

Abstract
Research over the past decade has confirmed that epigenetic alterations act in concert with genetic lesions to deregulate gene expression in acute myeloid leukemia and myelodysplastic syndromes. Epigenetic alterations may serve as markers of disease, and may potentially be used for classification, prognostication and to monitor minimal residual disease. In addition, we now have the capability to pharmaceutically target epigenetic modifications, and there is an urgent need for early validation of the efficacy of the drugs. Also, an improved understanding of the functionality of epigenetic modifications may further pave the road towards individualized therapy. The recent advances in biotechnology and bioinformatics provide a plethora of novel tools for characterizing the epigenome in clinical samples, but at this point the practical, clinical utility of these methodologies needs further exploration. Here, we provide the pros and cons of the currently most feasible methods used for characterizing the methylome in clinical samples, and give a brief introduction to novel approaches to sequencing that may revolutionize our abilities to characterize the genomes and epigenomes in acute myeloid leukemia and myelodysplastic syndrome patients.
Keywords: myelodysplastic syndrome, epigenetic, alterations, monitoring, biotechnology, bioinformatics
Introduction
Epigenetic alterations in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) have profound effects on gene expression and may cause tumor suppressor gene silencing. A large number of genes are inactivated by hypermethylation of CpG islands in the promoter regions. AML, and MDS in particular, have extensive aberrant DNA methylation when compared to normal CD34 positive bone marrow cells.1 In addition, genome-wide promoter DNA methylation profiling reveals unique AML subgroups and methylation patterns that are associated with clinical outcome, opening up new avenues for improved diagnostics and prognostication.2 Since thousands of gene promoters typically undergo DNA hypermethylation, there is a rationale for using DNA methyltransferase inhibitors (DNMTI), which are the first epigenetically targeted therapies to have shown efficacy in the treatment of MDS and AML.3–5 New drugs that target different epigenetic changes, i.e. histone modifications, are under development. To offer an individualized therapy, reliable methods to monitor the efficacy of these novel therapies are needed. Here, we summarize currently available analyses used for this purpose and highlight important issues in the choice of methodology.
Clinical utility of DNA methylation profiling
Over the last couple of decades, many studies have addressed the use of promoter methylation status of individual genes for prognostication in myeloid malignancies. Early studies showed that promoter methylation of the CDKN2B (p15) gene was associated with disease progression in MDS6,7 and secondary AML,8 and that the CDKN2B promoter was demethylated during treatment with 5-aza-2’deoxycytidine (5-aza-CdR).9 However, other studies imply that the clinical importance of CDKN2B methylation in AML is controversial.10,11 Nonetheless, methylation of different individual genes and combinations of genes (e.g. CDH1, HIC1, SOCS1, BCL2L10) were shown to have prognostic impact in MDS and AML,11–15 increasing expectations that a global screening approach would improve the clinical utility of DNA methylation profiling.
At present, the majority of global methylation profiling studies have been performed in AML, with only a few studies in MDS and chronic myeloproliferative disorders (CMPD). In AML, several studies have shown a link between global methylation profiles and distinct chromosomal rearrangements such as t(8;21), inv(16), t(15;17), MLL rearrangements or complex karyotype but, in addition, new prognostic sub-groups have been identified based only on their methylation profile.2,11,16,17 Furthermore, recent studies show a correlation between increased DNA methylation and mutations in enzymes involved in DNA demethylation (IDH1/2 and TET2),17,18 while another study shows association of TET2 mutations to global hypomethylation.19 Whether this relates to diversities in methodology or biology is a subject for further investigation. Furthermore, global and gene-specific DNA methylation levels have been associated with the expression level of miR29b, a microRNA that targets the 3’UTR of DNMT3A and 3B, and the Sp1 promoter element of DNMT1, leading to their downregulation and thus hypomethylation.20
In MDS, only a few global methylation profiling studies have been published. In a screen of 807 promoters, increased methylation was associated with transformation from low-to high-risk MDS.21 In a different study, a comprehensive screen of 14,000 promoters revealed that the basic level of methylation in MDS and secondary AML is much higher than in de novo AML and in normal CD34+ cells.1 However, so far, no global methylation studies have addressed the prognostic impact of global methylation changes in MDS; however, a decrease in methylation was observed after 5-azacytidine (5-aza-CR) treatment.1 There have been no global methylation profiling studies in CMML published so far, and a single study of CMPDs (polycythemia vera and essential thrombocytosis) did not reveal differential methylation when compared to normal controls.22
Accordingly, only limited quantities of global methylation profiling data in myeloid malignancies are currently available, and those available are from studies based on different approaches to methylation detection. Some studies only analyze methylation at a few CpG sites at the promoters of selected cancer-associated genes,11,21 while others also include promoter methylation of genes that have not previously been implicated in cancer.1,2,10,17,18,22 But all of the above-mentioned studies focus on a variable number of selected CpG sites, predominantly at CpG island promoters. Since recent studies suggest that cytosine methylation outside gene promoters may also be involved in gene regulation, an unbiased approach may provide new information.23,24 The current technological advances allow for a global digital mapping of CpG methylation that may potentially change our view of the role of DNA methylation in myeloid malignancies. However, at this point, the technology and data management needed is not to be underestimated. Hopefully, some of this new knowledge may translate into clinically useful tools.
