

Abstract
MicroRNAs (miRNAs) comprising 19–25 nucleotides are highly conserved small non-coding RNAs which regulate normal gene expression during development, cell proliferation and apoptosis by targeting mRNAs of protein-coding genes at the post-transcriptional level. Prevalent studies suggest that some human miRNAs, such as miRNA-16, are deregulated in human cancer and behave as tumor suppressors. The overall objective of our investigation was to assess whether miRNA-16 (miR-16) is involved in the regulation of critical genes, such as BCL2, that control the sensitivity of pancreatic cancer cells to apoptosis. This study showed that the ectopic overexpression of miR-16 may be therapeutically beneficial as is evidenced by impaired cell survival with concomitant attenuation of anti-apoptotic protein Bcl-2. Moreover, the luciferase reporter assay suggested that miR-16 post-transcriptionally regulates Bcl-2 expression in pancreatic cancer cells through the target sites of the 3′ untranslated region of this gene.
Keywords: pancreatic cancer, microRNA, Bcl-2, antiproliferative effect
Introduction
Pancreatic cancer is one of the leading causes of cancer-related mortality in the US, since the majority of affected patients present with advanced stages of surgically inoperable disease (1,2). The identification of effective therapeutic and prevention strategies (3–5) against this metastatic disease is of high priority and has public health significance.
Previous studies revealed the link between microRNAs (miRNAs) and human cancer, thus yielding interest in using them as targets for cancer therapies. miRNAs are a class of highly conserved, non-coding RNAs that are approximately 19–25 nucleotides long (6–8). Biogenesis of miRNA results from primary miRNAs (pri-miRNAs), endogenously expressed transcripts of hairpin RNAs. These pri-miRNAs are cleaved by the ribonuclease Drosha in the nucleus in order to generate 72–100 nucleotide precursor miRNAs (pre-miRNAs). The nuclear protein Exportin 5 translocates pre-miRNAs, in the fold-back structure, to the cytoplasm. Cytoplasmic ribonuclease Dicer then digests the pre-miRNAs to form short double-stranded miRNA duplexes. The miRNA duplexes separate into mature, single-stranded miRNAs of 19–25 nucleotides. These mature miRNAs are able to bind the RNA-induced silencing complex to align with target miRNAs at their 3′ untranslated region (UTR). Such interaction leads to translational repression or cleavage of miRNAs and the subsequent down-modulation of the respective genes.
Several miRNAs are involved in the regulation of gene expression, a critical aspect of many biological processes, including cell development, differentiation, apoptosis and proliferation (7–12). Recent studies have implicated miRNAs in the development of human malignancies (9–12) and have documented the oncogenic (overexpressed) and tumor-suppressor (underexpressed) roles of miRNAs in several neoplasms. Notably, previous findings have established the role of miRNA-16 (miR-16) as a tumor suppressor in a variety of human cancers (13–15). Moreover, Lu et al (16) demonstrated that the expression of miR-16 was globally higher in normal cells compared to cancer cells. miR-16 is involved in cell growth and apoptosis pathways in human malignancies, including chronic lymphocytic leukemia (13) and gastric carcinoma (14), by negatively regulating Bcl-2. The anti-apoptotic protein, Bcl-2, is also overexpressed in human pancreatic cancer (17,18). Previously, Bold et al (18) suggested that an increased BCL2 expression correlates with apoptotic resistance and metastatic potential, since the deregulation of BCL2 expression may be involved in the metastatic progression of pancreatic carcinoma. Findings of this study showed that the enforced overexpression of miR-16 mimic in pancreatic adenocarcinoma cells attenuates cell growth with the simultaneous repression of the BCL2 gene.
Materials and methods
Cell culture and treatment
Human pancreatic adenocarcinoma BxPC-3 cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were grown in RPMI with supplements of 10% fetal bovine serum and 50 μg/ml of gentamicin at 37°C in a 5% CO2 humidified atmosphere.
