

Background: The cytoplasmic adaptor protein CCM2 interacts with the TrkA receptor tyrosine kinase to induce pediatric tumor cell death.
Results: STK25 interacts with CCM2, and its kinase activity is necessary for TrkA-dependent death of medulloblastoma cells.
Conclusion: STK25 mediates TrkA-CCM2 death signaling in medulloblastoma cells.
Significance: These findings identify a downstream catalytic effector for receptor tyrosine kinase death signaling.
Keywords: Cell Death, Neuroblastoma, Neurotrophic Factor, Neurotrophins, Receptor Tyrosine Kinase, CCM2, GCKIII Kinase, NGF, TrkA, Medulloblastoma
Abstract
The TrkA receptor tyrosine kinase induces death in medulloblastoma cells via an interaction with the cerebral cavernous malformation 2 (CCM2) protein. We used affinity proteomics to identify the germinal center kinase class III (GCKIII) kinases STK24 and STK25 as novel CCM2 interactors. Down-modulation of STK25, but not STK24, rescued medulloblastoma cells from NGF-induced TrkA-dependent cell death, suggesting that STK25 is part of the death-signaling pathway initiated by TrkA and CCM2. CCM2 can be phosphorylated by STK25, and the kinase activity of STK25 is required for death signaling. Finally, STK25 expression in tumors is correlated with positive prognosis in neuroblastoma patients. These findings delineate a death-signaling pathway downstream of neurotrophic receptor tyrosine kinases that may provide targets for therapeutic intervention in pediatric tumors of neural origin.
Introduction
The Trk family of neurotrophin receptors are critical regulators of cell survival in the nervous system (1), but paradoxically, TrkA has also been reported to induce cell death in pediatric tumor cells of neural origin (2). This atypical function of TrkA is still largely unknown and is of keen interest in light of the fact that high expression of TrkA is strongly correlated with positive prognosis in neuroblastoma patients (3). We recently identified the protein product of the cerebral cavernous malformation 2 (CCM2) gene as a cytoplasmic interactor of TrkA that mediates its death effects in neuroblastoma and medulloblastoma (4). CCM2 is one of three genes mutated in patients with cerebral cavernous malformations (CCM),3 a common class of vascular malformations in the central nervous system (5). The protein products of these three genes, CCM1, CCM2, and CCM3, interact with each other and with numerous other partners to form a diversity of complexes that may mediate diverse functions in different cell types (6). However, CCM2 was the only CCM-related gene that could be linked to positive prognosis in neuroblastoma patients, in correlation with TrkA, suggesting that the signaling pathways mediating TrkA-CCM2 death signaling in cells of neural origin might be distinct from the pathways leading to CCM effects on the vasculature (2, 4).
CCM2 is a cytoplasmic protein composed of two domains, an N-terminal phosphotyrosine binding (PTB) domain and a C-terminal region with no obvious structural homologies or known function, termed the Karet domain (4). CCM2 interacts with TrkA via its PTB domain, but the interacting epitope in TrkA lacks phosphorylatable residues, suggesting that CCM2 serves as an adaptor that links to downstream effectors rather than directly impacting on TrkA signaling. Because both the PTB and the Karet domains of CCM2 are required for mediation of TrkA death signaling (4), we hypothesized that the Karet domain links TrkA-CCM2 to downstream effectors. Here we describe a proteomics screen for interactors of the Karet domain of CCM2, leading to identification of STK25 as an effector of death signaling initiated by TrkA and CCM2.
EXPERIMENTAL PROCEDURES
Antibodies, DNA Constructs, shRNAs, and Cells
Primary antibodies were acquired as follows. TrkA-RTA was a kind gift of Dr. Louis Reichardt (University of California, San Francisco, CA); HA 16B12 was from Covance; STK25 ab56654, STK24 ab51137, CCM2 ab123930, and GFP ab6556 were from Abcam; phospho-Mst4(p178)/Mst3(pT190)/Stk25(pT174) were from Epitomics; and Ysk1 sc-6865 and Trk sc-11 were from Santa Cruz Biotechnology.