Clinical utility of histone modification profiling
Histone modifications play an important role in determining cell fate, temporal and special organization of transcription, DNA repair and multiple other cellular functions. In hematologic malignancies, aberrations occur in writers as well as readers of histone modifications. For example, mutations of the H3K27me3 histone methyl-transferase Enhancer of Zeste2 (EZH2) occur in malignant myeloid disorders.25,26 In addition, in myeloproliferative syndromes, the frequently mutated JAK2 kinase directly phosphorylates histones.27 As will be discussed below, the analyses of histone modifications in clinical specimens are currently more difficult than the analyses of DNA methylation. Nonetheless, a few studies have been published that indicate that the profiling of histone modifications in clinical specimens is feasible.28–31 These experiments were performed with ChIP-chip procedures. Importantly, recent improvements in methodology appear to make the use of primary patient samples more easily accessible. These procedures will be discussed below.
Methodologies for detection of DNA methylation
Approaches to screen for epigenetic alterations in myeloid malignancies have mainly taken two directions. The first approach relies on the evaluation of specific candidate cancer genes, frequently representing known tumor suppressor genes.32 The second strategy employs a global screen for epigenetic alterations. Although genome-wide DNA methylation analyses provide new insights into the methylation pattern of normal cells and cancer cells, there are limitations to the extent to which genome-wide techniques give full and accurate assessment of the methylation status of individual genes or CpG sites. As a result, validation of gene and/or site-specific methylation is usually recommended as a complement to genome-wide methods.
Until now, investigations have been designed to focus on promoter CpG-island hypermethylation. Recent data indicate that the methylation status of CpGs outside the classical CpG-island promoter regions also influence transcriptional regulation. Methylation at the so-called CpG-island shores,23 in enhancer regions,33 and non-CpG island promoters24 may also influence gene expression. In addition, methylation of the repetitive LINE1 sequences has been used as a proxy marker to monitor the overall methylation level of cancer epigenomes in AML and MDS.34–36
Methodologies for global DNA methylation profiling
Currently used methods for global DNA methylation profiling are listed in Table 1. Methods based on methylation-sensitive restriction enzymes are limited in their use due to the location of restriction sites. These approaches, as for example Restriction Landmark Genomic Scanning (RLGS)37 or HpaII tiny fragment enrichment by ligation-mediated PCR (HELP),38 are being replaced more and more by methods that to a lesser extent pre-select the investigated sequence. Instead, enrichment for methylated DNA molecules is being accomplished by: i) using enrichment of methylated DNA sequences by antibodies or proteins binding methylated DNAs; or ii) methods based on bisulfite conversion (Figure 1).
Table 1.
Methods for genome-wide methylation profiling.
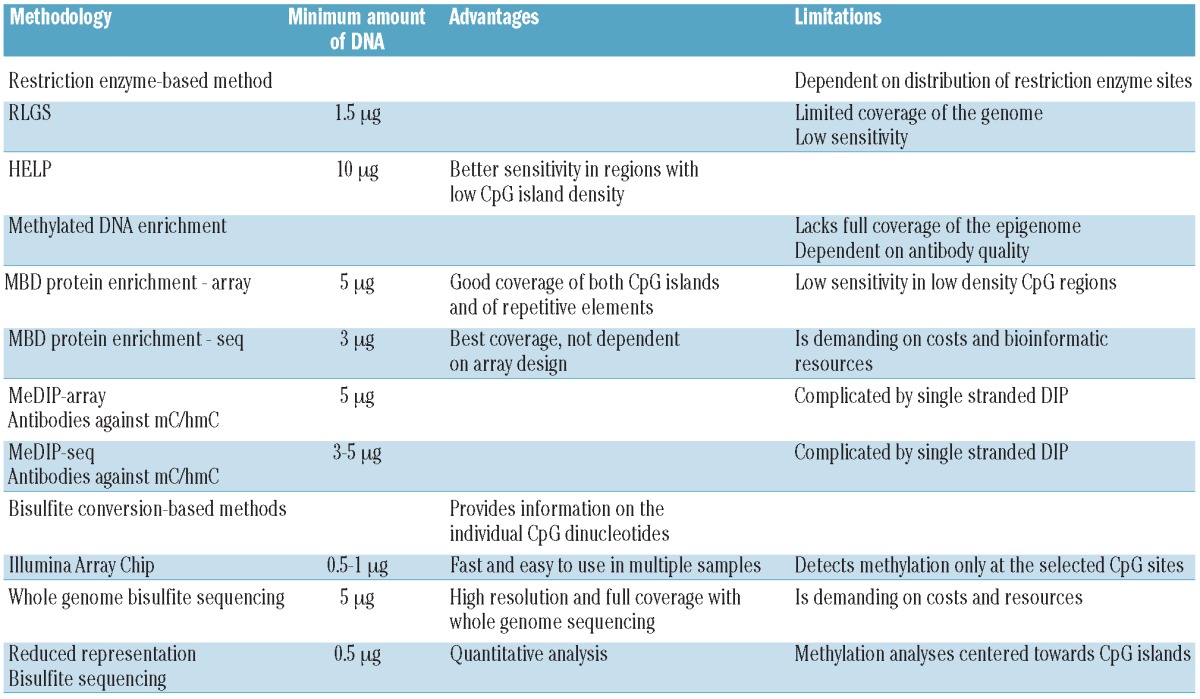
Figure 1.

Methods to detect global DNA methylation. (A) Methylated DNA immunoprecipitation (MeDIP). DNA is sonicated and subsequently denatured. Single stranded DNA is incubated with antibodies against either methylated (5mC) or hydroxymethylated (5hmC) cytosines followed by IP. The IP fraction (enriched for either methylated DNA or hydroxymethylated DNA) can either be labeled and hybridized to whole-genome tiling arrays or analyzed using next generation sequencing (NGS). (B) Chromatin immunoprecipitation (ChIP) of methylated DNA. DNA and protein are cross-linked by formalin followed by sonication. DNA is incubated with antibodies or proteins that bind MBDs and precipitated. The enriched fraction can be analyzed by the same methods as for MeDIP. (C) The DNA is denatured and treated with sodium bisulfite. Unmethylated cytosine, ‘C’, is converted to uracil, ‘U’, while 5mC or 5hmC are unchanged. In the subsequent amplification, ‘U’ is amplified as thymine, ‘T’, while the unconverted 5mC or 5hmC are amplified as ‘C’. The amplification products are fragmented and hybridized to bead-based arrays or analyzed by NGS.
