccharomyces pombe) | 1.5 |
| Genes involved in DNA replication | ||
| Kitl | Kit ligand | 1.9 |
| Prim1 | DNA primase, p49 subunit | 1.7 |
| Mcm7 | Minichromosome maintenance-deficient 7 (S. cerevisiae) | 1.7 |
| Ccne2 | Cyclin E2 | 1.7 |
| Pola1 | Polymerase (DNA directed), alpha 1 | 1.7 |
| Dtl | Denticleless homolog (Drosophila) | 1.7 |
| Cdc6 | Cell division cycle 6 homolog (S. cerevisiae) | 1.7 |
| Chtf18 | CTF18, chromosome transmission fidelity factor 18 homolog (S. cerevisiae) | 1.7 |
| Nfib | nuclear factor I/B | 1.6 |
| Prim1 | DNA primase, p49 subunit | 1.6 |
| Orc1l | Origin recognition complex, subunit 1-like (S. cerevisiae) | 1.6 |
| Rrm1 | Ribonucleotide reductase M1 | 1.6 |
| Rpa1 | Replication protein A1 | 1.6 |
| Cdt1 | Chromatin licensing and DNA replication factor 1 | 1.6 |
| Gins2 | GINS complex subunit 2 (Psf2 homolog) | 1.5 |
| Rbbp4 | Retinoblastoma-binding protein 4 | 1.5 |
| Chaf1b | Chromatin assembly factor 1, subunit B (p60) | 1.5 |
| Tk1 | Thymidine kinase 1 | 1.5 |
Ada3 Deletion Leads to Defects in Cell Division and Accumulation of Abnormal Nuclei
Based on our microarray analyses where several mitotic genes were affected upon deletion of Ada3 and a recent study showing the role of Ada3 in mitosis upon shRNA deletion (14), we examined the effect of Ada3 deletion on mitotic phase of cell cycle. These analyses showed that Cre-mediated Ada3 deletion led to increased accumulation of cells with abnormal nuclei when compared with control MEFs. Ada3-deficient MEFs showed various nuclear abnormalities such as fragmentation, lobulation, and multinucleation (Fig. 8A). When compared with 13.08 ± 2.39 S.E. % control MEFs, 83.41 ± 3.45 S.E. % of Ada3-deficient MEFs showed abnormal nuclei (Fig. 8B). Live imaging of cells for 24 h showed that the majority of Ada3-deleted cells failed to divide normally. Some of the cells snapped back while attempting to undergo cytokinesis, leading to the formation of binucleated cells, whereas other cells that had normal nucleus before mitosis showed fragmented nuclei afterward and were unable to divide. In other cases, cell division resulted in the formation of anucleated daughter cells (Representative images shown in supplemental Fig. S6). Taken together, these results demonstrate an indispensable role of Ada3 in normal cell cycle progression. The cell division defect results reported here corroborate with an earlier published study showing similar defects upon shRNA knockdown of Ada3 (14). Mitotic defects observed in their study were attributed to acetylation of a non-histone substrate cyclin A, and no changes in histone acetylation upon knockdown of Ada3 were reported. In contrast, we observed a dramatic change in global histone acetylation and expression of various genes involved in mitosis. Although at present we cannot explain this discrepancy, the differences in the results may be partly attributable to the use of different cellular systems and differences in approaches followed such as shRNA or Cre-mediated to delete Ada3.
Deletion of Ada3 Leads to Delay in G2/M to G1 Progression
As deletion of Ada3 in MEFs led to defects in cell division, we reasoned that the disruption of Ada3 should exert an effect on G2/M to G1 transition. To examine this effect, we synchronized control- and Cre-adenovirus-infected Ada3FL/FL MEFs at G2/M checkpoint by treating them with nocodazole and released them from synchrony followed by cell cycle analysis using flow cytometry (Fig. 8C). Nocodazole-synchronized Ada3-deleted MEFs showed a lower percentage of cells in G2/M phase (61%) at the 0-h time point when compared with control MEFs (80%) (Fig. 5C). On the contrary, we observed a higher percentage (20%) of Ada3-deleted MEFs in G1 phase when compared with control MEFs (7%) after synchronization. We speculate that Ada3-deficient MEFs that are exhibiting a delay in G1 to S transition were unable to get completely synchronized at G2/M checkpoint as these cells are potentially moving slowly through the G1 to S transition and require a prolonged treatment with nocodazole to show a complete synchronization as seen in control MEFs. When we compared the percentage of cells moving into G1 phase on release from nocodazole treatment in both Ada3-deficient and control MEFs, a significant impairment in G2/M to G1 transition in Ada3-deleted MEFs was observed (Fig. 8D). Taken together, these results demonstrate a critical role of Ada3 in both G1 to S transition as well as G2/M to G1 transition in MEFs, indicating that the cell proliferation defect observed in Ada3-deficient MEFs is due to a combined defect in G1 to S as well as G2/M to G1 transition.