Transfection of miRNA mimic and inhibitor
Cells were plated at a density of 1.8×105 cells/ml in T75 flasks and incubated overnight at 37°C. The following day, cells were either mock-transfected or transfected with 27 nM miR-16 miRIDIAN miRNA hairpin inhibitor or miR-16 miRNA mimic and their appropriate negative controls, using the Dharmafect2 transfection reagent (Dharmacon, Lafayette, CO, USA) in accordance with the manufacturer’s protocol. miRIDIAN mimic and inhibitor oligos were prepared by dissolution in 1X small interfering RNA (siRNA) buffer. Diluted oligos were placed in Tube 1, and transfection reagent was maintained in Tube 2 in serum-free medium. Reagents were mixed gently, followed by incubation at room temperature (RT) for 5 min. The contents in Tubes 1 and 2 were then combined and incubated at RT for 30 min. Subsequently, complete medium with 10% serum was added to the mixture followed by addition to the cells. The transfected cells were allowed to grow overnight. The transfection medium was replaced after 24 h with fresh medium.
miRNA Northern blot analysis
Total RNA (5 μg) was loaded and separated on a 15% denatured urea gel. After electrophoresis and transfer, the blot was hybridized with corresponding biotin pre-labeled miRNA probe (Signosis Inc., Sunnyvale, CA, USA). To normalize the hybridization, a biotin pre-labeled RNU48 probe was mixed with miRNA probe for co-hybridization. The hybridized probes were then monitored with streptavidin-HRP and chemiluminescent detection.
Luciferase activity assay
pGL3-BCL2 3′ UTR sense and antisense constructs were prepared as previously described (13). For luciferase reporter assay, human pancreatic cancer cells were co-transfected in a 96-well plate with 100 ng of the firefly luciferase reporter vector and 10 ng of the pEGFPC2 expression vector (transfection control; Clonetech) either alone or in combination with 50 nM miR-16 mimic oligo or 50 nM mimic negative control using the Dharmafect Duo transfection reagent. Luciferase activity was normalized to GFP expression. The relative expression was determined for the mimic negative control and miR-16-transfected samples.
Western immunoblotting
Following the designated treatment, total cellular proteins were extracted. Equal protein from each sample was fractionated by SDS-PAGE and blotted onto a nitrocellulose membrane (GE Healthcare, NJ, USA). Membranes were probed with the Bcl-2 antibody (Upstate Biotechnology, Syracuse, NY, USA) followed by incubation with a HRP-conjugated secondary antibody (3,4). Finally, immunodetection was carried out by the enhanced chemiluminescence method (GE Healthcare). Immunodetection with the β-actin antibody (Sigma, St. Louis, MO, USA) served as a protein loading control.
Clonogenic cell survival assay
Cells were washed 24 h post-transfection with phosphate-buffered saline and then seeded in triplicate (20,000 cells/10-cm dish). The cells were allowed to grow for an additional 2 weeks. The medium was changed every 4 days. The cells were then fixed with 4% buffered formalin (Electron Microscopy Sciences, Hatfield, PA, USA) and stained with 0.1% crystal violet for visualization and photography (19,20).
Results
Overexpression of miR-16 impairs pancreatic cancer cell clonal growth
To understand whether miR-16 regulates pancreatic adenocarcinoma cell proliferation, we used a commercially available miR-16 mimic (mature). miR-16 mimics and inhibitors as well as the respective negative controls were transfected into BxPC-3 cells. The overexpression or silencing of miR-16 was verified by Northern blotting of the transfected cells (Fig. 1B). At the 48-h transfection, we achieved a significant expression of miR-16 over mock-transfected and scrambled control-containing cells (Fig. 1B). The clonogenic cell survival assay (19,20) was employed to determine the long-term survival ability of BxPC-3 cells following transfection with miR-16 and its inhibitor oligos. Notably, the overexpression of miR-16 significantly decreased the clonogenic cell survival of pancreatic tumor cells when compared to the mimic negative control (Fig. 1, panel II vs. panel I), whereas silencing of the same miR-16 had no effect on cell survival (Fig. 1A, panel III vs. panel IV). Due to the low endogenous level, silencing of miR-16 did not enhance cell survival when compared to the control.
Figure 1.

miR-16 impairs the colony formation ability of pancreatic tumor cells and down-regulates Bcl-2/FGF-2. The transfection of miR-16 mimic, inhibitor and the respective negative controls was carried out using Dharmafect2 transfection reagent as described in Materials and methods. (A) Clonogenic cell survival assay. Cells were washed 2 times with phosphate-buffered saline and seeded in triplicate (20,000 cells/10-cm dish) 24 h post-transfection. Cells were allowed to grow for an additional 2 weeks, followed by staining with 0.1% crystal violet for visualization and photography. Panel I, mimic negative control; panel II, miR-16 mimic oligonucleotide; panel III, inhibitor negative control; panel IV, miR-16 inhibitor. (B) Northern blot analysis. RNA was isolated from the negative control, miR-16 mimic- and inhibitor-transfected BxPC-3 cells and subjected to Northern blotting. Lane 1, mock; lane 2, mimic negative control; lane 3, miR-16 mimic; lane 4, inhibitor negative control; lane 5, miR-16 inhibitor; lane M, molecular weight markers. To normalize the hybridization, RNU48 probe was used as an internal control.