Human Stk24 (BC035578) and Stk25 (BC007852) cDNAs were cloned into pcDNA3-FLAG using EcoRI and NotI sites. The STK25 mutants T174A, T174D, and D158A were cloned into pcDNA5/FRT/TO (Invitrogen). pLC-Stk25wt-GFP and pLC-Stk25K49R-GFP were a kind gift of Brian Howell (Upstate Medical University, Syracuse, NY). HA-tagged CCM2, PTB, and Karet expression constructs were as described (4). shRNAs for human STK25 and STK24 were from Sigma.
Medulloblastoma D283MED-TrkA (MB-TrkA) (7) and HEK293 cells were grown and viability assays were performed as described (4). Lentiviral preparations and cell transduction were as described (8).
Proteomics
HEK293 Flp-In T-REx derivatives expressing FLAG-tagged versions of CCM2 domains (supplemental Fig. S1A) were generated as described previously (9), except that pools of cells rather than individual clones were used. Immunopurification with FLAG M2-agarose coupled to mass spectrometry was performed as described previously (10). High confidence interactions were further defined as those identified with at least 20 spectra after removal of promiscuously interacting proteins (supplemental Tables S1 and S2).
Phosphorylation Assays
STK25 was incubated at 30 °C for 1 h with equimolar amounts of GST-CCM2, CCM3, or myelin basic protein in kinase assay buffer (20 mm MOPS, pH 7.2, 25 mm β-glycerophosphate, 5 mm EGTA, 1 mm NaVO4, 1 mm DTT, 10 mm MgCl2, 10 mm MnCl2, 200 μm ATP, and 1.25 μCi of [γ-32P]ATP), resolved by SDS-PAGE, and imaged. Phosphorylation sites on CCM2 were identified by mass spectrometry as described in supplemental Table S3.
Neuroblastoma Expression Analyses
Gene expression data from 478 primary neuroblastoma were analyzed as described previously (4), except for the threshold setting (11) for high versus low risk tumors. A 10-fold cross-validation was performed. Thresholds for high versus low expression were derived from the mean values of the cross-validation.
RESULTS
GCKIII Kinases Interact with CCM2
We sought new effectors of the TrkA-CCM2 death pathway. To this end, we generated tetracycline-inducible isogenic HEK cells stably expressing either full-length CCM2 or its PTB or Karet domains, fused to a FLAG epitope at the N terminus (supplemental Fig. S1A). Stable transfectants were selected and tested for expression of FLAG-CCM2, FLAG-Karet, and FLAG-PTB. Expression was induced in subconfluent cells for 24 h followed by FLAG immunoprecipitation of cell lysates, proteolytic digestion, and peptide identification by mass spectrometry. High confidence interactors for CCM2 or its subdomains are detailed in Fig. 1A and supplemental Tables S1 and S2. In addition to the previously reported interactors CCM1, CCM3, and ICAP1 (6), we found CCM2 or its subdomains interacting with MST4, STK24, and STK25, all members of the germinal center kinase class III (GCKIII) family. STK24 and STK25 appeared to co-precipitate specifically with the Karet domain in these assays and were previously implicated in death signaling (12, 13); hence we focused on these two kinases.
FIGURE 1.

Identification of STK24 and STK25 as CCM2 interactors. A, schematic representation of CCM2 interactions identified by affinity purification and mass spectrometry. Intact lines indicate high confidence interactions, whereas dashed lines indicate putative interactions that did not meet all filtering criteria. For additional details, please see supplemental Tables S1 and S2. B, anti-FLAG IP of HA-CCM2 and TrkA with FLAG-STK24 and FLAG-STK25 from co-transfected HEK cells. C, reverse IP from co-transfected HEK cells using anti-HA antibody. D, co-IP of endogenous STK25 from HEK cells stably expressing FLAG-CCM2. E, co-IP of FLAG-STK24 or FLAG-STK25 from HEK cells co-transfected with HA-tagged CCM2 subdomains PTB or Karet. White arrowhead indicates Karet, and black arrowhead indicates PTB.
