

Abstract
Smoldering myeloma is an asymptomatic plasma cell dyscrasia with a heterogeneous propensity to progress to active myeloma. In order to investigate the biology of smoldering myeloma patients with high risk of progression, we analyzed the genomic characteristics by FISH, SNP-arrays and gene expression profile of a group of patients with high-risk smoldering myeloma included in a multicenter randomized trial. Chromosomal abnormalities detected by FISH and SNP-arrays at diagnosis were not associated to risk of progression to symptomatic myeloma. However, the overexpression of four SNORD genes (SNORD25, SNORD27, SNORD30 and SNORD31) was correlated with shorter time to progression (P<0.03). When plasma cells from high-risk smoldering patients who progressed to symptomatic myeloma were sequentially analyzed, newly acquired lesions together with an increase in the proportion of plasma cells carrying a given abnormality were observed. These findings suggest that gene expression profiling is a valuable technique to identify smoldering myeloma patients with high risk of progression. (Clinical Trials NCT00443235)
Keywords: gene expression profile, SNP array, FISH, smoldering myeloma
Introduction
Smoldering multiple myeloma (SMM) is an asymptomatic plasma cell dyscrasia with a high propensity to progress to symptomatic myeloma (MM).1,2 It meets all the diagnostic criteria for MM, but without lytic bone disease, anemia, renal failure, or hypercalcemia (CRAB symptoms).3 Due to the risk of progression to symptomatic MM (approximately 10% per year), SMM patients need strict and frequent follow up.2 Globally, the median time to progression (TTP) is approximately four years. However, this figure is highly variable as some patients have MGUS-like clinical behavior and others progress to active MM in a short time, which could be considered to be early MM. Therefore, identifying reliable biological markers to predict which SMM patients will progress and which not is of genuine importance.4–9 In this context, the Mayo Clinic model underscores clonal plasma cell (PC) burden in bone marrow (BM) (>10%) with high monoclonal component (MC) values (>3 g/dL) and skewed free light-chain ratios as high-risk factors with a median TTP of less than two years. 2,10 Our group has added the role of the multiparametric flow cytometry techniques for identifying aberrant PC populations (>95% of total PC) together with the immunoparesis.11
Currently, SMM patients do not receive any treatment until progression to symptomatic MM. This ‘watch and wait’ approach is based on clinical trials in which early treatment intervention was not associated with survival benefit.12–14 However, it should be pointed out that patients included in those trials were not stratified with respect to their risk of progression to symptomatic MM. Due to the heterogeneous course of SMM patients, it does not seem appropriate to adopt a homogeneous approach because it is possible that while low-risk patients would not benefit from early treatment, those at high risk might benefit from early antimyeloma therapy. In line with this hypothesis, our group is conducting a multicenter randomized clinical trial designed to assess the TTP to symptomatic MM, and the efficacy and toxicity of a lenalidomide-dexamethasone (Len-Dex) schedule versus no treatment (abstention arm) in patients with high-risk SMM (HR-SMM).15,16
This trial has given us the opportunity to investigate the genomic characteristics of a group of patients with HR-SMM accurately defined on the basis of inclusion criteria described in the trial. Accordingly, FISH studies, genome-wide profiling using SNP-mapping arrays and gene expression analysis were used to ascertain whether the genomic abnormalities allow us to better define SMM patients with different clinical outcomes.
Design and Methods
Patients
A total of 123 HR-SMM patients were randomized to receive nine Len-Dex cycles plus maintenance (intervention arm) versus no treatment (abstention arm).15,16 HR-SMM were defined by the presence of more than 10% PCs in BM and an MC of IgG more than 3 g/dL, IgA more than 2 g/dL, or Bence Jones proteinuria more than 1 g/24 h together with the absence of CRAB.2 Patients meeting either, but not both, of these two criteria were also included in the study if they met the additional criterion of having 95% or more phenotypically aberrant PCs from the total BMPC compartment (aPC/BMPC) plus immunoparesis.11 The study was approved by the research ethics committees of all participating centers and written informed consent was obtained from all patients in accordance with the Declaration of Helsinki.