DISCUSSION
Regulated cell cycle entry and progression are essential for precise developmental programs as well as to maintain organ homeostasis in adult animals. Although the basic components of cell cycle have been largely defined, regulatory control mechanisms that ensure orderly proliferative responses to physiological cues and whose aberrations underlie the vast instances of altered proliferation in cancer continue to be elucidated. We previously identified the ADA complex component Ada3 as a human papillomavirus E6 oncoprotein partner as well as a coactivator of cell cycle checkpoint regulator and tumor suppressor p53 (15, 20). Several in vitro studies have shown that Ada3 is an essentially universal component of a multitude of HAT-based transcriptional regulatory complexes (8, 9), and it has become essential to define its physiological roles using in vivo animal models.
Here, we demonstrate that Ada3 is essential for embryonic development in mice and that Ada3-null embryos undergo very early lethality. As an essential component of the transcriptional coactivator complexes that include HATs and promote histone acetylation of key gene targets, Ada3 is known to be essential for growth in yeast (16) as well as in model metazoan organisms such as Drosophila where Ada3 deficiency is associated with arrest in early development (36). However, this study is the first direct demonstration of an essential role of Ada3 in mammalian embryonic development. Notably, the embryonic developmental block imposed by Ada3 deletion occurs very early, resulting in arrest of development at the blastocyst stage, the stage of embryonic development at which extensive cell proliferation occurs (37). Notably, studies that employed gene knockouts of subunits of several chromatin-modifying complexes, including Gcn5, Trrap, Ep300, CBP, Hdac3, or Atac2, also lead to early embryonic lethality (34, 38–42), consistent with an essential role of chromatin modification machinery in mammalian growth and development. However, except for Trrap knock-out, which produces lethality at the blastocyst stage (42), knockouts of other genes produce embryonic developmental arrest at much later stages: for example, Gcn5 (E9.5–E11.5), Ep300 (E9.5–E10.5), and Atac2 (E11.5) in comparison with E3.5 block observed in Ada3-null mice. The relatively early developmental arrest of Ada3-null mice when compared with other regulators could reflect the role of Ada3 as a component of multiple chromatin-remodeling complexes (see Introduction and below). The distinct times of arrest seen with Gcn5-null and Ada3-null embryos are somewhat surprising and suggest the possibility that Ada3 may mediate early developmental roles through complexes in which Gcn5 is not a critical component or is functionally redundant with other HATs. Consistent with this hypothesis, we observed that Ada3-deleted cells exhibit defects in multiple histone acetylations and show decrease in the levels of PCAF and p300 proteins.
We used the conditional deletion feature of the mouse model to assess the critical functional roles of Ada3 by utilizing Cre-dependent gene deletion in MEFs from Ada3FL/FL mice. This system provided a clear evidence that Ada3 plays an essential role in cell proliferation by promoting G1 to S as well as G2/M to G1 cell cycle progression. Furthermore, the proliferative arrest imposed by conditional deletion of Ada3 was reversed by ectopic expression of human Ada3, indicating that the loss of Ada3 itself, rather than alteration of any neighboring gene product, was responsible for the observed cell cycle phenotype.