We verified STK24 and STK25 interactions with CCM2 by co-immunoprecipitations (co-IPs) from transfected HEK or HeLa cells. HA tagged-CCM2 co-precipitated with FLAG-STK24 or FLAG-STK25 (Fig. 1B). The STK24 and STK25 interactions with CCM2 were confirmed in reverse co-IPs using HA antibody for immunoprecipitation (IP) and FLAG for Western blots (Fig. 1C). It is noteworthy that TrkA is also co-precipitated with STK24 or STK25 (Fig. 1B), either in the presence or in the absence of transfected HA-CCM2 (HEK cells express endogenous CCM2). Finally, we used isogenic HEK cells expressing FLAG-tagged CCM2 under the control of a tetracycline-regulated promoter to further assess CCM2-STK25 interactions at close to endogenous protein levels. STK24 was expressed at much lower levels than STK25 in these cells; hence the experiment was restricted to STK25. In the absence of tetracycline, these cells expressed low basal levels of FLAG-CCM2. Indeed, an interaction between basal levels of FLAG-CCM2 and endogenous STK25 could be detected under these conditions (Fig. 1D), and both STK25-GFP and CCM2-RFP were localized in the cytoplasm in transfected cells (supplemental Fig. S1B).
The affinity proteomics approach used in our screen identified an interaction between the Karet domain of CCM2 and STK24 or STK25. To determine whether this interaction is specific for the Karet domain, we co-transfected the two CCM2 subdomains into HEK cells together with FLAG-tagged-STK24 or STK25. Interestingly, both CCM2 subdomains were co-precipitated with either STK24 or STK25 (Fig. 1E), indicating that there may be multiple points of interaction between CCM2 to GCKIII kinases, or alternatively, that these interactions occur within the framework of a larger complex.
STK25, but Not STK24, Is Implicated in TrkA-dependent Medulloblastoma Cell Death
We tested whether STK24 and STK25 might play a role in TrkA-induced cell death by down-regulating their expression in the medulloblastoma cell line MB-TrkA, which undergoes cell death upon NGF treatment (7). Effective reduction of expression levels was achieved using lentiviral shRNAs against STK24 or STK25 in MB-TrkA cells (Fig. 2A and supplemental Fig. S2). Down-regulation of STK24 did not affect NGF-induced death of MB-TrkA cells, and similar levels of viability were observed in control and in STK24-down-regulated cells (Fig. 2, A and B, and supplemental Fig. S2). In contrast, down-regulation of STK25 in MB-TrkA cells protected them from NGF-induced TrkA-dependent cell death (Fig. 2, A and B, and supplemental Fig. S2). The anti-STK25 shRNA targets the 3′-UTR region in the endogenous transcript; hence we used a construct containing the ORF of STK25 fused to GFP to express STK25 that is not affected by the shRNA (supplemental Fig. S2). Expression of STK25-GFP in MB-TrkA cells did not affect viability of the cells in the absence of NGF treatment (data not shown); however, transfection with the STK25-GFP construct restored sensitivity of shRNA-treated MB-TrkA cells to NGF-induced death (Fig. 2B), confirming that the shRNA effect is specific for STK25. Moreover, NGF treatment caused an increase in STK25 protein levels in MB-TrkA cells, but had no effect on STK24 levels (supplemental Fig. S2). Finally, we found that STK25 transfection induces death in HEK cells, as reported previously (12), and that STK25-induced death is increased upon co-transfection with TrkA and CCM2 (supplemental Fig. S2). In contrast, we did not observe death of HEK cells upon STK24 transfection either alone or in combination with TrkA and CCM2 (supplemental Fig. S2). Thus, although both STK24 and STK25 can interact with CCM2, only STK25 mediated TrkA-CCM2-dependent death.
FIGURE 2.