Exogenous expression of miR-16 reciprocally regulates Bcl-2 expression in BxPC-3 cells
As mentioned previously, miR-16 has the ability to down-modulate anti-apoptotic protein Bcl-2 in diverse cancer types. We attempted to ascertain whether the transfection of miR-16 mimic oligonucleotide exerts any effect on the Bcl-2 level in pancreatic cancer cells. A decrease in the Bcl-2 protein level in the BxPC-3 cells (Fig. 2, lane 2) was clearly evident due to the ectopic overexpression of miR-16. However, the level of Bcl-2 did not decrease when the hairpin inhibitor oligonucleotide of miR-16 was transfected into these cells (Fig. 2, lane 4).
Figure 2.
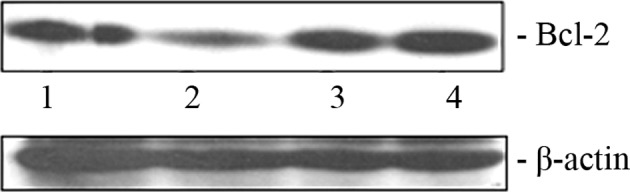
Bcl-2 expression correlates with the miR-16 level. A cell-free extract was subjected to Western blotting with the Bcl-2 antibody 48 h post-transfection. Lane 1, mimic negative control; lane 2, miR-16 mimic oligonucleotide; lane 3, inhibitor negative control; lane 4, miR-16 inhibitor.
Post-transcriptional repression of BCL2 by miR-16 in pancreatic cancer cells
The decrease in Bcl-2 protein in miR-16-overexpressed cells may be the direct effect of miRNA::mRNA complementarity or by an indirect interaction with other unknown targets. To address this issue, we used luciferase reporter constructs where the 3′ UTR sequence of the human BCL2 gene contained a presumed miR-16 complementary site that was fused into a luciferase reporter plasmid in both sense and antisense orientations (immediately downstream of the stop codon of luciferase). The resulting plasmids and control plasmid (pEGFPC2) were introduced into BxPC-3 pancreatic cancer cells together.
Notably, as shown in Fig. 3A, the co-transfection of the sense orientation constructs (pGL3 BCL2) with miR-16 mimic oligo resulted in a significant reduction in firefly luciferase reporter activity when normalized against GFP (P<0.002). In contrast, the transfection of miR-16 oligo in the presence of the luciferase vector containing BCL2 3′ UTR in the antisense orientation terminated this suppression (Fig. 3B). Moreover, the co-transfection of mimic negative control miRNA had no effect on the luciferase activity of pGL3 BCL2 and pGL3 BCL2 AS. Thus, our observation suggests that miR-16 directly regulates Bcl-2 expression in pancreatic cancer cells through the target site of 3′ UTR of BCL2 mRNA at the post-transcriptional level.
Figure 3.
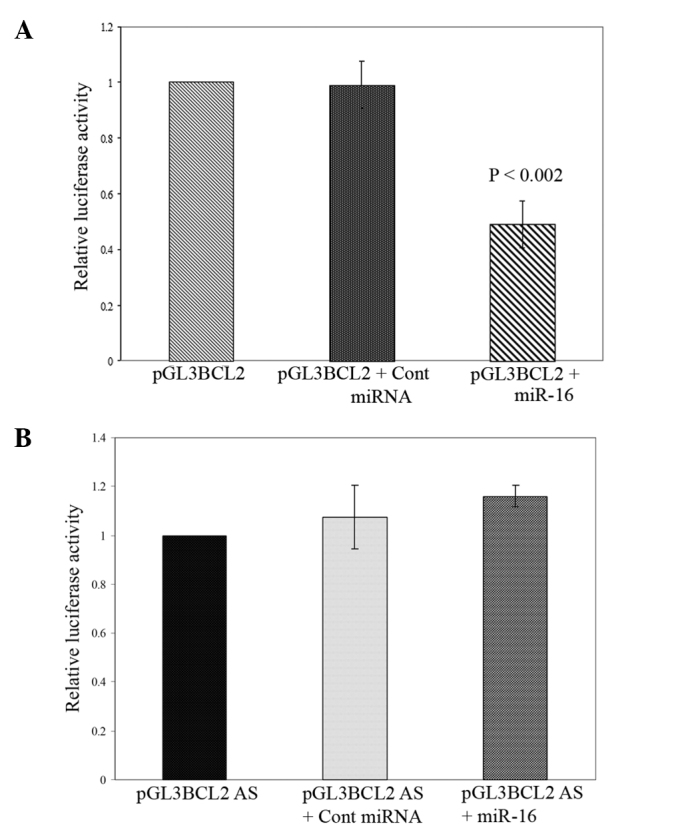
Direct identification of BCL2 3′ UTR by miR-16 in pancreatic cancer cells. (A and B) 3′ UTR (220 bp) of the BCL2 gene cloned in pGL3 control vectors in both sense (pGL3 BCL2) and antisense (pGL3 BCL2 AS) orientations. For luciferase reporter assay, human pancreatic cancer BxPC-3 cells were co-transfected in a 96-well plate with 100 ng of firefly luciferase reporter vector (pGL3-BCL2 UTR sense and antisense) and 10 ng of the pEGFPC2 expression vector in the presence of 50 nM miR-16 mimic oligo or 50 nM mimic negative control. The luciferase activity was measured (Allele Biotechnology, San Diego, CA, USA), 48 h post-transfection, and normalized to GFP expression. Notably, the luciferase activities of the control plasmids were considered to be 1.