Cell cycle progression is a tightly regulated process and is dependent on sequential and stringently controlled, concerted activation of Cdks and their inhibition by Cdk inhibitors. The novel cell cycle regulatory pathway downstream of Ada3 was suggested by our initial analyses of alterations in the levels of core components of mammalian cell cycle machinery. These analyses revealed a dramatic reduction in the key propeller of G1/S phase transition, hypophosphorylated Rb when Ada3 was deleted. Association of this defect with reduced Cdk2 activity without a reduction in Cdk2 levels suggested the role of elevated p27, which we established directly by demonstrating that shRNA knockdown of p27 substantially alleviated the G1/S block imposed by Ada3 deficiency. Further biochemical connections were suggested by recent findings that STAGA complex, which includes Ada3 as a component, enhances c-myc transcription (32, 33). Because c-Myc is shown to regulate the transcription of Skp2, an essential component of the SCF(Skp2) cell cycle-associated E3 ligase that regulates p27 levels, we sought and established evidence that cell cycle-associated Myc transcription is Ada3-dependent and that Ada3 is required for Skp2 transcription (which is a downstream target of Myc) and p27 stability (regulated by SCF(Skp2)). We provided direct evidence for key elements of this model, including ChIP analyses that demonstrated the cell cycle-associated early recruitment of Ada3 to c-myc enhancer elements. This result is consistent with independent findings from two groups that STAGA complex is recruited to c-Myc enhancer and regulates c-myc transcription (32, 33). In addition to control of c-myc gene transcription by Ada3-containing STAGA complex, studies have shown that STAGA associates with c-Myc on c-Myc target gene promoters and is required for efficient transcription activation by c-Myc (43, 44). This provides an additional mechanism by which Ada3 could control c-Myc-driven target genes that regulate cell proliferation. Thus, Ada3 might be involved in controlling both c-myc transcription as well as c-Myc function. Consistent with our observations, it is noteworthy that c-myc knock-out mice are embryonic lethal (45). Defective regulation of c-Myc transcription by Ada3-containing (STAGA or other) complexes might contribute to the early embryonic lethality seen in Ada3-null mice; further analyses of Myc-dependent pathways upon germline or conditional deletion of Ada3 during embryogenesis should help establish whether this is the case.
Although regulation of p27 protein stability by Ada3 through control of c-myc transcription forms an important basis for G1/S transition defects, we were not able to fully rescue the defect in cell cycle by using p27 shRNA, suggesting the involvement of other cellular pathways. To this end, examining global histone acetylations in Ada3-deficient cells revealed dramatic defects in histone acetylation. Because Ada3 forms a core structural component of various different HAT complexes in the cell, the presence of Ada3 is highly essential for structural maintenance and proper functioning of these complexes in cells. Additionally, loss of Ada3 led to substantial depletion of important HATs, p300, and PCAF proteins but not mRNA, which further explains the profound defects in histone acetylation seen upon loss of Ada3. This is consistent with the fact that PCAF and p300 are present in Ada3-containing protein complexes (8–11). These defects in histone acetylation could explain the partial rescue upon knockdown of p27 as histone acetylation has been shown to have an important role in the process of DNA replication (34, 35).
Given the role of Ada3 in regulating global histone acetylation and that histone acetylation is important in transcriptional activation of genes, we performed microarray analysis and showed that several genes were deregulated upon Ada3 deletion. Analysis of these genes by ingenuity pathway analysis revealed the RNA post-transcriptional modification and cellular assembly and organization network as the top affected network, with the cell cycle, endocrine system development and function, and cancer network as the third most affected. The top network affected in the microarray data is consistent with an earlier study, which showed that Ada3-containing STAGA complex interacts with pre-mRNA splicing machinery, components suggesting a role for this complex in mRNA splicing (46). Importantly, the top biological functions affected upon deletion of Ada3 included those involved in cell growth and proliferation with 386 deregulated genes involved in this process. Thus, our microarray data confirmed a role of Ada3 in cell cycle progression. Additionally, some of the top physiological functions affected upon deletion of Ada3 were those involving tissue development and organismal survival (supplemental Table S2), which could be linked to the early embryonic lethality observed upon knock-out of Ada3 in mouse.
