

Abstract
Background
Patients with gene expression profiling-defined high-risk myeloma in relapse have poor outcomes with current therapies. We tested whether natural killer cells expanded by co-culture with K562 cells transfected with 41BBL and membrane-bound interleukin-15 could kill myeloma cells with a high-risk gene expression profile in vitro and in a unique model which recapitulates human myeloma.
Design and Methods
OPM2 and high-risk primary myeloma tumors were grown in human fetal bone implanted into non-obese diabetic severe combined immunodeficiency mice with a deficient interleukin-2 receptor gamma chain. These mice are devoid of endogenous natural killer and T-cell activity and were used to determine whether adoptively transferred expanded natural killer cells could inhibit myeloma growth and myeloma-associated bone destruction.
Results
Natural killer cells from healthy donors and myeloma patients expanded a median of 804- and 351-fold, respectively, without significant T-cell expansion. Expanded natural killer cells killed both allogeneic and autologous primary myeloma cells avidly via a perforin-mediated mechanism in which the activating receptor NKG2D, natural cytotoxicity receptors, and DNAX-accessory molecule-1 played a central role. Adoptive transfer of expanded natural killer cells inhibited the growth of established OPM2 and high-risk primary myeloma tumors grown in the murine model. The transferred, expanded natural killer cells proliferated in vivo in an interleukin-2 dose-dependent fashion, persisted up to 4 weeks, were readily detectable in the human bone, inhibited myeloma growth and protected bone from myeloma-induced osteolysis.
Conclusions
These studies provide the rationale for testing expanded natural killer cells in humans.
Keywords: myeloma, expansion, natural killer cells
Introduction
Multiple myeloma (MM) is a malignant plasma cell disorder with debilitating symptoms related to anemia, immunosuppression, bone destruction, and renal failure.1 Significant advances have been made by combining novel agents with autologous peripheral blood stem cell transplantation which allows for long-term disease-free survival in the majority of transplant-eligible patients. However, the prognosis remains poor in approximately 15% of patients who have aggressive disease best captured by a high-risk gene expression profile (GEP).2 The vast majority of these patients enjoy only short-term remissions due to the persistence of chemotherapy-refractory MM cells. Therapeutic modalities that are non-cross-resistant with chemotherapy could offer new hope for this subset of patients with a very grave prognosis.
Human natural killer (NK) cells, which contribute to host antitumor defense reactions, represent a population of large granular lymphocytes that express a CD3−/CD56+ phenotype.3 NK cells discriminate between normal cells and transformed cells via a balance of inhibitory and activating signals induced by interactions between NK cell receptors and target cell ligands, with the inhibitory signals being dominant.4–6 NK cells from killer immunoglobulin-like receptor-ligand (KIR-L)-mismatched donors can exert a potent anti-leukemic effect and are highly effective in preventing relapse after haplo-identical T-cell-depleted allogeneic peripheral blood stem cell transplantation in patients with relapsed acute myeloid leukemia with a low tumor load after re-induction therapy.7,8 Although KIR-L-mismatched NK cells from haplo-identical donors are also cytotoxic to MM cells in vitro, infusion of an adequate dose of alloreactive NK cells is a challenge and therapy is limited to patients with a suitable donor.9
Irradiated K562 cells transfected with 41BBL and membrane-bound interleukin (IL)15 (K562-mb15-41BBL) led to the expansion of large numbers of highly activated NK cells capable of controlling K562 leukemia cell growth in a murine model.10 We tested whether we could apply this system to expand NK cells from both MM patients and healthy donors (HD) and investigated whether these expanded-NK (exp-NK) cells could kill MM cell lines and primary MM cells in vitro. Furthermore, we established aggressive human OPM2 cells and high-risk primary MM tumors in a protective human bone microenvironment in non-obese diabetic severe combined immunodeficiency mice with a deficient IL2 receptor gamma chain (NOD/scid/IL2Rγnull), which are devoid of endogenous NK and T-cell activity, and studied whether adoptively transferred exp-NK cells could inhibit tumor growth and consequently modulate myeloma-associated bone destruction.
Design and Methods
Details on cell lines, samples, cell selection, NK cell expansion, and gene expression profiling can be found in the Online Supplementary Design and Methods.
Phenotyping of cell subsets by flow cytometry analysis
Paired non-exp and exp-NK cells were cryopreserved on day 0 or day 10–14, respectively, then thawed and analyzed simultaneously for cell surface protein expression as determined by flow cytometry using the viability stain 7AAD (Sigma, St Louis, MO, USA) and the phenotyping antibodies listed in Online Supplementary Table S1. A FACSCalibur flow cytometer with CellQuest software was used for data acquisition (BD Biosciences, Mountain View, CA, USA). For each sample, 3000 7AAD−/CD56+/CD3− events were collected. Median fluorescence intensity (MFI) ratios (MFI day 10 to 14/MFI day 0) and percent positive cell ratios (post-/pre-expansion) were calculated for each cell surface protein stained.
51Cr release cytotoxicity assays
Details on 51Cr release assays are provided in the Online Supplementary Design and Methods.