STK25 is required for NGF-induced death in pediatric tumor cells of neural origin. A, representative fields of shRNA-transduced MB-TrkA cells after 72 h in culture with or without NGF (100 ng/ml). Scale bar, 40 μm. B, viability of shRNA-transduced MB-TrkA cells as measured by XTT after 72 h of NGF treatment. n = 4, average ± S.D., *** indicates p < 0.001 (Student's t test). C, overall survival (OS) of neuroblastoma patients as correlated with STK24 and STK25 mRNA levels in the tumors. 4.6e-4, 4.6 × 10−4; “6.8e-11, 6.8 × 10−11.
We then examined mRNA expression levels of STK24 and STK25 in correlation with clinical outcome in 478 neuroblastoma patients. TrkA and CCM2 expression was previously shown to correlate with good prognosis in this cohort (4). Expression levels of STK25, but not STK24, were significantly correlated with those of TrkA and CCM2 (Pearson's correlation, r(STK25,CCM2) = 0.41, p < 2 × 10−16; r(STK25,TrkA) = 0.23, p < 2 × 10−7). We then defined tumor expression level thresholds for STK25 and STK24, as described previously (4), and determined correlation with patient survival. A significantly better outcome was observed for patients with STK25 expression values above the threshold, whereas in contrast, higher expression levels of STK24 correlated with unfavorable clinical outcome (Fig. 2C). These data support a role for STK25, but not STK24, in TrkA-dependent retreat of neuroblastoma in patients in vivo.
NGF Modulates STK25 Activity and STK25 Binding with TrkA and CCM2
To determine whether NGF regulates STK25 activity, we examined phosphorylation of Thr-174 in STK25 after NGF stimulation of MB-TrkA cells. STK25 phosphorylation increased upon NGF treatment, in parallel with an increase in co-precipitation of STK25 with TrkA (Fig. 3). NGF treatment also increased CCM2 co-precipitation with STK25 (Fig. 3).
FIGURE 3.
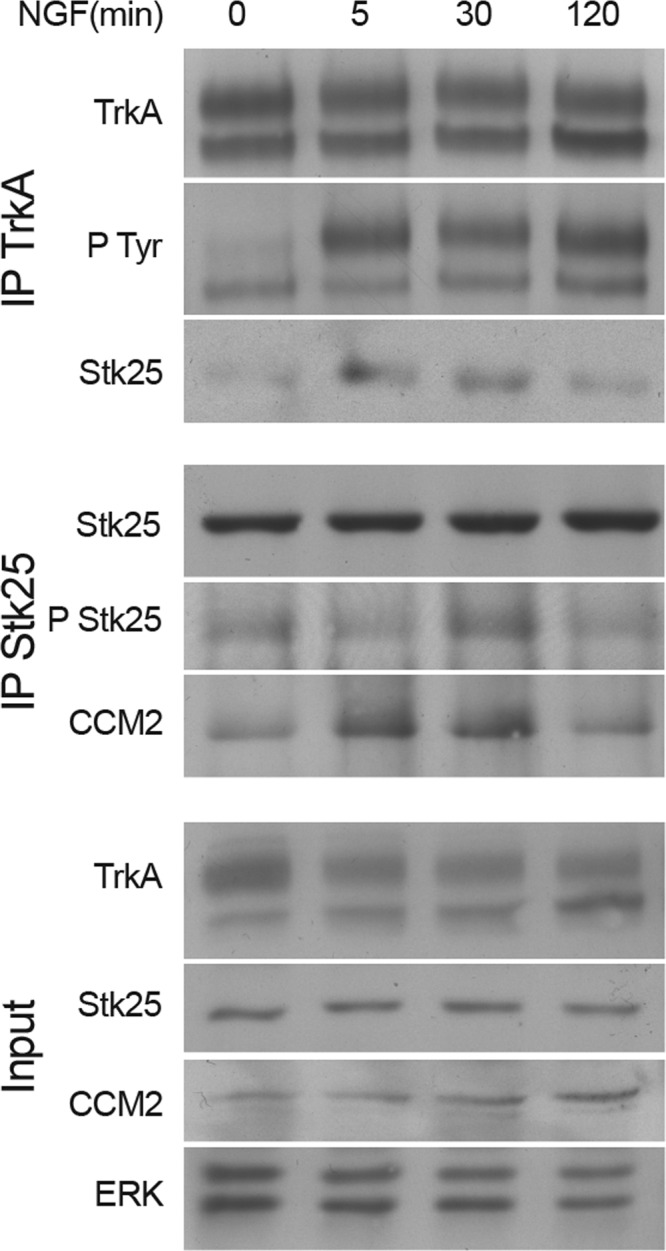
NGF modulates STK25 phosphorylation and interaction with TrkA and CCM2. MB-TrkA cells were treated with NGF (100 ng/ml) for the indicated times, and cell lysates were then subjected to IP and Western blots as indicated. n = 3 in all cases. P Tyr, phospho-Tyr; P Stk25, phospho-STK25.