Sample preparation
BM samples were collected at the time of inclusion, at symptomatic progression and after nine months of either treatment or abstention. In all the BM samples, CD138-positive PC selection (purity >95%) was carried out using the AutoMACs separation system (Miltenyi-Biotec, Auburn, CA, USA). PCs were frozen in RLT buffer (Quiagen, Valencia, CA, USA) and nucleic acids were then extracted using commercially available kits (Allprepkit, Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. RNA and DNA quality and quantity were determined using 2100 Bioanalyzer (Agilent, Palo Alto, CA, USA) and ND-1000 Spectrophotometer (Nano-Drop Technologies, Wilmington, DE, USA), respectively.
Interphase fluorescence in situ hybridization analysis
Fluorescence in situ hybridization (FISH) analysis was performed in all samples at diagnosis and in 11 samples at symptomatic progression. The systematic screening for genomic aberrations in our institution includes FISH studies for detecting IGH rearrangements t(11;14)(q13;q32), t(4;14)(p16;q32) and t(14;16)(q32;q23) with the corresponding dual-color, dual-fusion translocation probes (Abbott Molecular/Vysis), 13q (LSI 13, RB1 13q14) and 17p deletions (LSI p53, 17p13.1) (Abbott Molecular/Vysis), and 1q gains (on 1q21/SRD 1p36, Kreatech Diagnostics, Amsterdam).17 FISH results of 80% of the patients included in the present study have been reported previously.18
Gene expression profiling and SNP-array studies
The gene expression profiling (GEP) was investigated using Human Gene 1.0 ST (Affymetrix, Santa Clara, CA, USA) in 33 samples at the time of inclusion and 6 at the time of progression. Differentially expressed genes were identified using Significant Analysis of Microarrays (SAM). Genome-wide detection of copy number abnormalities and loss of heterozygosity (LOH) was also performed in 20 samples at diagnosis using the Genome-Wide Human SNP Array 6.0 assay protocol (Affymetrix, Santa Clara, CA, USA). The complete dataset was analyzed by visual inspection using the Genotyping Console 4.0 (Affymetrix) and Chromosome Analysis Suite (ChAS) software (Affymetrix).
Statistical analysis
SPSS version 15.0 (SPSS Inc.) was used for statistical analysis. The χ2 and Fisher’s exact tests were used to examine the association between genetic abnormalities and other categorical variables. TTP distribution curves were plotted using the Kaplan-Meier method; the log rank test was used to estimate the statistical significance of differences between the curves. P<0.05 was considered statistically significant.
Results and Discussion
MM is often the final step in the transformation of two premalignant conditions, MGUS and SMM, which have a very variable progression rate.19,20 Given the current treatment paradigm that consists of delaying treatment until the onset of CRAB symptoms, MM is typically incurable using the available drugs. Therefore, an early therapeutic intervention, particularly in SMM, could prevent or delay progression to MM, changing its adverse outcome. However, the lack of reliable clinical and biological criteria to predict the patients who will progress to MM hinder the development of individualized treatment for this group of patients. Even though the patients analyzed in the present study are SMM with a high risk of progression to MM, the time to progression still varied.15,16 In an attempt to identify additional biological parameters that could be useful in characterizing the natural history of SMM progression, we explored genomic abnormalities by using FISH and SNP-arrays, together with GEP.