Blocking experiments
To test the role of Fas-L and TRAIL-mediated killing, 5 μg/mL anti-Fas-L and/or 2.5 μg/mL anti-TRAIL or control mouse IgG1 antibody (Online Supplementary Table S1) was added to effectors at the time of plating and left in the wells for the rest of the assay. To block perforin-mediated cytotoxicity, effectors were pretreated for 2 h in 100 nM concanamycin A (CMA; Sigma).11 For NK cell receptor blocking experiments, we first incubated NK cells with human IgG (1 μg/105 cells) (Invitrogen) on ice for 20 min to prevent antibody-dependent cell-mediated cytotoxicity and then at room temperature with various antibodies (Online Supplementary Table S1) before adding the target cells. NK cell ligands on myeloma cells were blocked by incubating recombinant human fusion proteins or antibodies (Online Supplementary Table S1) for 2 h prior to plating cells at a final concentration of 10 μg/mL.
Murine model
NOD.Cg-PrkdcscidIL2rgtm1Wjl/SzJ (NOD/scid/IL2Rγnull) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Human (hu) fetal bones (Advanced Bioscience Resources; Alameda, CA, USA) were implanted subcutaneously into the mice as previously reported.12 After engraftment (4–5 weeks), 3×105 OPM2 cells transfected with luciferase (OPM2-Lu) or MM patients’ cells in 50 μL phosphate-buffered saline (PBS) were injected into the hu bone. MM cells were allowed to grow until the tumor was visible by bio-imaging (7 days) or detectable by a human immunoglobulin (huIg) ELISA, at which time mice with comparable baseline tumor burden were randomized to treatment groups. For OPM2-Lu experiments the groups included: (i) PBS vehicle; (ii) 40×106 exp-NK cells divided into two doses, 48 h apart; (iii) 160×106 exp-NK cells divided into four doses, each 48 hours apart. Exp-NK cells were transferred through the lateral tail vein. For primary MM experiments only treatment groups (i) and (iii) were used. Weekly imaging and/or ELISA was performed to assess tumor burden. Details on live animal imaging and blood/plasma collection are described in the Online Supplementary Design and Methods.
Weekly expanded natural killer cell persistence and phenotyping
Peripheral blood cells from mice were re-suspended in PBS and stained with a cocktail of antibodies comprising CD3-FITC and CD56-PE (Online Supplementary Table S1).
In vivo expansion assays
Exp-NK cells were labeled with 10 μM carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen) for 6 min then washed. Some of the cells were analyzed immediately to obtain a baseline level of fluorescence. CFSE-labeled exp-NK cells (40×106) were injected into the myelomatous mice through the tail vein. Peripheral blood mononuclear cells collected after adoptive transfer of exp-NK were stained with CD3 and CD56 antibodies. CD3−/CD56+ gated cells were analyzed with ModFit LT (Verity Software, Topsham, ME, USA).
Histology and immunohistochemistry
Implanted bone tissues were recovered from mice at the time points indicated and fixed in 10% phosphate-buffered formalin for 24 h, decalcified with 5% ethylenediaminetetraacetic acid (pH 7.0) and embedded in paraffin. Sections of 5 μm were stained with hematoxylin and eosin (H&E) for general histology. Osteoclasts were stained with tartrate-resistant acid phosphatase (TRAP) (Sigma-Aldrich, St Louis, MO, USA), and osteoblasts were stained for osteocalcin (QED Bioscience, San Diego, CA, USA). Static histomorphometric analysis was performed as previously described.13
Natural killer cell homing to myelomatous bone
Anti-CD57 was used to identify NK cells since OPM2 MM cells are CD56+. Paraffin sections were stained with CD3 to confirm the absence of T-cells and CD138 to identify plasma cells (Online Supplementary Table S1). Both positive (normal tonsil) and negative (isotype-matched) controls were included. The Online Supplementary Design and Methods provides further details.
Micro-computed tomography analysis
The implanted hu bones were excised at the end of the experiments and fixed in phosphate-buffered 10% formalin (pH 7.4) for 24 h. Bones were analyzed as described previously to obtain information on volume, trabecular thickness, number, and spacing.14
Statistical analysis
Significance levels were determined by two-tailed Student’s t test analysis. A P value of 0.05 or less was considered statistically significant.
Results
K562-mb15-41BBL cells expand natural killer cells, but not T-cells
Peripheral blood mononuclear cells from both HD (n=15) and MM patients (n=30) were co-cultured with K562-mb15-41BBL cells, resulting in dramatic NK cell expansion. By day 7, we observed a median 19-fold NK cell expansion (range, 4–87) and by the day of harvest (day 10–14) the fold expansion had increased to 447 (range, 20–10,430) (Online Supplementary Figure S1A). In contrast, T-cell fold expansion was low (day 7: median, 0.7, range, 0.1 to 5.5; day 14: median, 3.92, range, 0.2 to 60). Differences in NK or T-cell fold expansion were not significant for samples derived from HD versus MM patients, or for newly diagnosed (n=22) versus previously-treated (n=8) MM patients. Online Supplementary Figure S1B demonstrates that cultures were highly enriched with NK cells by day 7 and, on average, comprised 88% of NK cells by day 14. Other cell populations were not highly represented at the time of harvest, with median percentages being 2.2% for T-cells (of which less than 0.2% were γδ T-cells), 7.4% for NKT cells, and less than 1% each for B lymphocytes, monocytes, and myeloid cells. In agreement with Fujisaki et al.,10 cultures with either non-transfected K562 with IL2 or IL2 alone failed to induce significant (≥ 2-fold) NK cell proliferation (data not shown). The exp-NK cells were >95% viable at harvest.