STK25 Kinase Activity Is Required for NGF-induced Medulloblastoma Cell Death
We then sought to further characterize the STK25-CCM2 interaction with the help of specific constructs mutated at residues affecting kinase activity of the protein (14). Two activity-reducing mutants (T174A and K49R) and one constitutively active kinase mutant (T174D) co-precipitated with CCM2 in a similar manner as wild type STK25, whereas another kinase-reducing mutation, D158A, revealed a much reduced interaction with CCM2 (Fig. 4, A and B). Thus, although kinase activity per se is not required for the interaction, Asp-158 in STK25 facilitates efficient CCM2 co-precipitation. A kinase activity assay demonstrated CCM2 phosphorylation by STK25 (Fig. 4C). Mass spectrometric analyses further revealed Ser-384 as the major site for CCM2 phosphorylation by STK25 (Fig. 4D), in addition to four less prominent sites (supplemental Table S3). We therefore tested whether STK25 kinase activity is required for TrkA-dependent death. MB-TrkA cells with reduced STK25 expression were transduced with lentiviral vectors expressing shRNA-resistant STK25 constructs, either the wild type or the K49R kinase-dead mutant (supplemental Fig. S2D). There was no reduction in the viability of NGF-treated cells expressing the K49R mutant (Fig. 4E), indicating that STK25 kinase activity is required for TrkA-CCM2-dependent death in medulloblastoma cells.
FIGURE 4.

STK25 kinase activity is required for NGF-induced death. A, co-IP of HA-CCM2 from HEK cells co-transfected with FLAG-tagged STK25 and mutants. Relative amounts of HA-CCM2 co-precipitated with the different STK25 mutants are shown in the graph below (n = 3). B, co-IP of GFP fusions of STK25 or STK25K49R with HA-CCM2 from co-transfected HEK cells. Relative levels of co-precipitation are shown below (n = 3). C, in vitro kinase assay. STK25 was incubated with either GST-CCM2 or the known substrates CCM3 or myelin basic protein (MBP). Autoradiography (32p) and Coomassie Blue staining are shown. D, representative spectrum for a phospho-serine 384 CCM2 peptide. For further details, please see supplemental Table S3. AMU, atomic mass units. E, viability of shRNA-transduced MB-TrkA cells after 72 h of culture with NGF (100 ng/ml). Note that the kinase-dead K49R STK25 mutant cannot restore cell death response in MB-TrkA cells silenced for STK25 (n = 4). Average ± S.D., *** indicates p < 0.001, ** indicates p < 0.01 for all panels (t test).
DISCUSSION
We have previously shown that CCM2 is a cytoplasmic interactor of TrkA that is necessary for NGF-induced cell death in pediatric tumor cells (4). Here we found that the GCKIII serine-threonine kinases STK24, STK25, and MST4 are new molecular partners of CCM2, and we identified a role for STK25 in TrkA-CCM2-dependent cell death in medulloblastoma cells. Although all three GCKIII kinases were identified as CCM2 interactors in our screen, initial results with the Karet domain directed our focus to STK24 and STK25. Further experiments revealed that both the PTB and the Karet domains of CCM2 interact with STK24 and STK25 and that STK25 can phosphorylate CCM2. Interestingly, GCKIII kinases are known to interact with CCM3 (10, 15, 16), and CCM3 was also reported to be an STK25 substrate (15). A recent phosphorylation mapping study on a CCM1-associated complex demonstrated multiple phosphorylation sites on CCM2 (17); however, to date, the kinases involved in CCM2 phosphorylation are not known. Our results suggest that STK25 is likely one of the serine/threonine kinases responsible for CCM2 phosphorylation.