Methods based on enrichment of methylated DNA
One approach uses an anti 5-methyl cytosine (anti-5mC) antibody for methylome profiling in a technique called methylated DNA immunoprecipitation (MeDIP).39,40 First, the DNA is sonicated, denatured to single-strand DNA and then incubated with the anti-5mC antibody. Alternative methods enrich for methylated DNA by binding to methyl CpG binding domain proteins (MBDs). This may be done using MBD3L1, a binding partner of MBD2 that increases the affinity of MBD2 for methylated DNA for enrichment (Methylated-CpG island recovery assay (MIRA).41,42 In an alternative approach, Methyl CpG immunoprecipitation (MCIp) was developed utilizing antibody-like recombinant protein (MBD2-Fc) and A-Sepharose columns for the enrichment of methylated DNA from genomic DNAs.43 Enriched DNA can either be labeled and hybridized to whole-genome tiling DNA arrays (ChIP-on-chip) or analyzed using next generation sequencing (ChIP-seq; see below)44 (Figure 1A and B). These protocols allow a comprehensive analysis of the methylation pattern across the entire genome.
Methods based on bisulfite conversion
At present, most of the commonly used methods for methylation detection at the single gene and whole genome levels are based on the initial treatment of the DNA template by sodium bisulfite. Sodium bisulfite converts C to uracil (U) while it leaves 5mC intact.45 In the subsequent PCR reactions, U will be amplified as thymine (T), while the static and unconverted 5mC is amplified as C.
One of the most commonly used methods for global methylation detection uses the bead-based arrays from Illumina. After bisulfite treatment, the DNA is amplified, fragmented and hybridized to a large set of short oligonucleotides bound to beads. Each unique genomic region is represented by two homologous oligonucleotides that only differ by a specific C/T, and thus gives a relative measure of the amounts of mC versus C in the template (Figure 1C). This method has been able to record the methylation status of approximately 27,500 promoter island CpGs, but a new chip now enables the recording of more than 485,000 CpGs, including CpGs at non-CpG islands, in non-coding regions and at miRNA promoters.46 However, this is still much below the estimated number of approximately 28 million CpG dinucleotides present in the whole genome.
Next generation sequencing of the methylome
As for chromatin immunoprecipitated DNA, bisulfite-converted DNA may be analyzed by next generation sequencing (NGS). In this process, simultaneous amplification of hundreds of sequences from bisulfite-converted or immunoprecipitated DNA is followed by the sequencing of up to several hundred million reads. Since the value of the data is directly proportional to the number of times a particular genomic region is sequenced in parallel (so-called coverage), a complete sequencing is currently cost-prohibitive for studies of multiple samples. Despite these issues, several groups have recently published whole DNA methylome data from various hematopoietic cell types including human pluripotent and differentiated stem cells,47 and peripheral blood mononuclear cells.48
Reduced Representation Bisulfite Sequencing (RRBS) is a modification of this technology. The MspI restriction enzyme that cuts the sequence ‘CCGG’ is used to enrich for genomic regions that show two of these sites within a distance of 300 bp or less. As a consequence, CpG islands and promoters are significantly enriched. The subsequent sequencing allows quantitative assessment of more than 1×106 independent CpGs.49 This procedure is relatively cost-effective and allows a higher degree of quantitative DNA methylation analysis than MeDIP based assays.
Methods for confirmation of methylation status at single gene or CpG site level
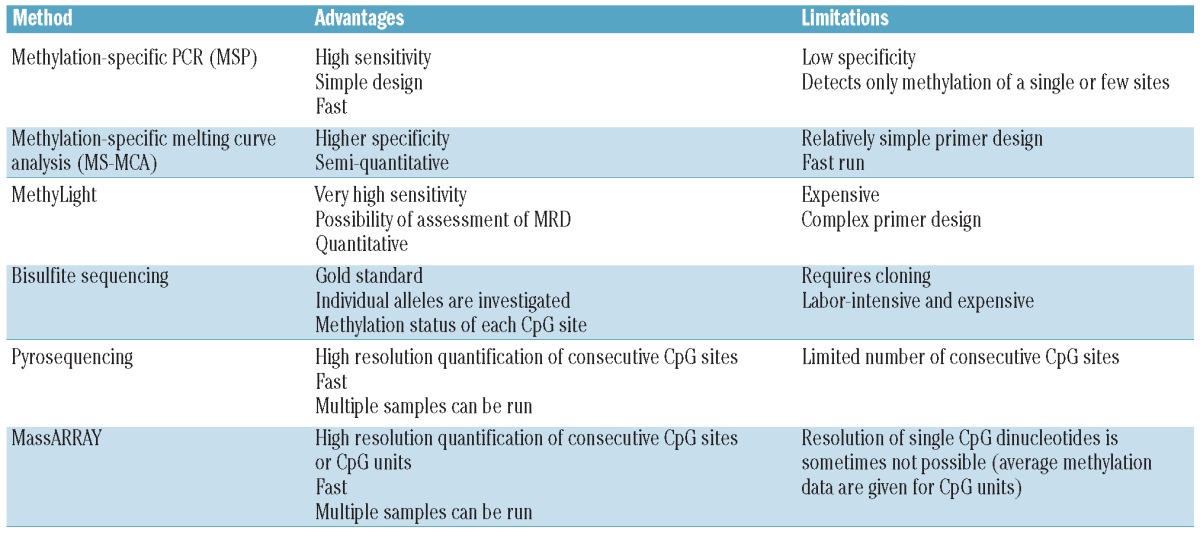
The methods reviewed below are summarized in Table 2. Most of the currently used methods for DNA methylation detection at the single gene level rely on the initial bisulfite conversion of the DNA. This will allow separation of methylated and unmethylated alleles by two major mechanisms: i) by specifically amplifying methylated and unmethylated alleles in two different PCR reactions; or ii) by a physical separation of a PCR product containing a mixture of methylated and unmethylated DNA.