Gene expression profiling and phenotyping analysis of expanded natural killer cells
The GEP of non-exp-NK cells from HD and MM patients was remarkably similar with only one gene being differentially expressed, PRKCι (false discovery rate <0.05; P<3×10−10), a member of the protein kinase C family. Similarly, GEP analysis comparing exp-NK from HD versus MM patients did not reveal differential expression of genes between these two groups. In contrast, the GEP of exp-NK cells from both HD and MM patients was very different when compared to that of non-exp-NK cells (n=16 pairs) with over-expression of 10,639 and under-expression of 26,057 probes by exp-NK cells. The exp-NK cells from HD and MM patients had up-regulated expression of genes associated with cytolytic activity, cytokines and chemokines, activating receptors, adhesion molecules, cell cycle regulators, and genes involved in multiple pathways. The up- or down-regulation of selected cell surface molecules was confirmed by flow cytometry analysis (Figure 1). The change in cell surface expression is shown both as a ratio of post-/pre-expansion MFI values on the receptor positive subset of NK cells and the ratio of the percent positive cells post-/pre-expansion (Online Supplementary Figure S2A, B). There was increased expression of NKG2D and natural cytotoxicity receptors, NKp30, NKp44, and other activating/co-stimulating molecules such as CD26, CD69 and CD70. The adhesion molecules CD54 and CD56 and chemokine receptors CXCR3, 4, and 6 were also increased. Reduced cell surface expression of CD16 was observed on exp-NK cells, consistent with the observed increase in natural cytotoxicity against tumor targets.15 Significant up-regulation of the inhibitory receptors NKG2A and KIR2DL1 was observed. Increases in KIR2DL2/3 and 3DL1 cell surface density were not significant, although the percentage positive cells did increase significantly for KIR2DL2/3.
Figure 1.

Exp-NK cells have an activated phenotype. Flow cytometry confirms the increased cell surface density of NKG2D, NKp30, NKp44, CD26, CD56, CD54, CD69, CD70, and the chemokine receptor CXCR3. Open peaks represent non-exp-NK, shaded peaks represent exp-NK. One representative result from 12 experiments is shown.
Expanded natural killer cells have enhanced cytotoxicity against both primary allogeneic and autologous multiple myeloma cells
The in vitro anti-myeloma potential of exp-NK cells is demonstrated in Figure 2. The NK cell sensitive cell line K562 does not express HLA-class I, and was used as a positive control for maximum lysis. Figure 2A shows the cytolytic capacity of exp-NK cells compared with non-exp-NK against K562 and the MM cell line U266. Exp-NK cells killed K562 and U266 cells significantly more avidly than non-exp-NK cells. Time lapse photography demonstrated the ability of one exp-NK cell to kill multiple targets (Online Supplementary Movie). Similarly, as shown in Figure 2B, primary MM cells were avidly killed by exp-NK cells from HD compared to lower level killing by non-exp-NK cells. Patients’ non-myeloma cells (phytohemagglutinin blasts) were included as a negative control. Purified CD34+ cells were not killed by allogeneic exp-NK cells (data not shown). Importantly, significant lysis of primary MM cells, albeit at a lower level than that of allogeneic MM cells, was also observed with autologous exp-NK cells (Figure 2C) indicating that the exp-NK cells were able to partially overcome KIR-mediated inhibition. MM patient-derived exp-NK cells and HD-derived exp-NK cells killed primary allogeneic MM cells equally well (Figure 2D) and MM cells from GEP-defined high-risk and low-risk patients were killed equally well by exp-NK cells (data not shown). The primary mechanism of exp-NK cell killing of primary MM cells was perforin-mediated, as the perforin inhibitor CMA greatly reduced killing, while blocking TRAIL and Fas-L did not have a significant impact on killing (Figure 2E). More than 80% of exp-NK cells have high expression of perforin, granzyme A and granzyme B (Online Supplementary Figure S3). To determine which activating molecules on exp-NK cells were responsible for increased NK cell activity, we performed receptor blocking experiments by using neutralizing antibodies, alone or in combination, to NKG2D, NCRs, DNAX-accessory molecule (DNAM)-1, CD27, and CD69 receptors. The contribution of these molecules to exp-NK cell cytotoxicity is presented in Figure 2F. Inhibition of NK cell-mediated killing of patients’ MM cells was observed when using anti-NKG2D, anti-NCRs, anti-NKG2D+NCRs, or anti-DNAM-1 antibody, which yielded a mean inhibition of 35%, 47%, 62% and 35%, respectively, at 10:1 effector:target (E:T) ratios. We found that CD27 together with CD69 also played an important role in exp-NK cell lysis of primary MM cells (mean inhibition: 35%, P<0.01). We confirmed these findings by blocking the corresponding ligand on patients’ MM cells and observed similar inhibition when compared to blocking of activating receptors on exp-NK cells (Figure 2G). Almost total abrogation of primary MM killing was observed when all these antibodies were combined, suggesting that these activating receptors function in concert to produce marked increases in exp-NK cell antitumor activity.