Our data specifically implicate STK25 in NGF-induced cell death in medulloblastoma cells. The apparent specificity of the pathway for STK25 over STK24 is intriguing, given the fact that both these kinases have been implicated in cell death in different systems (18). STK25 depletion by RNAi rescues TrkA-expressing medulloblastoma cells from NGF-induced death, whereas similar depletion of STK24 had no effect. Introduction of shRNA-resistant STK25 restored cell death in STK25-silenced cells, whereas the kinase-dead K49R STK25 mutant had no such effect. NGF treatment of TrkA-expressing medulloblastoma cells activates STK25 and increases its interactions with both TrkA and CCM2. Finally, STK25 expression levels in tumors correlate with those of CCM2 and TrkA and with good prognosis in neuroblastoma patients. Thus, active STK25 kinase participates in TrkA- and CCM2-mediated death in pediatric tumor cells of neural origin.
STK25 was originally identified as an oxidant stress-activated kinase (13) and subsequently implicated in apoptotic death induced by chemical anoxia (12). Nonapoptotic roles have been described for STK25 in hippocampal neurons (19), as indeed have been described for TrkA or CCM2 in other cell types (2). The extent of cell death induced by STK25 was suggested to depend on its expression levels (12), and indeed, we observed an increase in STK25 levels upon NGF stimulation of TrkA-expressing medulloblastoma cells and a correlation between STK25 expression levels and neuroblastoma prognosis, as previously shown for CCM2 and TrkA (4). Overexpression of TrkA from the tau locus has been shown to cause extensive neuronal death in vivo (20). Caspase activation is required for both CCM2-mediated death in medulloblastoma cells (4) and STK25-mediated cell death after chemical anoxia (12). Thus, the TrkA-CCM2-STK25 pathway likely represents a switch mechanism activated by correlated and increased expression, leading to activation of canonical apoptosis pathways. Future elucidation of the mechanisms underlying this selectivity should provide new avenues to target pediatric tumors of neural origin.
Supplementary Material
Acknowledgments
We thank Louis Reichardt, Frank Sicheri, and Brian Howell for generous gifts of reagents.
This work was supported by the European Union (Endocyte Network), the German-Israeli Cooperation in Cancer Research (CA 140), the Canadian Institutes of Health Research, Nationales Genom-Forschungs-Netz (NGFN+) project ENGINE (01GS0898), and the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie CancerSys-Verbundprojekt: MYCNET (0316076C), a Lombroso postdoctoral fellowship (to B. C.), and a Banting and Best Canada Graduate Scholarship (to M. J. K.).

This article contains supplemental Figs. S1 and S2 and Tables S1–S3.
- CCM
- cerebral cavernous malformation protein
- PTB
- phosphotyrosine binding
- GCKIII
- germinal center kinases group III
- MB-TrkA
- medulloblastoma cell line D283MED-TrkA
- IP
- immunoprecipitation
- XTT
- 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt.