Discussion
Pancreatic cells often become malignant during tumorigenesis when cells acquire traits such as the ability to evade apoptosis, replicate indefinitely and engage in persistent angiogenesis. These malignant cells acquire resistance to therapy due to multiple genetic alterations at each stage of tumorigenesis, thereby imparting a selective advantage of growth over normal cells. In this respect, the ability of miR-16 to attenuate the growth of pancreatic adenocarcinoma cells by down-modulation of the anti-apoptotic gene BCL2 is significant. Our finding is potentially significant for utilizing miRNA-16 as a therapeutic tool for pancreatic cancer treatment. The role of miR-16 as a tumor suppressor was primarily documented (13) in B-cell chronic lymphocytic leukemia, which is a common form of adult leukemia. Among other studied potential tumor suppressor miRNAs, let-7 was found to be consistently under-expressed in lung cancers compared to normal adjacent tissues (21). Similar to our finding, the overexpression of tumor suppressor let-7g miRNA was shown to impair lung cancer cell proliferation and promote cell death in vitro and in vivo (22,23).
Notably, the effect of anticancer drugs and chemopreventive agents that modulate cell proliferation and apoptotic signaling on miRNA expression profiles has also been explored. In this context, Blower et al (24) suggested the potential role of miRNAs, such as let-7i, miR-16 and miR-21, in the anticancer drug response when tested in NCI-60 human cancer cell lines. Recently, the pro-apoptotic action of EGCG, a significant component of green tea, has been shown to be mediated by the up-regulation of miR-16 and down-regulation of Bcl-2 in hepatocellular carcinoma cells (25). Previously, Scott et al (26) demonstrated a functional link between histone deacetylase inhibition and the presence of miR-27a/27b miRNA. In addition, all-trans-retinoic acid treatment of leukemic cells resulted in the differential expression of a number of miRNAs, including let-7 and the miR-16 family (27). More importantly, Weidhaas et al (22) suggested that miRNAs may be potential targets for altering resistance to cytotoxic anticancer therapy.
Since miRNA-16 overexpression diminishes the proliferation of human pancreatic cancer cells, it can be used in conjunction with chemotherapeutic agents such as gemcitabine to combat pancreatic cancer. miRNAs have multiple target genes and may decrease the expression levels of genes with various biological functions. As opposed to the silencing of individual genes, this multi-targeted approach can revolutionize therapeutic strategies against pancreatic cancer by exerting a stronger inhibitory effect on tumor growth. Therefore, this novel observation implicating the antiproliferative effect of miR-16 on pancreatic adenocarcinoma cells, may provide new insights into treating pancreatic cancer.
Acknowledgements
This study was supported, in part, by NIH Grants CA137476 (A. Basu) and CA 109181 (S. Haldar). We also thank D. Haldar and K. Haas for the experimental assistance.