Notably, many of the genes that were involved in regulating cell growth and proliferation were those involved in mitosis and some that were involved in DNA replication. This led us to examine cell division upon deletion of Ada3. Consistent with the microarray data, we observed massive nuclear abnormalities, cell division defects, and delay in G2/M to G1 phase progression upon deletion of Ada3. Our observed phenomenon of cell division defects upon deletion of Ada3 is consistent with a recently published study (14). The authors showed that ATAC HAT complex is specifically involved in regulating mitosis and that shRNA-mediated knockdown of Ada3 or Ada2a led to defects in cell division, which were attributed to stabilization of cyclin A upon disruption of ATAC complex. Although we did not observe an increase in cyclin A levels (in fact the converse) in our system, we did observe a similar effect on nuclear abnormalities and a clear defect in mitosis. Furthermore, the authors did not observe any changes in histone acetylation defects upon depletion of Ada3, which is not consistent with our results. Of note, Ada2a is a component of only ATAC complex; however, Ada3 has been shown to be a core component of a number of HAT complexes. The authors used depletion of Ada3 as an indication of disruption of only ATAC complex; however, deletion of Ada3 would affect several HAT complexes and not just ATAC complex. Thus, deletion of Ada3 would cause disruption of several HAT complexes that function in different phases of the cell cycle leading to defects in various phases of the cell cycle. Based on these findings, we propose the following working model of Ada3 regulation of cell cycle progression. As part of a chromatin-remodeling complex, likely the STAGA complex, Ada3 is recruited to and modifies the c-myc transcriptional regulatory elements to enhance Skp2 transcription. This leads to destabilization of p27 by the SCF(Skp2) E3 ligase, resulting in increased Cdk2 activity and Rb phosphorylation to promote G1/S progression. Additionally, Ada3, by regulating the number of genes involved in mitosis, regulates cell division. Lastly, Ada3 as part of ATAC and STAGA complex regulates transcription of various genes by recruiting HATs and acetylating histones. Combination of these functions led to severe cell cycle defect and embryonic lethality upon Ada3 deletion (Fig. 9). Finally, although our studies here have focused on the role of Ada3 in cell cycle progression, future studies using cell type- or stage-specific conditional deletion of Ada3 in mouse to assess its role in functions other than transcriptional activation, including optimal transcription elongation, mRNA export, and nucleotide excision repair, need to be explored (8, 46, 47).
FIGURE 9.
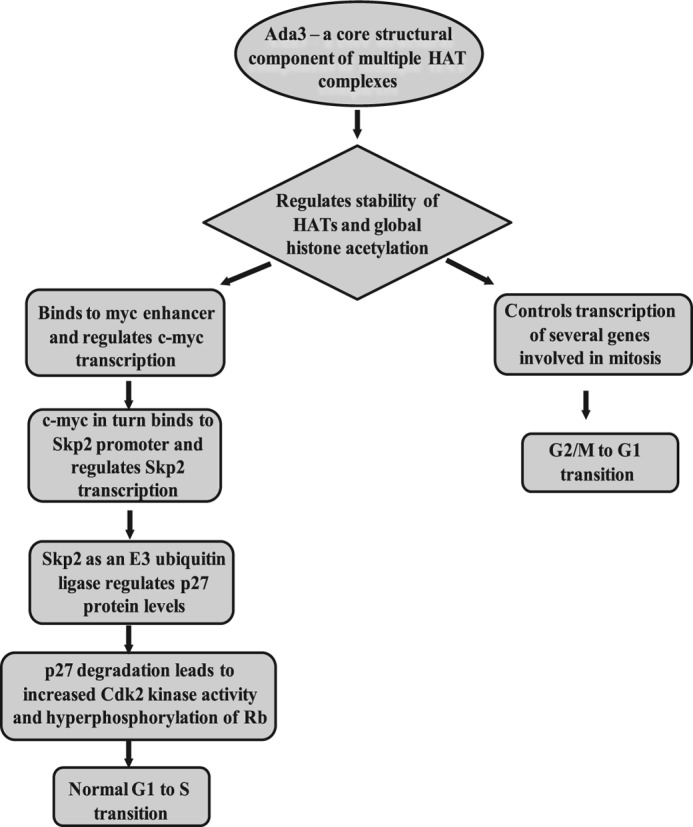
Proposed model for the role of Ada3 in cell cycle progression. As a core structural component of various HAT complexes, Ada3 maintains the integrity of HAT complexes and thus regulates global histone acetylation. Ada3 regulates G1 to S transition by controlling transcription of c-myc gene, which in turn controls Skp2 gene expression by binding to its promoter. Skp2 as an E3 ubiquitin ligase causes timely degradation of p27 protein so that cells can enter into S phase by increasing Cdk2 kinase activity, thus inducing hyperphosphorylation of Rb and cell progression from G1 to S phase of cell cycle. Additionally, through controlling global histone acetylation, Ada3 controls transcription of various genes involved in cell division and is required for cells to undergo normal mitosis and G2/M to G1 progression.