REFERENCES
- 1. Huang E. J., Reichardt L. F. (2003) Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642 [DOI] [PubMed] [Google Scholar]
- 2. Harel L., Costa B., Fainzilber M. (2010) On the death Trk. Dev. Neurobiol. 70, 298–303 [DOI] [PubMed] [Google Scholar]
- 3. Brodeur G. M., Minturn J. E., Ho R., Simpson A. M., Iyer R., Varela C. R., Light J. E., Kolla V., Evans A. E. (2009) Trk receptor expression and inhibition in neuroblastomas. Clin. Cancer Res. 15, 3244–3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harel L., Costa B., Tcherpakov M., Zapatka M., Oberthuer A., Hansford L. M., Vojvodic M., Levy Z., Chen Z. Y., Lee F. S., Avigad S., Yaniv I., Shi L., Eils R., Fischer M., Brors B., Kaplan D. R., Fainzilber M. (2009) CCM2 mediates death signaling by the TrkA receptor tyrosine kinase. Neuron 63, 585–591 [DOI] [PubMed] [Google Scholar]
- 5. Riant F., Bergametti F., Ayrignac X., Boulday G., Tournier-Lasserve E. (2010) Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J. 277, 1070–1075 [DOI] [PubMed] [Google Scholar]
- 6. Faurobert E., Albiges-Rizo C. (2010) Recent insights into cerebral cavernous malformations: a complex jigsaw puzzle under construction. FEBS J. 277, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muragaki Y., Chou T. T., Kaplan D. R., Trojanowski J. Q., Lee V. M. (1997) Nerve growth factor induces apoptosis in human medulloblastoma cell lines that express TrkA receptors. J. Neurosci. 17, 530–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tiscornia G., Singer O., Verma I. M. (2006) Production and purification of lentiviral vectors. Nat. Protoc. 1, 241–245 [DOI] [PubMed] [Google Scholar]
- 9. Dunham W. H., Larsen B., Tate S., Badillo B. G., Goudreault M., Tehami Y., Kislinger T., Gingras A. C. (2011) A cost-benefit analysis of multidimensional fractionation of affinity purification-mass spectrometry samples. Proteomics 11, 2603–2612 [DOI] [PubMed] [Google Scholar]
- 10. Goudreault M., D'Ambrosio L. M., Kean M. J., Mullin M. J., Larsen B. G., Sanchez A., Chaudhry S., Chen G. I., Sicheri F., Nesvizhskii A. I., Aebersold R., Raught B., Gingras A. C. (2009) A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol. Cell Proteomics 8, 157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hothorn T., Lausen B. (2003) On the exact distribution of maximally selected rank statistics. Comput Stat. Data Anal. 43, 121–137 [Google Scholar]
- 12. Nogueira E., Fidalgo M., Molnar A., Kyriakis J., Force T., Zalvide J., Pombo C. M. (2008) SOK1 translocates from the Golgi to the nucleus upon chemical anoxia and induces apoptotic cell death. J. Biol. Chem. 283, 16248–16258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pombo C. M., Bonventre J. V., Molnar A., Kyriakis J., Force T. (1996) Activation of a human Ste20-like kinase by oxidant stress defines a novel stress response pathway. EMBO J. 15, 4537–4546 [PMC free article] [PubMed] [Google Scholar]
- 14. Preisinger C., Short B., De Corte V., Bruyneel E., Haas A., Kopajtich R., Gettemans J., Barr F. A. (2004) YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3ζ. J. Cell Biol. 164, 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Voss K., Stahl S., Schleider E., Ullrich S., Nickel J., Mueller T. D., Felbor U. (2007) CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 8, 249–256 [DOI] [PubMed] [Google Scholar]
- 16. Ma X., Zhao H., Shan J., Long F., Chen Y., Chen Y., Zhang Y., Han X., Ma D. (2007) PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol. Biol. Cell 18, 1965–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim J., Sherman N. E., Fox J. W., Ginsberg M. H. (2011) Phosphorylation sites in the cerebral cavernous malformations complex. J. Cell Sci. 124, 3929–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takeda K., Naguro I., Nishitoh H., Matsuzawa A., Ichijo H. (2011) Apoptosis signaling kinases: from stress response to health outcomes. Antioxid. Redox Signal. 15, 719–761 [DOI] [PubMed] [Google Scholar]
- 19. Matsuki T., Matthews R. T., Cooper J. A., van der Brug M. P., Cookson M. R., Hardy J. A., Olson E. C., Howell B. W. (2010) Reelin and Stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell 143, 826–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nikoletopoulou V., Lickert H., Frade J. M., Rencurel C., Giallonardo P., Zhang L., Bibel M., Barde Y. A. (2010) Neurotrophin receptors TrkA and TrkC cause neuronal death, whereas TrkB does not. Nature 467, 59–63 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.