Chromosomal abnormalities assessed by FISH were identified in 91 (72%) of the 123 SMM patients. IGH translocations (tx) were observed in 52 of 123 (42%), 1q gains in 47 of 114 (41%), 13q deletions in 51 of 122 (42%), whereas 17p deletions were present in 9 of 123 (7%). The distribution of IGH tx according to 14q32 partners were: t(4;14) in 15 of 123 (12%), t(11;14) in 21 of 123 (17%), t(14;16) in 7 of 122 (6%) and IGH rearrangements with another unknown partner in 10 of 123 (8%). After a median follow up of 24 months, 23 of 61 (38%) patients progressed to symptomatic MM in the abstention arm. When we analyzed the frequency of each of the chromosomal abnormalities in the group of patients who progressed to active MM, we did not find any statistically significant differences with regard to the SMM patients who did not progress. Even when genetic abnormalities were grouped as high or standard risk, no statistical differences were observed. Analysis using the Kaplan-Meier method revealed no differences in the TTP in the aforementioned comparisons. For the patients allocated to the treatment arm, we tested whether chromosomal abnormalities detected by FISH were capable of discriminating Len-Dex response. To do this, we categorized patients in two ways: 1) responders (stringent complete remision, sCR; complete remision, CR; very good partial response, VGPR; and partial response, PR) versus non-responders (stable disease, SD; progression disease, PD); and 2) CR responders (sCR and CR) versus non-CR responders (VGPR, PR, SD and PD). There were no significant differences in either comparison. However, it should be pointed out that the percentage of t(4;14) was higher in non-responders and non-CR responders than in responders and CR responders, respectively (17% vs. 7% and 25% vs. 12%), although the differences were without statistical significance. Thus, we could not identify a high-risk group using FISH. Nevertheless, as the FISH analysis only examined a small number of chromosomal abnormalities, the next step was to perform a high-resolution analysis of genomic imbalances by high-density 6.0 SNP-array, which scans the genome at 680-bp intervals on average. Therefore, 20 patients at diagnosis were analyzed by SNP-arrays. The number and type of copy number abnormalities (CNA), the presence of homozygous deletions (HZD) and copy number-neutral LOH (CNN-LOH) were evaluated. Chromosomal imbalances were identified in 17 (85%) of the 20 patients analyzed. Overall, 166 DNA CNA were identified with a median of 7.5 imbalances per abnormal case (range 0–23). CNN-LOH was detected in 5 of the 20 patients and HZD in 2 of the 20 patients. There were no significant differences in these DNA abnormalities between the 13 patients who progressed to symptomatic MM and the 7 who remained stable, although further studies with a larger number of patients could be needed to confirm these results.
GEP was investigated in 33 patients (18 and 15 in the abstention and treatment arm, respectively). After a median follow up of 29 months, 13 of 18 (72%) patients from the abstention arm progressed to symptomatic MM, with a median TTP of 25 months. When we compared the GEP of these patients to those without CRAB, we found no differentially expressed genes between the two groups. However, this stratification of patients by progression status has the potential bias of including within the progression group both early (≤12 months) and late (> 3 years after diagnosis) progressions. Furthermore, within the group that did not progress to symptomatic MM, the follow up of patients was also heterogeneous (13 vs. 41 months from inclusion in the trial). Therefore, to compare the two extremes of these responses, we selected the 4 patients without progression after more than 30 months follow up and the 4 patients with symptomatic progression in the first ten months since inclusion. Overexpression of five genes (RADD17, SNORD25, SNORD27, SNORD30 and SNORD31) in the SAM analysis was observed in the group with the most unfavorable response (Figure 1). We then tried to validate these results in the global group using the Kaplan-Meier method. To define high and low expression levels of these five genes, we selected a cut-off value close to the median for each of them. We found that the median TTP was significantly shorter in SMM patients with high expression levels of SNORD25, SNORD27, SNORD30 and SNORD31 (Figure 2). Although overexpression of RAD17 was associated with short TTP, these differences were without statistical significance. In conclusion, although GEP was very homogeneous independently of the clinical evolution, it should be noted that SMM patients who developed symptomatic MM displayed high expression levels of SNORD25, SNORD27, SNORD30, SNORD31 and RAD17. Interestingly, four of the five differentially expressed genes were small nucleolar RNA molecules (snoRNA). These non-coding RNAs (ncRNA), although less well known than other ncRNAs, such as miRNAs and siRNAs, could be actively involved in the development of cancer.21 For example, SNORA42 has been shown to act as a potential oncogene in the development and progression of lung cancer.22 Furthermore, homozygous and heterozygous deletions in U50, a C/D snoRNA, have been described in prostate and breast cancer tissues, respectively.23,24 On the other hand, there are some preliminary data showing that the genes that host snoRNAs might also contribute to cancer pathogenesis. Thus, the snoRNA host gene GAS5 controls apoptosis and is down-regulated in breast cancer.21 In addition, RAD17 is a cell cycle checkpoint gene required for cell cycle arrest and DNA damage repair. Regarding the 15 patients treated with Len-Dex, we did not find a characteristic gene signature that predicted response to therapy.