In conclusion, we demonstrate that the evolutionarily conserved Ada3 protein as an essential component of HAT complex plays an important role in embryogenesis and cell division. Thus, our studies identify Ada3 as a novel component of the physiological regulation of mammalian cell cycle progression and set the stage for future studies to assess the role of Ada3 in cell cycle progression during in vivo physiological and pathological settings. Use of Ada3FL/FL mice should facilitate these analyses to functionally dissect the in vivo roles of Ada3.
Supplementary Material
Acknowledgments
We acknowledge technical assistance from Valerie Tran and Poonam Joshi as well as assistance with time-lapse microscopy from Tom Dao. The University of Nebraska Medical Center (UNMC) DNA Microarray Core facility is supported by grants from the National Center for Research Resources (5P20RR016469) and the NIGMS (8P20GM103427), a component of the National Institutes of Health.
This work was supported, in whole or in part, by National Institutes of Health Grant CA96844 and CA144027 (to V. B.) and CA87986, CA99163, CA105489, CA116552 and NCI 5U01CA151806-02 (to H. B.). This work was also supported by Department of Defense Grants W81XWH-07-1-0351 and W81XWH-11-1-0171 (to V. B.).

This article contains supplemental Materials and Methods, Figs. S1–S6, and Tables S1–S3.
- HAT
- histone acetyltransferase
- Ada3
- alteration/deficiency in activation 3
- hAda3
- human Ada3
- MEF
- mouse embryonic fibroblast
- Cdk
- cyclin-dependent kinase
- Gcn5
- general control nonderepressible 5
- PCAF
- p300/CBP-associated factor
- CBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- STAGA
- Spt3/Taf9/Gcn5 acetyltransferase complex
- ATAC
- Ada2a-containing complex
- adeno-Cre
- adenovirus expressing the Cre recombinase
- Rb
- retinoblastoma protein
- E
- embryonic days
- PI
- propidium iodide
- TBP
- TATA-binding protein
- TAF
- TBP-associated factor.
REFERENCES
- 1. Schafer K. A. (1998) The cell cycle: a review. Vet. Pathol 35, 461–478 [DOI] [PubMed] [Google Scholar]
- 2. Li B., Carey M., Workman J. L. (2007) The role of chromatin during transcription. Cell 128, 707–719 [DOI] [PubMed] [Google Scholar]
- 3. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 4. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 5. Strahl B. D., Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 6. Roth S. Y., Denu J. M., Allis C. D. (2001) Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120 [DOI] [PubMed] [Google Scholar]
- 7. Carrozza M. J., Utley R. T., Workman J. L., Côté J. (2003) The diverse functions of histone acetyltransferase complexes. Trends Genet. 19, 321–329 [DOI] [PubMed] [Google Scholar]
- 8. Lee K. K., Workman J. L. (2007) Histone acetyltransferase complexes: one size doesn't fit all. Nat. Rev. Mol. Cell Biol. 8, 284–295 [DOI] [PubMed] [Google Scholar]
- 9. Nagy Z., Tora L. (2007) Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 26, 5341–5357 [DOI] [PubMed] [Google Scholar]
- 10. Wang T., Kobayashi T., Takimoto R., Denes A. E., Snyder E. L., el-Deiry W. S., Brachmann R. K. (2001) hADA3 is required for p53 activity. EMBO J. 20, 6404–6413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Germaniuk-Kurowska A., Nag A., Zhao X., Dimri M., Band H., Band V. (2007) Ada3 requirement for HAT recruitment to estrogen receptors and estrogen-dependent breast cancer cell proliferation. Cancer Res. 67, 11789–11797 [DOI] [PubMed] [Google Scholar]
- 12. Vernarecci S., Ornaghi P., Bâgu A., Cundari E., Ballario P., Filetici P. (2008) Gcn5p plays an important role in centromere kinetochore function in budding yeast. Mol. Cell. Biol. 28, 988–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paolinelli R., Mendoza-Maldonado R., Cereseto A., Giacca M. (2009) Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nat. Struct. Mol. Biol. 16, 412–420 [DOI] [PubMed] [Google Scholar]