Table 2.
Common methods for determining single gene or CpG site-specific DNA methylation
Screening for methylation at individual gene promoters
Methylation-specific PCR (MSP) is without doubt the most frequently used method used to detect methylation at individual genes. The advantage of MSP is the relatively simple experimental design and its high sensitivity with a detection rate of 1 methylated allele in 1,000 unmethylated alleles.50 However, the major disadvantages are its relatively low specificity, the fact that methylation is only investigated at the binding sites of the primers, and that it only detects methylation on a single or a few cytosine molecules.
More detailed information on the pattern and density of methylation can be achieved by methylation-specific melting curve analysis (MS-MCA). This method is based on the fact that the melting properties of bisulfite-treated methylated and unmethylated DNA species differ considerably and can, therefore, be separated by melting the different PCR products in a thermal cycler (LightCycler).51,52 The advantages of these methods are their high specificity, and that they allow semi-quantification of methylation and resolution of variable methylation patterns at different molecules in the same specimen.
The MethyLight assay is a TaqMan fluorescence-based, real-time PCR method, which gives a quantitative measure of the amount of methylated alleles in a mixture with unmethylated alleles. The average level of methylated CpG sites in the amplified region is calculated; however, methylation at individual CpGs is not resolved. The advantage of this method is its high sensitivity that allows for detection of one methylated allele among 10,000 unmethylated alleles. Accordingly, MethyLight is efficient in situations where a limited number of methylated alleles are detected in a mixture with abundant wild-type DNA, e.g. in the detection of minimal disease in body fluids such as sputum, serum and urine. Also, it gives a quantitative measurement of the amount of methylated alleles. The disadvantages are the cost and the relative complexity of the assay.
Quantitative analysis of methylation at individual CpG sites
To obtain detailed information about the methylation status at every single cytosine in a particular sequence, bisulfite genomic sequencing has long been the gold standard. However, methylation detection by pyrosequencing has recently been accepted as a robust alternative.
In bisulfite genomic sequencing, the bisulfite-converted DNA sequence is cloned into a plasmid vector in order to obtain substantial amounts of template for direct sequencing. In this way, individual alleles are examined for their specific methylation pattern, and the relative methylation density of individual cytosines can be estimated.45 The method is rather laborious, and requires the sequencing of at least 10 clones per transcript in order to obtain reliable results.
By contrast, the pyrosequencing technology allows direct, high-resolution quantification of methylation density over several consecutive CpG sites. The method is based on the release of pyrophosphate (PPi, P2O74-) during nucleotide extension.53 Typically, protocols recommend that the sequence analyzed should be no longer than 300 bp; however, in reality, much is resolved by analyzing sequences no longer than approximately 100 bp. The advantages of this method are that it is fast and allows for the detection of 96 samples in a single run, but in contrast to bisulfite sequencing, methylation is not resolved at individual alleles.
In the MassARRAY assay, bisulfite-converted DNAs are amplified with PCR primers that do not discriminate between methylated or unmethylated sequences. The resulting PCR product is transcribed into RNA, which is subsequently cleaved by RNase A. The resulting fragments are then quantitatively tested for their DNA methylation status using matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry.54
Current challenges and novel approaches to DNA methylation detection
Methods that distinguish 5-hydroxymethylcytosine (5hmC) from 5-methylcytosine (5mC)
The discovery that the frequently mutated TET2 proteins can convert 5mC to 5hmC has challenged the use of bisulfite-based methods for DNA methylation detection.55,56 Recent data suggest that TET proteins survey DNA methylation fidelity at transcription start sites of TET target genes,57 but at present the abundance and distribution of 5hmC in MDS and AML, and its exact role in transcriptional regulation, is still a question of debate. However, it has clearly been shown that bisulfite conversion does not distinguish 5mC and 5hmC, and that 5hmC containing regions are poorly amplified by PCR after bisulfite conversion.58 In order to discriminate these two functionally different species, MeDIP-based technologies may be applied. DNA immunoprecipitation by antibodies specific to 5hmC and 5mC are followed by PCR for detection of methylation at the single gene level, and by whole genome sequencing or array analysis for global methylation detection. A major obstacle has been the specificity of the antibodies that discriminate 5mC and 5hmC, respectively, but recently developed alternative methods using enzymatic and chemical steps to isolate a few or single molecule hmCs may improve separation of the two species.59
’Third generation’ sequencing of methylated nucleotides
In the near future, it is envisioned that the cost of sequencing will be further reduced and the sequencing of multiple cancer genomes is undertaken in international consortia. Multiple reads per sequence are required due to the heterogeneity of prior DNA methylation patterns representing a certain sequence. Initial bisulfite treatment or ChIP would no longer be required as single molecules such as 5mC and 5hmC can now be detected directly from the sequencing reads. Novel technologies, such as single molecule real-time (SMRT) sequencing,60 single-molecule nanopore sequencing with protein pores,61 or ‘Ion-chip’ technology that directly senses the ions released during DNA synthesis62 may replace current technologies. However, for the moment, further documentation of their power and specificity for methylation detection in clinical samples is needed.