Figure 1.

Expression of RAD17, SNORD25, SNORD27, SNORD30 and SNORD31 by microarrays in high-risk SMM patients who progressed to symptomatic MM and those who remained with stable disease.
Figure 2.

Progression-free survival in SMM patients with respect to SNORD 25, SNORD 27, SNORD 30 and SNORD 31 gene expression levels.
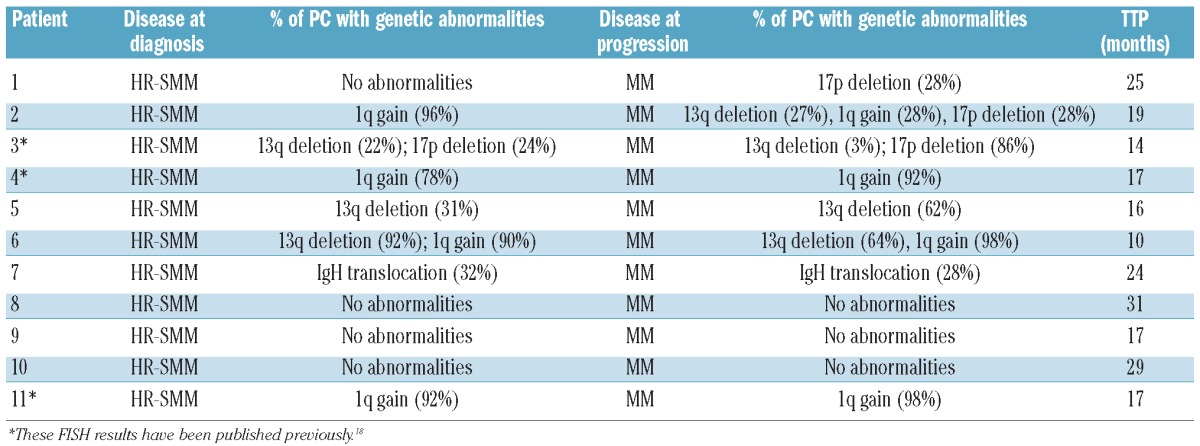
We also wanted to investigate whether myeloma cells obtained at the time of progression had genomic modifications with regard to myeloma cells from the same patient at the time of diagnosis. For this purpose, we sequentially analyzed 11 SMM patients who progressed to symptomatic MM. Newly acquired lesions detected by FISH, together with an increase in the proportion of PCs carrying a given abnormality were observed. The genetic abnormalities of these patients are described in detail in Table 1. For this analysis we have focused on patients with genetic abnormalities in less than 90% of the malignant PCs, since otherwise an increase in the percentages cannot be evaluated. Accordingly, 3 of the 4 patients who already carried chromosomal abnormalities in less than 90% of PCs had a clearly higher percentage of PCs bearing such a genetic aberration at the time of transformation into symptomatic MM. The proportion was doubled in one case, trebled in another, and increased from 68% to 92% in the third. Additionally, 2 SMM patients acquired a 17p deletion at the time of progression. When GEP was performed in 12 samples belonging to 6 control-arm patients before and after symptomatic progression, the paired SAM analysis did not detect significantly deregulated genes between the two groups.
Table 1.
FISH evaluation of 11 patients who developed symptomatic disease.