Figure 2.

Exp-NK cells (unselected, purity >92%) kill MM cells and this killing is mediated by the perforin pathway and critical activating receptor-ligand interactions. (A) Exp-NK cells kill K562 cells and the MM cell line U266 better than non-exp NK cells (P<0.001; the mean and SEM for nine assays are shown). (B) Exp-NK cells derived from HD kill primary MM cells while killing of patients’ phytohemagglutinin blasts remains low (P<0.001; the mean and SEM for six assays are shown). (C) Exp-NK cells derived from MM patients can induce significant killing of autologous MM cells (P<0.001; the mean and SEM for eight assays are shown). (D) Killing of primary MM cells by exp-NK is similar whether NK cells are derived from MM patients or HD (P>0.2), whereas killing is lower when targets are autologous. (E) HD-derived exp-NK cells kill primary MM cells via a perforin pathway. Blocking effectors with anti-TRAIL or anti-FAS-L antibodies did not significantly reduce the level of killing whereas the addition of the perforin inhibitor concanamycin A (CMA) to the assay reduced killing appreciably. One of three representative assays is shown. Blocking critical NK cell receptors (F) or their ligands on MM cells (G) can inhibit killing of primary MM cells. The mean ±SEM for three independent experiments is shown for (F) and (G).
Cytokine secretion of expanded natural killer cells
After 72 h of co-incubation with non-modified K562 cells, CD56+CD3− exp-NK cells secreted a significant amount of interferon-γ (mean, 488 pg/mL; range, 291–792), compared to cultures without K562 (Online Supplementary Figure S4). The level of the inflammatory cytokine tumor necrosis factor-α level (mean, 16 pg/mL; range, 12–21) following 72 h of stimulation was not altered. Production of IL4 (mean, 4 pg/mL), IL6 (mean, 6 pg/mL), or IL10 (mean, 6 pg/mL) was also not changed. Hence, both the GEP and cytometric bead array analysis revealed increased interferon-γ production compatible with a cytolytic-effector exp-NK cell phenotype.
Expanded natural killer cells inhibit myeloma growth and osteolysis in vivo
We studied the effect of exp-NK cell treatment on NOD/scid/IL2Rγnull mice bearing human MM tumors derived from the luciferase-transfected plasma cell leukemia cell line OPM2. Figure 3A shows bio-images of three representative mice from the untreated, group and the group given 160×106 exp-NK cells. There was a dramatic progression of the OPM2 tumors in the control group whereas significant myeloma growth inhibition was observed in the cohort given 160×106 exp-NK cells. Figure 3B graphically illustrates the fold increase in tumor volume relative to baseline by bio-imaging for all data points. Tumor growth inhibition was observed in an exp-NK cell dose-dependent manner, and the group treated with 160×106 exp-NK cells had a statistically lower tumor burden than the control group (P<0.04).
Figure 3.

Human exp-NK cells significantly inhibit luciferase-transfected OPM2 myeloma tumor growth in the NOD/scid/IL2Rγnull-hu model. (A) Imaging of OPM2 tumors expressing luciferase illustrates the myeloma burden in mice receiving PBS (control) or a total dose of 160×106 exp-NK cells (given by four intravenous injections 48 h apart). Day 0 = date of NK cell injection #1. IL2 (100 U) was given intraperitoneally twice weekly to support NK cell survival in vivo. Three representative mice from each group are shown. (B) The fold tumor volume relative to baseline values was significantly lower in the cohort of mice that received 160×106 exp-NK cells than in either the control group or the group that received a lower NK cell dose (40×106). Each symbol represents one mouse in the different groups at the days indicated and median values are noted for each data set (−).
H&E staining and micro-computed tomography revealed gross loss of bone in the untreated, control group (Figure 4A-E) compared to that in the mice given exp-NK cells (Figure 4F–J). Paraffin sections from the control and exp-NK cell treated groups were stained with H&E for normal histology, TRAP for osteoclasts and osteocalcin to detect osteoblasts. High numbers of osteoclasts (suggestive of increased osteoclastogenesis) were observed in the control group and the numbers were markedly reduced by treatment with exp-NK cells (Figure 4B, G). Conversely, higher numbers of osteoblasts were observed in the exp-NK cell-treated group (Figure 4H) in which bone was preserved (Figure 4I–J).
Figure 4.