Analyses of histone modifications
Posttranslational modifications of the core histones constitute an important part of the epigenetic memory.63 Each of the core histones is strongly conserved throughout phylogenesis and can be modified at multiple residues primarily on its N-terminal tails by multiple enzymes.
Chromatin-IP analysis to identify histone modifications
Histone modification analyses in AML have been fuelled by the development of ChIP as a suitable method to identify posttranslational histone modifications at specific gene promoters. Initially introduced in yeast, this method has matured over the last ten years and is currently the standard approach.64 ChIP coupled with real-time PCR allows the identification of histone modification patterns of single promoters and enhancers. The possibility of covering a region of several kilobases with interspaced amplicons has enabled researchers to clearly define binding patterns of transcription factors and of regions of his-tone modifications with high sensitivity and specificity.65 Nonetheless, this approach still requires an a priori hypothesis of relevant genes and loci.
The development of unbiased methods, such as ChIP-chip and ChIP-seq, has vastly improved the potential of chromatin IP-based analyses of histone modifications.66 The basic principle underlying ChIP-chip and ChIP-seq is very similar: sheared chromatin with specific histone modifications is enriched by antibody-based precipitation. The DNA is subsequently isolated. For ChIP-chip analyses, the usually very small amount of DNA needs to be amplified and labeled. Finally, a hybridization step onto specific microarrays is needed. In contrast, for ChIP-seq, libraries can be generated from small amounts of DNA available after ChIP and these can be sequenced using NGS. Nonetheless, there are several critical points that are common to both procedures. First of all, the antibody used for ChIP is of vital importance. Specificity of the antibody needs to be rigorously controlled. For the ChIP procedure itself, it is necessary to have smaller fragments of chromatin with DNA between 400 and 1,000 base pairs long. The widely used sonication bears the problem that intensity needs to be adjusted for each cell type. It is also possible to use enzymatic digestion and this can be performed by several methods. Another important problem and critical point in the analyses of histone modifications in leukemia samples relates to the controls used. One control for changes in leukemia is made up of CD34+ progenitor cells, but in specific instances it may be feasible to use a cell population that more closely resembles the transformed cells as control.
A general problem is the variety of histone modifications found in vivo and their potential crosstalk. Given that two alleles are usually present, it might be difficult to assess whether the histone modifications shown as more or less well defined peaks are found on the same chromosome or on different chromosomes within the same cell. Sequential ChIP has been described as one solution to this problem.66,67 In this method, the immunoprecipitated histones are used for a subsequent ChIP to ensure that the resulting sample is enriched for both modifications on the same histones.
Standard chromatin IP methods require high numbers of cells as starting material. In contrast, most clinical specimens have a rather limited number of cells available. An important issue is, therefore, to identify the minimum number of cells that are required for a ChIP-seq experiment. In the last few years, several procedures have been developed that make use of a limited number of available cells. In 2007, Attema et al. used about 50,000 purified stem cells for H3K4 methylation and acetylation analyses.68 Recently, protocols developed by Bradley Bernstein’s group have developed protocols (so-called nano-ChIP-seq) in which cell numbers as low as 10,000 can be successfully used.69–71 These protocols contain significant changes in the chromatin IP procedure itself but especially also in the library preparation. Typically, such protocols also involve a separate amplification step to obtain sufficient DNA.
In addition to genome-wide histone modification analyses with identified loci, it is also possible to quantify total histone modification levels. One recent approach used mass spectrometry-based methods.72 Due to the abundance of histones in normal cells, mass spectrometry can be used to quantify and identify the posttranslational modification patterns.73,74 This approach might be especially interesting for control of epigenetic therapy whereby global alterations of histone modifications can be analyzed. Since ChIP-seq and ChIP-chip analyses are unlikely to be involved in diagnostic procedures in the near future, it might also be interesting to look at approaches to identify histone modification changes at a few specific loci. One possibility here is to couple a chromatin IP with subsequent PCR amplification. However, problems with standardization and the amount of work involved in this procedure make such approaches currently unsuitable for use with clinical specimens on a larger scale. Technologies such as nano-string might help to improve throughput and reproducibility.75,76 This technology is based on complementary sequence-specific probes coupled to biotin and molecular tags that can be easily identified and quantified. It is possible that the coupling of this nano-string technology to chromatin IP material might help to improve quantification.
An important problem for ChIP-chip and ChIP-seq relates to the bioinformatic analyses of the data. Excellent reviews have been published on this topic.77 The bioinformatic analyses are currently being further improved, especially for ChIP-seq.
Future developments in histone analysis
The rise of NGS has provided novel opportunities to decipher epigenetic changes. With the further increase in sequencing capacity most groups now focus on ChIP-seq of histone modifications in leukemia. But several points remain to be clarified. Of note, histone modifications as analyzed by ChIP-based techniques leads to relative enrichment data only. This is in contrast to DNA methylation studies including methods that allow determination of the exact methylation status of each CpG dinucleotide. It is increasingly clear that the histone code is very complex and that the same histone modification might have a different impact in different regions of the genome. Finally, only the integration of histone modification pattern data, along with transcription factor binding studies, RNA-expression and large genome sequencing studies will reveal the true relevance of these patterns for leukemogenesis.
Technical requirements of methods used to monitor epigenetic therapy
Since technologies for epigenetic monitoring are constantly evolving, the recommendations for sample handling and requirements may change rapidly. However, for the time being, a few issues may be worth considering.