To summarize, the present study identifies four SNORD genes up-regulated in those SMM patients with rapid progression to symptomatic MM. Despite our failure to find chromosomal lesions associated to risk of progression, we observed an increase in the proportion of clonal PCs carrying a given abnormality, supporting the hypothesis that the number of genetically abnormal PC increases from high-risk SMM to active MM. Since it is difficult to gather a large number of SMM patients with long-term follow up, this study is statistically limited by the relatively small number of patients. However, our findings are promising enough to encourage us to validate them in further series and perform functional studies. In addition, the genomic analysis of those SMM with low-risk of progression to active MM could help to find a biological support to the low/high-risk SMM clasification.
Acknowledgments
The authors thank “Grupo Español de Mieloma” clinicians for providing MM samples and S. González, I. Rodriguez, I. Isidro, T. Prieto y V. Gutiérrez for technical assistance.
Footnotes
Funding: this study was partially supported by Spanish FIS (PI080568, PS09/01450 and PS0901897), “Gerencia Regional de Salud, Junta de Castilla y León” (GRS 702/A/11) grant, and the Spanish Myeloma Network Program (RD06/0020/0006).
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. Hematol Oncol Clin North Am. 2007;21(6):1093–113. doi: 10.1016/j.hoc.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356(25):2582–90. doi: 10.1056/NEJMoa070389. [DOI] [PubMed] [Google Scholar]
- 3.IMWG. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2010;121(5):749–57. [PubMed] [Google Scholar]
- 4.Dimopoulos MA, Moulopoulos A, Smith T, Delasalle KB, Alexanian R. Risk of disease progression in asymptomatic multiple myeloma. Am J Med. 1993;94(1):57–61. doi: 10.1016/0002-9343(93)90120-e. [DOI] [PubMed] [Google Scholar]
- 5.Rajkumar SV. MGUS and smoldering multiple myeloma: update on pathogenesis, natural history, and management. Hematology Am Soc Hematol Educ Program. 2005:340–5. doi: 10.1182/asheducation-2005.1.340. [DOI] [PubMed] [Google Scholar]
- 6.Cesana C, Klersy C, Barbarano L, Nosari AM, Crugnola M, Pungolino E, et al. Prognostic factors for malignant transformation in monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. J Clin Oncol. 2002;20(6):1625–34. doi: 10.1200/JCO.2002.20.6.1625. [DOI] [PubMed] [Google Scholar]
- 7.Dimopoulos MA, Moulopoulos LA, Maniatis A, Alexanian R. Solitary plasmacytoma of bone and asymptomatic multiple myeloma. Blood. 2000;96(6):2037–44. [PubMed] [Google Scholar]
- 8.Rosiñol L, Blade J, Esteve J, Aymerich M, Rozman M, Montoto S, et al. Smoldering multiple myeloma: natural history and recognition of an evolving type. Br J Haematol. 2003;123(4):631–6. doi: 10.1046/j.1365-2141.2003.04654.x. [DOI] [PubMed] [Google Scholar]
- 9.Rosinol L, Carrio A, Blade J, Queralt R, Aymerich M, Cibeira MT, et al. Comparative genomic hybridisation identifies two variants of smoldering multiple myeloma. Br J Haematol. 2005;13085:729–32. doi: 10.1111/j.1365-2141.2005.05673.x. [DOI] [PubMed] [Google Scholar]
- 10.Dispenzieri A, Kyle RA, Katzmann JA, Therneau TM, Larson D, Benson J, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood. 2008;111(2):785–9. doi: 10.1182/blood-2007-08-108357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Persona E, Vidriales MB, Mateo G, Garcia-Sanz R, Mateos MV, de Coca AG, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110(7):2586–92. doi: 10.1182/blood-2007-05-088443. [DOI] [PubMed] [Google Scholar]