Exp-NK cell treatment protects bone from myeloma-induced osteolysis. On day 29, at the termination of the experiment depicted in Figure 3, implanted fetal bones were excised and subjected to immunohistochemistry and micro-computed tomography (CT). H&E, TRAP, and osteocalcin staining revealed gross loss of bone, increased numbers of osteoclasts, and reduced numbers of osteoblasts in the control group (A–C) compared to those in the mice treated with 160×106 exp-NK cells (F–H). Micro-CT further confirmed the dramatic difference in implanted human bone structure between control and exp-NK cell–treated groups. Three-dimensional reconstructions of the bones are shown (D, I), as well as longitudinal sections through the midpoint from each specimen (E, J). Heavy bone resorption is clearly seen in the control group (D, E), while bone resorption is not prominent in the exp-NK cell–treated group (I, J).
In a separate experiment, we demonstrated that allogeneic exp-NK cells (160×106) also inhibited primary MM cell growth in vivo. The MM cells used in this experiment were derived from a patient with high-risk, highly proliferative MM relapsing after high-dose chemotherapy and autologous peripheral blood stem cell transplantation. ELISA for huIg showed a lower tumor burden in mice treated with exp-NK cells followed by 1000 U IL2 daily, and this difference was statistically significant at day 21 (Figure 5).
Figure 5.
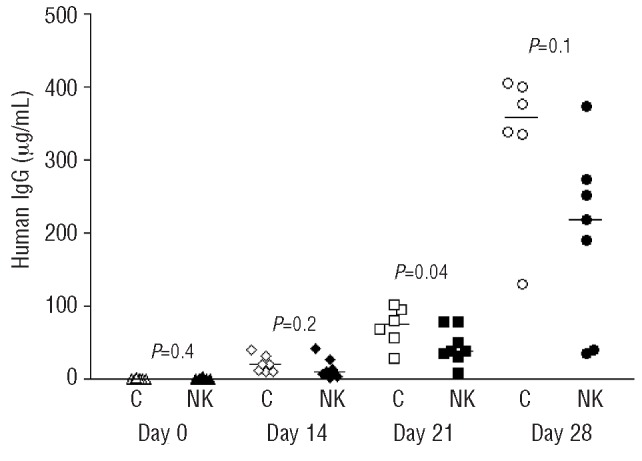
Exp-NK cells inhibit primary MM cell growth in a NOD/scid/IL2Rγnull-hu model. ELISA for huIg demonstrates significant tumor growth inhibition in vivo at day 21. Each symbol represents one mouse and the median values are also shown (−).
Expanded natural killer cells persist and proliferate up to one month after adoptive transfer and are detected in the myeloma tumor bed
We observed that NK-cell persistence after adoptive transfer was both exp-NK cell dose- (data not shown) and IL2-dependent (Figure 6A). In our experiment targeting OPM2 tumors in vivo, we utilized support with IL2 100 U injected twice weekly. We subsequently found that a dose of 1000 U daily dramatically increased the number of detectable exp-NK cells in the peripheral blood of the murine hosts and also increased the time over which they could be detected to 1 month. This dose of IL2 is comparable to that used in our previous clinical trial.9 We were further able to show that these exp-NK cells can proliferate after adoptive transfer (Figure 6B). We injected CFSE-labeled exp-NK cells and observed that 6 days after transfer, the exp-NK cells had undergone up to nine doublings in vivo. Importantly, we could detect CFSE-labeled NK cells in the cryosections of the MM tumor bed within the hu bone implant collected 7 days after injection of exp-NK (data not shown). Furthermore, we were able to detect exp-NK cells by immunohistochemistry for CD57 in hu bone implants harvested 28 days after infusion (Figure 6C). The exp-NK cells had been stained prior to adoptive transfer with CD3, CD56, and CD57 which showed that CD3−CD56+ NK cells co-expressed CD57, allowing for the use of CD57 rather than CD56, which is frequently co-expressed by plasma cells, for the identification of NK cells in hu bone (Online Supplementary Figure S5). Finally, we confirmed the absence of human T-cells in the myelomatous bone via immunohistochemistry for CD3 (data not shown).
Figure 6.

Human exp-NK cells persist for up to 28 days, proliferate further in the peripheral blood of OPM2 myeloma-bearing NOD/scid/IL2Rγnull mice and can be detected in the MM tumor bed. (A) NOD/scid/IL2Rγnull-hu hosts bearing OPM2 tumors were dosed with 160×106 exp-NK (given by four i.v. injections 48 h apart) followed by either “low IL2” (100 U IL2 twice weekly, open bars), or “high IL2” (1000 U IL2 daily, filled bars). Peripheral blood was collected weekly and subjected to flow cytometry. Exp-NK cells were detectable 28 days after infusion in mice given high IL2. One representative set of dot plots is shown for days 7, 14, 21, 28 for a mouse receiving high IL2. *Exp-NK high IL2 versus low IL2 P<0.005. Day 0 = date of NK cell injection #1. (B) Exp-NK cells proliferated in vivo in NOD/scid/IL2Rγnull mice receiving high IL2. CFSE-labeled exp-NK cells (4×107) were injected i.v., and peripheral blood was obtained for flow cytometry analysis 6 days after injection. (C) Exp-NK cells were detectable in appreciable numbers in the MM tumor bed 28 days after adoptive transfer. In paraffin sections, exp-NK cells were stained brown (arrows) with anti-human CD57 and MM cells were stained red (arrowhead) with CD138 antibodies in conjunction with DAB and Fast red for dual immunohistochemistry staining. Nuclei are stained blue with hematoxylin.