- 14. Orpinell M., Fournier M., Riss A., Nagy Z., Krebs A. R., Frontini M., Tora L. (2010) The ATAC acetyl transferase complex controls mitotic progression by targeting non-histone substrates. EMBO J. 29, 2381–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar A., Zhao Y., Meng G., Zeng M., Srinivasan S., Delmolino L. M., Gao Q., Dimri G., Weber G. F., Wazer D. E., Band H., Band V. (2002) Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Mol. Cell. Biol. 22, 5801–5812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piña B., Berger S., Marcus G. A., Silverman N., Agapite J., Guarente L. (1993) ADA3: a gene, identified by resistance to GAL4-VP16, with properties similar to and different from those of ADA2. Mol. Cell. Biol. 13, 5981–5989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Horiuchi J., Silverman N., Marcus G. A., Guarente L. (1995) ADA3, a putative transcriptional adaptor, consists of two separable domains and interacts with ADA2 and GCN5 in a trimeric complex. Mol. Cell. Biol. 15, 1203–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saleh A., Lang V., Cook R., Brandl C. J. (1997) Identification of native complexes containing the yeast coactivator/repressor proteins NGG1/ADA3 and ADA2. J. Biol. Chem. 272, 5571–5578 [DOI] [PubMed] [Google Scholar]
- 19. Eberharter A., Sterner D. E., Schieltz D., Hassan A., Yates J. R., 3rd, Berger S. L., Workman J. L. (1999) The ADA complex is a distinct histone acetyltransferase complex in Saccharomyces cerevisiae. Mol. Cell. Biol. 19, 6621–6631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nag A., Germaniuk-Kurowska A., Dimri M., Sassack M. A., Gurumurthy C. B., Gao Q., Dimri G., Band H., Band V. (2007) An essential role of human Ada3 in p53 acetylation. J. Biol. Chem. 282, 8812–8820 [DOI] [PubMed] [Google Scholar]
- 21. Zeng M., Kumar A., Meng G., Gao Q., Dimri G., Wazer D., Band H., Band V. (2002) Human papilloma virus 16 E6 oncoprotein inhibits retinoic X receptor-mediated transactivation by targeting human ADA3 coactivator. J. Biol. Chem. 277, 45611–45618 [DOI] [PubMed] [Google Scholar]
- 22. Zhao Y., Lang G., Ito S., Bonnet J., Metzger E., Sawatsubashi S., Suzuki E., Le Guezennec X., Stunnenberg H. G., Krasnov A., Georgieva S. G., Schüle R., Takeyama K., Kato S., Tora L., Devys D. (2008) A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol Cell 29, 92–101 [DOI] [PubMed] [Google Scholar]
- 23. Todaro G. J., Green H. (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weinberg R. A. (1995) The retinoblastoma protein and cell cycle control. Cell 81, 323–330 [DOI] [PubMed] [Google Scholar]
- 25. Dyson N. (1998) The regulation of E2F by pRB family proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 26. Nourse J., Firpo E., Flanagan W. M., Coats S., Polyak K., Lee M. H., Massague J., Crabtree G. R., Roberts J. M. (1994) Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature 372, 570–573 [DOI] [PubMed] [Google Scholar]
- 27. Reynisdóttir I., Polyak K., Iavarone A., Massagué J. (1995) Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev. 9, 1831–1845 [DOI] [PubMed] [Google Scholar]
- 28. Sherr C. J., Roberts J. M. (1999) CDK inhibitors: positive and negative regulators of G1 phase progression. Genes Dev. 13, 1501–1512 [DOI] [PubMed] [Google Scholar]
- 29. Besson A., Gurian-West M., Chen X., Kelly-Spratt K. S., Kemp C. J., Roberts J. M. (2006) A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 20, 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carrano A. C., Eytan E., Hershko A., Pagano M. (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1, 193–199 [DOI] [PubMed] [Google Scholar]