References
- 1.Jamal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer Statistics. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Goggins M. Identifying molecular markers for the early detection of pancreatic neoplasia. Semin Oncol. 2007;34:303–310. doi: 10.1053/j.seminoncol.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basu A, Haldar S. 2-Methoxyestradiol mediated signaling network in pancreatic cancer. Front Biosci. 2009;14:2170–2178. doi: 10.2741/3369. [DOI] [PubMed] [Google Scholar]
- 4.Basu A, Castle VP, Bouziane M, Bhalla K, Haldar S. Crosstalk between extrinsic and intrinsic cell death pathways in pancreatic cancer: synergistic action of estrogen metabolite and ligands of death receptor family. Cancer Res. 2006;66:4309–4318. doi: 10.1158/0008-5472.CAN-05-2657. [DOI] [PubMed] [Google Scholar]
- 5.Qanungo S, Das M, Haldar S, Basu A. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis. 2005;26:958–967. doi: 10.1093/carcin/bgi040. [DOI] [PubMed] [Google Scholar]
- 6.Zeng Y. Principles of micro-RNA production and maturation. Oncogene. 2006;25:6156–6162. doi: 10.1038/sj.onc.1209908. [DOI] [PubMed] [Google Scholar]
- 7.Jovanovic M, Hengartner MO. miRNAs and apoptosis: RNAs to die for. Oncogene. 2006;25:6176–6187. doi: 10.1038/sj.onc.1209912. [DOI] [PubMed] [Google Scholar]
- 8.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 9.Dalmay T, Edwards DR. MicroRNAs and the hallmarks of cancer. Oncogene. 2006;25:6170–6175. doi: 10.1038/sj.onc.1209911. [DOI] [PubMed] [Google Scholar]
- 10.Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006;25:6188–6196. doi: 10.1038/sj.onc.1209913. [DOI] [PubMed] [Google Scholar]
- 11.Negrini M, Nicoloso MS, Calin GA. MicroRNAs and cancer – new paradigms in molecular oncology. Curr Opin Cell Biol. 2009;21:470–479. doi: 10.1016/j.ceb.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 12.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 13.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia L, Zhang D, Du R, et al. miR15-b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer. 2008;123:372–379. doi: 10.1002/ijc.23501. [DOI] [PubMed] [Google Scholar]
- 15.Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L, D’Urso L, Pagliuca A, Biffoni M, Labbaye C, Bartucci M, Muto G, Peschle C, De Maria R. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–1277. doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- 16.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 17.Galante JM, Mortenson MM, Bowles TL, Virudachalam S, Bold RJ. ERK/BCL-2 pathway in the resistance of pancreatic cancer to anoikis. J Surg Res. 2009;152:18–25. doi: 10.1016/j.jss.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 18.Bold RJ, Virudachalam S, McConkey DJ. BCL2 expression correlates with metastatic potential in pancreatic cancer cell lines. Cancer. 2001;92:1122–1129. doi: 10.1002/1097-0142(20010901)92:5<1122::aid-cncr1429>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 19.Munshi A, Hobbs M, Meyn RE. Clonogenic cell survival assay. Methods Mol Med. 2005;110:21–28. doi: 10.1385/1-59259-869-2:021. [DOI] [PubMed] [Google Scholar]
- 20.Basu A, Haldar S. Antiproliferative and proapoptotic effects of benzyl isothiocyanate on human pancreatic cancer cells is linked to death receptor activation and RasGAP/Rac1 down-modulation. Int J Oncol. 2009;35:593–599. doi: 10.3892/ijo_00000370. [DOI] [PubMed] [Google Scholar]
- 21.Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18:505–516. doi: 10.1016/j.tcb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M, Gillespie E, Slack FJ. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111–11116. doi: 10.1158/0008-5472.CAN-07-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, Jacks T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. 2008;105:3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blower PE, Chung JH, Verducci JS, Lin S, Park JK, Dai Z, Liu CG, Schmittgen TD, Reinhold WC, Croce CM, Weinstein JN, Sadee W. MicroRNAs modulate the chemosensitivity of tumor cells. Mol Cancer Ther. 2008;7:1–9. doi: 10.1158/1535-7163.MCT-07-0573. [DOI] [PubMed] [Google Scholar]
- 25.Tsang WP, Kwok TT. Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. J Nutr Biochem. 2010;21:140–146. doi: 10.1016/j.jnutbio.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 26.Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006;66:1277–1281. doi: 10.1158/0008-5472.CAN-05-3632. [DOI] [PubMed] [Google Scholar]
- 27.Garzon R, Pichiorri F, Palumbo T, Visentini M, Aqeilan R, Cimmino A, Wang H, Sun H, Volinia S, Alder H, Calin GA, Liu CG, Andreeff M, Croce CM. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene. 2007;26:4148–4157. doi: 10.1038/sj.onc.1210186. [DOI] [PubMed] [Google Scholar]