- 12.Hjorth M, Hellquist L, Holmberg E, Magnusson B, Rodjer S, Westin J, et al. Initial versus deferred melphalan-prednisone therapy for asymptomatic multiple myeloma stage I–a randomized study. Myeloma Group of Western Sweden. Eur J Haematol. 1993;50(2):95–102. doi: 10.1111/j.1600-0609.1993.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 13.Rajkumar SV, Dispenzieri A, Fonseca R, Lacy MQ, Geyer S, Lust JA, et al. Thalidomide for previously untreated indolent or smoldering multiple myeloma. Leukemia. 2001;15(8):1274–6. doi: 10.1038/sj.leu.2402183. [DOI] [PubMed] [Google Scholar]
- 14.Rajkumar SV, Gertz MA, Lacy MQ, Dispenzieri A, Fonseca R, Geyer SM, et al. Thalidomide as initial therapy for early-stage myeloma. Leukemia. 2003;17(4):775–9. doi: 10.1038/sj.leu.2402866. [DOI] [PubMed] [Google Scholar]
- 15.Mateos MV, Lopez-Corral L, Hernandez MT, de la Rubia J, Lahuerta JJ, Giraldo P, et al. Multicenter, Randomized, Open-Label, Phase III Trial of Lenalidomide-Dexamethasone (Len/dex) Vs Therapeutic Abstention in Smoldering Multiple Myeloma at High Risk of Progression to Symptomatic MM: Results of the First Interim Analysis. ASH Annual Meeting Abstracts. 2009;114(22):614. [Google Scholar]
- 16.Mateos MV, Lopez-Corral L, Hernandez M, Giraldo P, de la Rubia J, de Arriba F, et al. Smoldering Multiple Myeloma (SMM) at High-Risk of Progression to Symptomatic Disease: A Phase III, Randomized, Multicenter Trial Based On Lenalidomide-Dexamethasone (Len-Dex) as Induction Therapy Followed by Maintenance Therapy with Len Alone Vs No Treatment. ASH Annual Meeting Abstracts. 2010;116(21):1935. [Google Scholar]
- 17.Gutierrez NC, Castellanos MV, Martin ML, Mateos MV, Hernandez JM, Fernandez M, et al. Prognostic and biological implications of genetic abnormalities in multiple myeloma undergoing autologous stem cell transplantation: t(4;14) is the most relevant adverse prognostic factor, whereas RB deletion as a unique abnormality is not associated with adverse prognosis. Leukemia. 2006;21(1):143–50. doi: 10.1038/sj.leu.2404413. [DOI] [PubMed] [Google Scholar]
- 18.Lopez-Corral L, Gutierrez NC, Vidriales MB, Mateos MV, Rasillo A, Garcia-Sanz R, et al. The Progression from MGUS to Smoldering Myeloma and Eventually to Multiple Myeloma Involves a Clonal Expansion of Genetically Abnormal Plasma Cells. Cancer Res. 2011;17(7):1692–700. doi: 10.1158/1078-0432.CCR-10-1066. [DOI] [PubMed] [Google Scholar]
- 19.Kyle RA, Durie BG, Rajkumar SV, Landgren O, Blade J, Merlini G, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24(6):1121–7. doi: 10.1038/leu.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113(22):5418–22. doi: 10.1182/blood-2008-12-195008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams GT, Farzaneh F. Are snoRNAs and snoRNA host genes new players in cancer? Nat Rev Cancer. 2012;12(2):84–8. doi: 10.1038/nrc3195. [DOI] [PubMed] [Google Scholar]
- 22.Mei YP, Liao JP, Shen J, Yu L, Liu BL, Liu L, et al. Small nucleolar RNA 42 acts as an oncogene in lung tumorigenesis. Oncogene. 2012;31(22):2794–804. doi: 10.1038/onc.2011.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong XY, Guo P, Boyd J, Sun X, Li Q, Zhou W, et al. Implication of snoRNA U50 in human breast cancer. J Genet Genomics. 2009;36(8):447–54. doi: 10.1016/S1673-8527(08)60134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong XY, Rodriguez C, Guo P, Sun X, Talbot JC, Zhou W, et al. SnoRNA U50 is a candidate tumor-suppressor gene at 6q14.3 with a mutation associated with clinically significant prostate cancer. Hum Mol Genet. 2008;17(7):1031–42. doi: 10.1093/hmg/ddm375. [DOI] [PMC free article] [PubMed] [Google Scholar]