Optimal handling of patient material for epigenetic profiling
The correct selection and handling of patient material is crucial, since the generated data are only as good as the input material allows. Although new approaches imply that, for example, formalin-fixed paraffin-embedded (FFPE) tissue may be used for genome-wide analyses, it is recommended to use fresh or fresh frozen material in order to get as few artifacts as possible. For bone marrow samples with a high fraction of AML blasts, density centrifugation could be used for further enrichment of blasts, which might be sufficient to define global DNA methylation patterns and to compare the patterns of a particular histone modification. In cases in which specific subpopulations need to be analyzed, it might be worthwhile to use magnetic cell sorting or FACS sorting. For MDS samples, sorting of the malignant clone might be necessary, but often difficult. However, using granulocytic fraction may lead to an enrichment of malignant cells and clearer results.78
Any method that relies only on the use of DNA is particularly attractive in a clinical setting due to its stability that, for example, allows overnight shipment at room temperature. In contrast, methods involving ChIP typically require careful and relatively fast handling of material, and the cells must be analyzed fresh or fresh frozen. In addition, some analyses may be optimized by immediate crosslinking of the DNA and chromatin of fresh cells. Another issue is the amount of material needed which is typically larger in ChIP-based approaches. However, novel methods are being developed that allow ChIP experiments with smaller amounts of patient material, as described above. Requirements for the individual analyses are listed in Table 1.
Monitoring the effects of epigenetic therapy
Since the responses to epigenetic therapy are only observed well after treatment has started, there is a relentless demand to find markers that allow determination of treatment efficacy at an early time point. This has been investigated for 5-aza-CdR9,13 for combination treatment of 5-aza-CR and the selective Class I HDACI Entinostat,1,79 and for 5-aza-CR and valproic acid (VPA).80,81 These studies clearly show that DNA methylation can be reversed by epigenetic therapy at an early time point during treatment, and that this alteration persists onto the next treatment cycle.1,9 In neither of the studies could the baseline level of methylation be used to predict response to epigenetic therapy. However, in the 5-aza-CdR study, reduced methylation levels over time during treatment could predict response. In this study, the methylation level of 10 selected genes was measured quantitatively by pyrosequencing. In the 5-aza-CR/entinostat trial, neither gene re-expression, nor promoter methylation changes of 4 selected genes, as measured by MSP after the first treatment cycle, could predict response after 4 cycles.79 These observations may indicate that quantitative measurements of methylation may be crucial for the development of markers that can select patients upfront who will benefit from treatment with DNMTI. Another explanation might be that the effects of 5-aza-CdR are more directly linked to DNA hypomethylation, whereas methylation-independent effects, such as RNA damage, may play a larger role in 5-aza-CR mediated responses.82 However, in the combined 5-aza-CR/VPA trial, responders showed a relative demethylation and increased expression of the cell cycle regulator PI-PLCβ1 over time,81 and a recent study suggests that BCL2L10 methylation may be a useful prognostic marker for outcome in patients treated with 5-aza-CR with and without the addition of VPA.14
Finally, a special issue relates to the material used when monitoring the effects of epigenetic therapy; in the responders the malignant cells may have completely disappeared and molecular investigations are then performed on the residual healthy bone marrow. Thus, surrogate markers in normal tissue may be even more efficient in monitoring effects of epigenetic therapy. Obviously, these investigations should ideally be performed in large, prospective clinical trials.
Footnotes
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood. 2009;114(16):3448–58. doi: 10.1182/blood-2009-01-200519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17(1):13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–32. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Gattermann N, Germing U, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28(4):562–9. doi: 10.1200/JCO.2009.23.8329. [DOI] [PubMed] [Google Scholar]
- 5.Lubbert M, Suciu S, Baila L, Ruter BH, Platzbecker U, Giagounidis A, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol. 2011;29(15):1987–96. doi: 10.1200/JCO.2010.30.9245. [DOI] [PubMed] [Google Scholar]
- 6.Quesnel B, Guillerm G, Vereecque R, Wattel E, Preudhomme C, Bauters F, et al. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood. 1998;91(8):2985–90. [PubMed] [Google Scholar]
- 7.Tien HF, Tang JH, Tsay W, Liu MC, Lee FY, Wang CH, et al. Methylation of the p15(INK4B) gene in myelodysplastic syndrome: it can be detected early at diagnosis or during disease progression and is highly associated with leukaemic transformation. Br J Haematol. 2001;112(1):148–54. doi: 10.1046/j.1365-2141.2001.02496.x. [DOI] [PubMed] [Google Scholar]
- 8.Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Methylation of p15INK4B is common, is associated with deletion of genes on chromosome arm 7q and predicts a poor prognosis in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2003;17(9):1813–9. doi: 10.1038/sj.leu.2403054. [DOI] [PubMed] [Google Scholar]
- 9.Daskalakis M, Nguyen TT, Nguyen C, Guldberg P, Kohler G, Wijermans P, et al. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood. 2002;100(8):2957–64. doi: 10.1182/blood.V100.8.2957. [DOI] [PubMed] [Google Scholar]
- 10.Deneberg S, Grovdal M, Karimi M, Jansson M, Nahi H, Corbacioglu A, et al. Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia. Leukemia. 2010;24(5):932–41. doi: 10.1038/leu.2010.41. [DOI] [PubMed] [Google Scholar]