Discussion
We have previously reported that infusions of IL2-stimulated KIR-L-mismatched haplo-identical NK cells were safe and feasible in patients with relapsed/refractory MM.9 However, we also found that doses of alloreactive NK cells contained in T-depleted non-exp-NK cell leukapheresis collections may have been too low to eradicate the MM burden remaining after chemotherapy. In addition, we were unable to identify an appropriately KIR-L-mismatched donor for one third of otherwise eligible patients.
Recent studies have demonstrated that NK cells can be expanded to large numbers ex vivo using K562 cells transfected with IL15 and 4-1BBL, by 3 weeks of co-culture with EBV-LCL feeder cell lines, or by incubation with OKT3 and IL2.10,16–18 Such exp-NK cells exerted antitumor activity in vitro to a variety of cell lines and malignancies including MM, acute myeloid leukemia, Ewing’s sarcoma, and gastric, prostate, lung and breast cancer.10,18,19 We expanded CD56+/CD3− NK cells nearly 450-fold in only 10–14 days in 45 separate experiments. Peripheral blood mononuclear cells from MM patients and HD alike were expanded to a similar degree and their GEP showed a striking resemblance with up-regulation of genes involved in cytotoxicity, adhesion, proliferation, and migration. NK cells expanded from MM patients and HD also exhibited equal cytotoxicity toward MM cells. Furthermore, there was significant killing of autologous MM targets suggesting that KIR-mediated inhibition could be overcome, supporting the notion that it may be feasible to use autologous NK cells if no suitable family donor is available.
We demonstrated killing of MM cell lines and primary high-risk MM cell targets, well characterized by GEP, both in vitro and in vivo. Further, we tested the activity of exp-NK cells in a NOD/scid/IL2Rγnull-hu model, which is unique since it allows for the growth of human myeloma cells in a hu bone marrow microenvironment and does not have endogenous NK cell activity. Previously reported in vivo studies with exp-NK cells had been conducted with the NK-cell sensitive cell line K562 implanted subcutaneously in NOD-SCID mice.10 We observed significant, dose-dependent inhibition of aggressive OPM2 tumor growth in mice treated with exp-NK cells. Importantly, we also observed significant exp-NK cell-mediated inhibition of primary MM tumors derived from patients whose MM exhibited a high-risk GEP signature, although this inhibition was less strong than that observed with the aggressively growing OPM2 cell line. This could be due to differential read-out systems, imaging versus ELISA, respectively, but it cannot be ruled out that primary myeloma cells in this murine model were less susceptible to exp-NK cell therapy than the OPM2 cell line.
Fujisaki et al. demonstrated that exp-NK cells from HD continue to expand in vitro up to 21 days. We chose to harvest the exp-NK cells around day 10 to explore whether additional expansion might occur in vivo after the infusion.10,20 We found that the in vivo expansion of the adoptively transferred exp-NK cells was dependent on the dose of IL2, and persistence of NK cells was demonstrated up to 28 days with IL2 doses similar to those used in our previous human clinical trial utilizing non-exp-NK cells. The exp-NK cells did not only proliferate in vivo in the peripheral blood, but were readily detected in the implanted bone 1 month after infusion. Next, we demonstrated by micro-computed tomography and immunohistochemistry of the implanted hu bone that the exp-NK cell-mediated inhibition of tumor growth was accompanied by preservation of bone architecture and persistence of osteoblasts in mice treated with exp-NK cells. In contrast, control animals experienced considerable bone loss due to increased osteoclastogenesis.
We found that the primary mechanism of exp-NK cell cytolysis was perforin-mediated and not via the death-receptor pathway. Blocking studies of both receptors and ligands identified several interactions critical for killing, including NKG2D, natural cytotoxicity receptors, DNAM-1, CD27, and CD69, suggesting that multiple receptors working via different signaling pathways act in concert to achieve maximum cytolytic activity.18,19,21,22 Flow cytometry of the exp-NK cells showed a marked increase in the expression of the activating natural cytotoxicity receptors, NKp30 and NKp44, and NKG2D, all required for cancer target cell recognition. Other activating molecules with increased expression, such as CD69 and CD70, may have provided co-stimulatory signals to NK cells, while up-regulation of the adhesion molecules CD54 and CD56 enhanced NK:MM cell interactions. Although the cell density of some inhibitory receptors, including CD94, NKG2A, KIR2DL1, and KIR2DL2/3, also increased after expansion, the summation of the inhibitory and activating signals resulted in a significant increase in cytolysis of MM cell lines and primary MM cells. Normal targets, such as phytohemagglutinin-blasts and CD34+ cells, were not affected by the exp-NK cells.