- 31. Bretones G., Acosta J. C., Caraballo J. M., Ferrándiz N., Gómez-Casares M. T., Albajar M., Blanco R., Ruiz P., Hung W. C., Albero M. P., Perez-Roger I., León J. (2011) SKP2 oncogene is a direct MYC target gene, and MYC down-regulates p27(KIP1) through SKP2 in human leukemia cells. J. Biol. Chem. 286, 9815–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J., Luo Q., Yuan Y., Huang X., Cai W., Li C., Wei T., Zhang L., Yang M., Liu Q., Ye G., Dai X., Li B. (2010) Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Mol. Cell. Biol. 30, 5621–5635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang M., Waterman M. L., Brachmann R. K. (2008) hADA2a and hADA3 are required for acetylation, transcriptional activity, and proliferative effects of β-catenin. Cancer Biol. Ther. 7, 120–128 [DOI] [PubMed] [Google Scholar]
- 34. Bhaskara S., Chyla B. J., Amann J. M., Knutson S. K., Cortez D., Sun Z. W., Hiebert S. W. (2008) Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 30, 61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burgess R. J., Zhou H., Han J., Zhang Z. (2010) A role for Gcn5 in replication-coupled nucleosome assembly. Mol. Cell 37, 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grau B., Popescu C., Torroja L., Ortuño-Sahagún D., Boros I., Ferrús A. (2008) Transcriptional adaptor ADA3 of Drosophila melanogaster is required for histone modification, position effect variegation, and transcription. Mol. Cell. Biol. 28, 376–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ciemerych M. A., Sicinski P. (2005) Cell cycle in mouse development. Oncogene 24, 2877–2898 [DOI] [PubMed] [Google Scholar]
- 38. Yao T. P., Oh S. P., Fuchs M., Zhou N. D., Ch'ng L. E., Newsome D., Bronson R. T., Li E., Livingston D. M., Eckner R. (1998) Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93, 361–372 [DOI] [PubMed] [Google Scholar]
- 39. Yamauchi T., Yamauchi J., Kuwata T., Tamura T., Yamashita T., Bae N., Westphal H., Ozato K., Nakatani Y. (2000) Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc. Natl. Acad. Sci. U.S.A. 97, 11303–11306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kasper L. H., Fukuyama T., Biesen M. A., Boussouar F., Tong C., de Pauw A., Murray P. J., van Deursen J. M., Brindle P. K. (2006) Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol. Cell. Biol. 26, 789–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guelman S., Kozuka K., Mao Y., Pham V., Solloway M. J., Wang J., Wu J., Lill J. R., Zha J. (2009) The double-histone-acetyltransferase complex ATAC is essential for mammalian development. Mol. Cell. Biol. 29, 1176–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herceg Z., Hulla W., Gell D., Cuenin C., Lleonart M., Jackson S., Wang Z. Q. (2001) Disruption of Trrap causes early embryonic lethality and defects in cell cycle progression. Nat. Genet. 29, 206–211 [DOI] [PubMed] [Google Scholar]
- 43. Liu X., Tesfai J., Evrard Y. A., Dent S. Y., Martinez E. (2003) c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. J. Biol. Chem. 278, 20405–20412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu X., Vorontchikhina M., Wang Y. L., Faiola F., Martinez E. (2008) STAGA recruits Mediator to the MYC oncoprotein to stimulate transcription and cell proliferation. Mol. Cell. Biol. 28, 108–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davis A. C., Wims M., Spotts G. D., Hann S. R., Bradley A. (1993) A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 7, 671–682 [DOI] [PubMed] [Google Scholar]
- 46. Martinez E., Palhan V. B., Tjernberg A., Lymar E. S., Gamper A. M., Kundu T. K., Chait B. T., Roeder R. G. (2001) Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo. Mol. Cell. Biol. 21, 6782–6795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Torok M. S., Grant P. A. (2004) Histone acetyltransferase proteins contribute to transcriptional processes at multiple levels. Adv. Protein Chem. 67, 181–199 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.