- 11.Alvarez S, Suela J, Valencia A, Fernandez A, Wunderlich M, Agirre X, et al. DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia. PLoS One. 2010;5(8):e12197. doi: 10.1371/journal.pone.0012197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P. Promoter hypermethylation of p15INK4B, HIC1, CDH1, and ER is frequent in myelodysplastic syndrome and predicts poor prognosis in early-stage patients. Eur J Haematol. 2006;76(1):23–32. doi: 10.1111/j.1600-0609.2005.00559.x. [DOI] [PubMed] [Google Scholar]
- 13.Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X, et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol. 2010;28(4):605–13. doi: 10.1200/JCO.2009.23.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voso MT, Fabiani E, Piciocchi A, Matteucci C, Brandimarte L, Finelli C, et al. Role of BCL2L10 methylation and TET2 mutations in higher risk myelodysplastic syndromes treated with 5-Azacytidine. Leukemia. 2011;25(12):1910–3. doi: 10.1038/leu.2011.170. [DOI] [PubMed] [Google Scholar]
- 15.Brakensiek K, Langer F, Schlegelberger B, Kreipe H, Lehmann U. Hypermethylation of the suppressor of cytokine signalling-1 (SOCS-1) in myelodysplastic syndrome. Br J Haematol. 2005;130(2):209–17. doi: 10.1111/j.1365-2141.2005.05590.x. [DOI] [PubMed] [Google Scholar]
- 16.Bullinger L, Ehrich M, Dohner K, Schlenk RF, Dohner H, Nelson MR, et al. Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood. 2010;115(3):636–42. doi: 10.1182/blood-2009-03-211003. [DOI] [PubMed] [Google Scholar]
- 17.Deneberg S, Guardiola P, Lennartsson A, Qu Y, Gaidzik V, Blanchet O, et al. Prognostic DNA methylation patterns in cytogenetically normal acute myeloid leukemia are predefined by stem cell chromatin marks. Blood. 2011;118(20):5573–82. doi: 10.1182/blood-2011-01-332353. [DOI] [PubMed] [Google Scholar]
- 18.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcyto-sine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–43. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113(25):6411–8. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O'Keefe C, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009;113(6):1315–25. doi: 10.1182/blood-2008-06-163246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrio S, Gallardo M, Albizua E, Jimenez A, Rapado I, Ayala R, et al. Epigenomic profiling in polycythaemia vera and essential thrombocythaemia shows low levels of aberrant DNA methylation. J Clin Pathol. 2011;64(11):1010–3. doi: 10.1136/jclinpath-2011-200175. [DOI] [PubMed] [Google Scholar]
- 23.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41(12):1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han H, Cortez CC, Yang X, Nichols PW, Jones PA, Liang G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleo-some occupied promoter. Hum Mol Genet. 2011;20(22):4299–310. doi: 10.1093/hmg/ddr356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–6. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 26.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665–7. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 27.Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461(7265):819–22. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agrawal-Singh S, Isken F, Agelopoulos K, Klein HU, Thoennissen NH, Koehler G, et al. Genome wide analysis of histone H3 acetylation patterns in AML identifies PRDX2 as an epigenetically silenced tumor suppressor gene. Blood. 2012;119(10):2346–57. doi: 10.1182/blood-2011-06-358705. [DOI] [PubMed] [Google Scholar]
- 29.Muller-Tidow C, Klein HU, Hascher A, Isken F, Tickenbrock L, Thoennissen N, et al. Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood. 2010;116(18):3564–71. doi: 10.1182/blood-2009-09-240978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14(5):355–68. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brena RM, Huang TH, Plass C. Quantitative assessment of DNA methylation: Potential applications for disease diagnosis, classification, and prognosis in clinical settings. J Mol Med (Berl) 2006;84(5):365–77. doi: 10.1007/s00109-005-0034-0. [DOI] [PubMed] [Google Scholar]
- 33.You JS, Kelly TK, De Carvalho DD, Taberlay PC, Liang G, Jones PA. OCT4 establishes and maintains nucleosome-depleted regions that provide additional layers of epigenetic regulation of its target genes. Proc Natl Acad Sci USA. 2011;108(35):14497–502. doi: 10.1073/pnas.1111309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32(3):e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karimi M, Johansson S, Stach D, Corcoran M, Grander D, Schalling M, et al. LUMA (LUminometric Methylation Assay)--a high throughput method to the analysis of genomic DNA methylation. Exp Cell Res. 2006;312(11):1989–95. doi: 10.1016/j.yexcr.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 36.Romermann D, Hasemeier B, Metzig K, Gohring G, Schlegelberger B, Langer F, et al. Global increase in DNA methylation in patients with myelodysplastic syndrome. Leukemia. 2008;22(10):1954–6. doi: 10.1038/leu.2008.76. [DOI] [PubMed] [Google Scholar]
- 37.Plass C, Shibata H, Kalcheva I, Mullins L, Kotelevtseva N, Mullins J, et al. Identification of Grf1 on mouse chromosome 9 as an imprinted gene by RLGS-M. Nat Genet. 1996;14(1):106–9. doi: 10.1038/ng0996-106. [DOI] [PubMed] [Google Scholar]
- 38.Khulan B, Thompson RF, Ye K, Fazzari MJ, Suzuki M, Stasiek E, et al. Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res. 2006;16(8):1046–55. doi: 10.1101/gr.5273806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37(8):853–62. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 40.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39(4):457–66. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 41.Rauch T, Pfeifer GP. Methylated-CpG island recovery assay: a new technique for the rapid detection of methylated-CpG islands in cancer. Lab Invest. 2005;85(9):1172–80. doi: 10.1038/labinvest.3700311. [DOI] [PubMed] [Google Scholar]
- 42.Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66(16):7939–47. doi: 10.1158/0008-5472.CAN-06-1888. [DOI] [PubMed] [Google Scholar]
- 43.Gebhard C, Schwarzfischer L, Pham TH, Schilling E, Klug M, Andreesen R, et al. Genome-wide profiling of CpG methylation identifies novel targets of aberrant hypermethylation in myeloid leukemia. Cancer Res. 2006;66(12):6118–28. doi: 10.1158/0008-5472.CAN-06-0376. [DOI] [PubMed] [Google Scholar]