Optimization of NK-cell doses is critical in cytolytic assays and in dose-response curves in murine studies of adoptive lymphocyte transfer.9,23 It is presently not known which of two approaches, in vitro expansion prior to adoptive transfer or in vivo expansion post-adoptive transfer, is most efficacious. The present data suggest that a combination of ex-vivo expansion of NK cells and in vivo proliferation of the exp-NK cells supported by IL2 administration could be a viable alternative.
In conclusion, stimulation with K562 transfectants induced vigorous specific expansion of NK cells. ‘Supercharged’ exp-NK cells overcame inhibitory signals delivered by MM cells and had significantly augmented cytotoxic function compared to non-exp-NK cells. Exp-NK cells were capable of trafficking to MM tumors and persisting in the human bone microenvironment. The exp-NK cells exerted an anti-MM effect in vivo and protected from tumor-mediated bone destruction. Although the expanded cells mostly comprised NK cells, CD3 depletion would still be required in the haplo-identical setting to avoid graft-versus-host disease. As exp-NK cells from MM patients have significant cytolytic function against autologous primary MM cells, this treatment could be extended to patients who do not have a suitable haplo-identical donor. The anti-MM activity of exp-NK cells is exciting and may open new therapeutic avenues for patients with GEP-defined high-risk disease who have a dismal prognosis with presently available therapies. The activity of exp-NK cells could be further potentiated by adding a KIR blocking antibody,24 or re-targeting the exp-NK cells to the MM cells by transfecting chimeric receptors specific for cell surface molecules expressed on high-risk MM cells, as has been attempted in other malignancies.25–28 The activity and survival of human NK cells may be further enhanced by using IL15 which has not yet been applied in human trials, but has been shown to expand NK cell numbers in non-human primates.29 Bortezomib could be utilized to down-regulate HLA class I molecules,30 increase TRAIL-mediated apoptosis and up-regulate DNAM-1 and NKG2D ligands.31–33
A phase II clinical trial has been initiated at our institution which examines the therapeutic effect of both allogeneic and autologous exp-NK cells for relapsed/refractory GEP-defined high-risk MM. Expansion of NK cells in a GMP-compliant setting allows for the generation of 2×1010 exp-NK cells from a single steady state leukapheresis product.34 Exp-NK cells are administered after lymphodepletion with fludarabine and cyclophosphamide to facilitate further expansion by depleting T regulatory cells and increasing the availability of homeostatic cytokines such IL15. Bortezomib is also given to sensitize myeloma cells to NK cell-mediated lysis.
Acknowledgments
The authors would like to thank Emily Hansen for assistance with biostatistical analysis, and Emily Woods, Katie Stone and Mallory Jenkins-Coleman for excellent technical assistance. We would also like to acknowledge the UAMS Translational Research Institute and its associated cores including the Flow Cytometry Core Facility for assistance with Modfit software, the UAMS Orthopedic Core for Bioluminescent Imaging, the Experimental Pathology Core for histology, and the Digital and Confocal Microscopy Laboratory for time lapse microscopy.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding: this work was supported by the National Institutes of Health (CA55819, CA134522, CA134522-02-S1, UL1 RR029884), the Multiple Myeloma Research Foundation (Senior-28-06), and the Carl L. Nelson Chair in Orthopedic Creativity.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.van Rhee F, Anaissie E, Angtuaco E, Bartel TB, Epstein J, Nair B, et al. Myeloma. In: Lichtman MA, Beutler E, Kipps T, Seligsohn U, Kaushansky K, Prchal J, editors. Williams Hematology. New York: McGraw-Hill; Medical: 2010. pp. 1645–81. [Google Scholar]
- 2.Shaughnessy JD, Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–84. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 3.Caligiuri MA. Human natural killer cells. Blood. 2008;112(3):461–9. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purdy AK, Campbell KS. Natural killer cells and cancer: regulation by the killer cell Ig-like receptors (KIR) Cancer Biol Ther. 2009;8(23):2211–20. doi: 10.4161/cbt.8.23.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farag SS, Fehniger TA, Ruggeri L, Velardi A, Caligiuri MA. Natural killer cell receptors: new biology and insights into the graft-versus-leukemia effect. Blood. 2002;100(6):1935–47. doi: 10.1182/blood-2002-02-0350. [DOI] [PubMed] [Google Scholar]
- 6.McQueen KL, Parham P. Variable receptors controlling activation and inhibition of NK cells. Curr Opin Immunol. 2002;14(5):615–21. doi: 10.1016/s0952-7915(02)00380-1. [DOI] [PubMed] [Google Scholar]
- 7.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 8.Ruggeri L, Aversa F, Martelli MF, Velardi A. Allogeneic hematopoietic transplantation and natural killer cell recognition of missing self. Immunol Rev. 2006;214:202–18. doi: 10.1111/j.1600-065X.2006.00455.x. [DOI] [PubMed] [Google Scholar]