- 44.Palmke N, Santacruz D, Walter J. Comprehensive analysis of DNA-methylation in mammalian tissues using MeDIP-chip. Methods. 2011;53(2):175–84. doi: 10.1016/j.ymeth.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89(5):1827–31. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98(4):288–95. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 47.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471(7336):68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010;8(11):e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6(4):468–81. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- 50.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93(18):9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guldberg P, Worm J, Grønbæk K. Profiling DNA methylation by melting analysis. Methods. 2002;27(2):121–7. doi: 10.1016/s1046-2023(02)00063-4. [DOI] [PubMed] [Google Scholar]
- 52.Wojdacz TK, Dobrovic A. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007;35(6):e41. doi: 10.1093/nar/gkm013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat Protoc. 2007;2(9):2265–75. doi: 10.1038/nprot.2007.314. [DOI] [PubMed] [Google Scholar]
- 54.Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci USA. 2005;102(44):15785–90. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethyl-cytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838–42. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 57.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473(7347):343–8. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydrox-ymethylcytosine in bisulfite sequencing. PLoS One. 2010;5(1):e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473(7347):394–7. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7(6):461–5. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stoddart D, Heron AJ, Mikhailova E, Maglia G, Bayley H. Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proc Natl Acad Sci USA. 2009;106(19):7702–7. doi: 10.1073/pnas.0901054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475(7356):348–52. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 63.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 64.Pillai S, Dasgupta P, Chellappan SP. Chromatin immunoprecipitation assays: analyzing transcription factor binding and histone modifications in vivo. Methods Mol Biol. 2009:523323–39. doi: 10.1007/978-1-59745-190-1_22. [DOI] [PubMed] [Google Scholar]
- 65.Hoemme C, Peerzada A, Behre G, Wang Y, McClelland M, Nieselt K, et al. Chromatin modifications induced by PML-RARalpha repress critical targets in leukemogenesis as analyzed by ChIP-Chip. Blood. 2008;111(5):2887–95. doi: 10.1182/blood-2007-03-079921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120(2):169–81. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 67.Furlan-Magaril M, Rincon-Arano H, Recillas-Targa F. Sequential chromatin immunoprecipitation protocol: ChIP-reChIP. Methods Mol Biol. 2009;543:253–66. doi: 10.1007/978-1-60327-015-1_17. [DOI] [PubMed] [Google Scholar]
- 68.Attema JL, Papathanasiou P, Forsberg EC, Xu J, Smale ST, Weissman IL. Epigenetic characterization of hematopoietic stem cell differentiation using miniChIP and bisulfite sequencing analysis. Proc Natl Acad Sci USA. 2007;104(30):12371–6. doi: 10.1073/pnas.0704468104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adli M, Bernstein BE. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat Protoc. 2011;6(10):1656–68. doi: 10.1038/nprot.2011.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adli M, Zhu J, Bernstein BE. Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nat Methods. 2010;7(8):615–8. doi: 10.1038/nmeth.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goren A, Ozsolak F, Shoresh N, Ku M, Adli M, Hart C, et al. Chromatin profiling by directly sequencing small quantities of immunoprecipitated DNA. Nat Methods. 2010;7(1):47–9. doi: 10.1038/nmeth.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Su X, Ren C, Freitas MA. Mass spectrometry-based strategies for characterization of histones and their post-translational modifications. Expert Rev Proteomics. 2007;4(2):211–25. doi: 10.1586/14789450.4.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 74.Lin YH, Kakadia PM, Chen Y, Li YQ, Deshpande AJ, Buske C, et al. Global reduction of the epigenetic H3K79 methylation mark and increased chromosomal instability in CALM-AF10-positive leukemias. Blood. 2009;114(3):651–8. doi: 10.1182/blood-2009-03-209395. [DOI] [PubMed] [Google Scholar]
- 75.Strobl-Mazzulla PH, Sauka-Spengler T, Bronner-Fraser M. Histone demethylase JmjD2A regulates neural crest specification. Dev Cell. 2010;19(3):460–8. doi: 10.1016/j.devcel.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Payton JE, Grieselhuber NR, Chang LW, Murakami M, Geiss GK, Link DC, et al. High throughput digital quantification of mRNA abundance in primary human acute myeloid leukemia samples. J Clin Invest. 2009;119(6):1714–26. doi: 10.1172/JCI38248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008;26(12):1351–9. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Praulich I, Tauscher M, Gohring G, Glaser S, Hofmann W, Feurstein S, et al. Clonal heterogeneity in childhood myelodysplastic syndromes--challenge for the detection of chromosomal imbalances by array-CGH. Genes Chromosomes Cancer. 2010;49(10):885–900. doi: 10.1002/gcc.20797. [DOI] [PubMed] [Google Scholar]
- 79.Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114(13):2764–73. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Follo MY, Russo D, Finelli C, Mongiorgi S, Clissa C, Fili C, et al. Epigenetic regulation of nuclear PI-PLCbeta1 signaling pathway in low-risk MDS patients during azacitidine treatment. Leukemia. 2012;26(5):943–50. doi: 10.1038/leu.2011.300. [DOI] [PubMed] [Google Scholar]
- 81.Follo MY, Finelli C, Mongiorgi S, Clissa C, Chiarini F, Ramazzotti G, et al. Synergistic induction of PI-PLCbeta1 signaling by azacitidine and valproic acid in high-risk myelodysplastic syndromes. Leukemia. 2011;25(2):271–80. doi: 10.1038/leu.2010.266. [DOI] [PubMed] [Google Scholar]
- 82.Qiu X, Hother C, Ralfkiaer UM, Sogaard A, Lu Q, Workman CT, et al. Equitoxic doses of 5-azacytidine and 5-aza-2'deoxycytidine induce diverse immediate and overlapping heritable changes in the transcriptome. PLoS One. 2010;5(9):e12994. doi: 10.1371/journal.pone.0012994. [DOI] [PMC free article] [PubMed] [Google Scholar]