- 9.Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol. 2008;143(5):641–53. doi: 10.1111/j.1365-2141.2008.07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69(9):4010–7. doi: 10.1158/0008-5472.CAN-08-3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kataoka T, Shinohara N, Takayama H, Takaku K, Kondo S, Yonehara S, et al. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell-mediated cytotoxicity. J Immunol. 1996;156(10):3678–86. [PubMed] [Google Scholar]
- 12.Yaccoby S, Barlogie B, Epstein J. Primary myeloma cells growing in SCID-hu mice: a model for studying the biology and treatment of myeloma and its manifestations. Blood. 1998;92(8):2908–13. [PubMed] [Google Scholar]
- 13.Pennisi A, Li X, Ling W, Khan S, Zangari M, Yaccoby S. The proteasome inhibitor, bortezomib suppresses primary myeloma and stimulates bone formation in myelomatous and nonmyelomatous bones in vivo. Am J Hematol. 2009;84(1):6–14. doi: 10.1002/ajh.21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology. 2004;145(1):401–6. doi: 10.1210/en.2003-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Penack O, Gentilini C, Fischer L, Asemissen AM, Scheibenbogen C, Thiel E, et al. CD56dimCD16neg cells are responsible for natural cytotoxicity against tumor targets. Leukemia. 2005;19(5):835–40. doi: 10.1038/sj.leu.2403704. [DOI] [PubMed] [Google Scholar]
- 16.Berg M, Lundqvist A, McCoy P, Jr, Samsel L, Fan Y, Tawab A, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy. 2009;11(3):341–55. doi: 10.1080/14653240902807034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlens S, Gilljam M, Chambers BJ, Aschan J, Guven H, Ljunggren HG, et al. A new method for in vitro expansion of cytotoxic human CD3-CD56+ natural killer cells. Hum Immunol. 2001;62(10):1092–8. doi: 10.1016/s0198-8859(01)00313-5. [DOI] [PubMed] [Google Scholar]
- 18.Alici E, Sutlu T, Bjorkstrand B, Gilljam M, Stellan B, Nahi H, et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood. 2008;111(6):3155–62. doi: 10.1182/blood-2007-09-110312. [DOI] [PubMed] [Google Scholar]
- 19.Voskens CJ, Watanabe R, Rollins S, Campana D, Hasumi K, Mann DL. Ex-vivo expanded human NK cells express activating receptors that mediate cytotoxicity of allogeneic and autologous cancer cell lines by direct recognition and antibody directed cellular cytotoxicity. J Exp Clin Cancer Res. 2010;29:134–46. doi: 10.1186/1756-9966-29-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujisaki H, Kakuda H, Imai C, Mullighan CG, Campana D. Replicative potential of human natural killer cells. Br J Haematol. 2009;145(5):606–13. doi: 10.1111/j.1365-2141.2009.07667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006;214:73–91. doi: 10.1111/j.1600-065X.2006.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107(1):159–66. doi: 10.1182/blood-2005-04-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller J. Should natural killer cells be expanded in vivo or ex vivo to maximize their therapeutic potential? Cytotherapy. 2009;11(3):259–60. doi: 10.1080/14653240902888000. [DOI] [PubMed] [Google Scholar]
- 24.Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood. 2009;114(13):2667–77. doi: 10.1182/blood-2009-02-206532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83. doi: 10.1182/blood-2004-12-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann HG, et al. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100(4):1265–73. [PubMed] [Google Scholar]
- 27.Schirrmann T, Pecher G. Specific targeting of CD33(+) leukemia cells by a natural killer cell line modified with a chimeric receptor. Leuk Res. 2005;29(3):301–6. doi: 10.1016/j.leukres.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Demirtzoglou FJ, SP, GZ Cytolytic and cytotoxic activity of a human natural killer cell line genetically modified to specifically recognize HER-2/neu overexpressing tumor cells. Immunopharmacol Immunotoxicol. 2006;28:571–90. doi: 10.1080/08923970601066971. [DOI] [PubMed] [Google Scholar]
- 29.Berger C, Berger M, Hackman RC, Gough M, Elliott C, Jensen MC, et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood. 2009;114(12):2417–26. doi: 10.1182/blood-2008-12-189266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi J, Tricot GJ, Garg TK, Malaviarachchi PA, Szmania SM, Kellum RE, et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood. 2008;111(3):1309–17. doi: 10.1182/blood-2007-03-078535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, et al. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180(1):163–70. doi: 10.4049/jimmunol.180.1.163. [DOI] [PubMed] [Google Scholar]
- 32.Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113(15):3503–11. doi: 10.1182/blood-2008-08-173914. [DOI] [PubMed] [Google Scholar]
- 33.Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, et al. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006;66(14):7317–25. doi: 10.1158/0008-5472.CAN-06-0680. [DOI] [PubMed] [Google Scholar]
- 34.Lapteva N, Sun J, Durrett A, Vera J, Jackson K, Szmania S, et al. Robust ex vivo expansion of natural killer cells for clinical applications in gas-permable rapid expansion cell culture devices. Cytotherapy. 2011;13 Abstract 207. [Google Scholar]