

Abstract
In this article, I reflect on research on two ATPases. The first is F1F0-ATPase, also known as ATP synthase. It is the terminal enzyme in oxidative phosphorylation and famous as a nanomotor. Early work on mitochondrial enzyme involved purification in large amount, followed by deduction of subunit composition and stoichiometry and determination of molecular sizes of holoenzyme and individual subunits. Later work on Escherichia coli enzyme utilized mutagenesis and optical probes to reveal the molecular mechanism of ATP hydrolysis and detailed facets of catalysis. The second ATPase is P-glycoprotein, which confers multidrug resistance, notably to anticancer drugs, in mammalian cells. Purification of the protein in large quantity allowed detailed characterization of catalysis, formulation of an alternating sites mechanism, and recently, advances in structural characterization.
Keywords: ATP Synthase, ATPases, Enzyme Catalysis, Mitochondria, Phosphorylation
Introduction
I began undergraduate degree studies in September 1961. Thus, over the last half-century, I had the good fortune, as student, teacher, and researcher, to participate in one of the greatest advances in knowledge that has ever occurred in a single discipline in human history. That discipline is biochemistry, and its flagship journal is the Journal of Biological Chemistry. For most of the last fifty years, the editor-in-chief was Herbert Tabor. We owe to him a huge debt of gratitude. When Herb asked me to write a Reflections article, I felt supremely honored. Here goes.
Very Early Days
I was born in Sheffield, England, in 1943, so the Second World War dominated my early years. Rationing was in place; food and clothing were sometimes in short supply. Sheffield, the fifth largest city in England, was a center of steel manufacturing, world-renowned for its cutlery. It had been severely bombed; there were frequent desolate spaces interrupting rows of houses and factories. The buildings were uniformly black, and it was not until the 1960s, when use of soft coal was prohibited, that steam cleaning of civic buildings revealed to a delighted populace that they were in reality white, pink, yellow, and admirable. Dad served in the Royal Artillery from 1939 to 1945; virtually all of his contemporaries served in the armed forces or in war-related activity. Colorful Uncle Walt used to regale us with stories of how, as a steel mill foreman, he constantly thwarted the Luftwaffe's best efforts to shut down his operation. Mum had gone into retailing, but her department store was destroyed, and she became a bus conductor, a previously all-male occupation. Naturally, these experiences strongly affected all of their lives, but normality did return. By the mid-1950s, the economy was looking up, and by the 1960s, it was “swinging.” Of course, we young folk took full responsibility for this, but in retrospect, Britain owed its recovery to a strong resolve and astute leaders.
Dad had left school at age 14, and Mum at 16. Both understood that education was key, and they emphasized it. At age 11, I took the citywide entrance test for secondary schooling. Sheffield had historically been the venue of important innovations in steel technology; it respected education and had strong public schools, the best of which was the all-male King Edward VII School. When the results came out, I was admitted to King Edward's and was rewarded with a silver sixpence by an ecstatic Mum. King Edward's lived up to its billing. It was rigorous and demanding. The first four years covered a range of topics leading up to the nationwide O-level examinations at age 15. After that, one was required to specialize in classics, modern studies, or science. I had done well in Latin and Greek and had enjoyed greatly a course in ancient Greek history (who wouldn't?), so I was considering classics. I had also done well in science. At this point, a thoughtful classics teacher took me aside and recommended science. He did not feel, he told me, that I would get to the top in classics, but that I might in science, good advice for which I have been grateful. At age 17, I successfully passed the A-level exams in physics, chemistry, and biology, and at 18, I passed the more difficult S-level examinations in chemistry and biology. I left King Edward's in 1961, the recipient of a superb education.
Newcastle Years
The physiology department in the Medical School at King's College, Newcastle, offered me a place in its three-year program for a bachelor's degree with honors, which I gratefully accepted. In 1961, King's was part of Durham University, but the plan was to make it an independent university. There was a democratic approach to choosing a new name, with the students involved, but when it became apparent that “The Blue Star University” might win, the vice-chancellor stepped in smartly, and we became the University of Newcastle upon Tyne, shortened in recent years to Newcastle University. The blue star in question is to be found on the label of the students' favorite libation, which today is Newcastle's best known export. The physiology department at Newcastle combined strong research with excellent teaching. The local people were proud of their university and welcomed students from outside the region gladly. Altogether, it was an excellent choice.
In the first year, I took zoology, organic chemistry, and physiology. Zoology featured invertebrate species starting at Amoeba, ending at Amphioxus, and describing everything in between. This involved lots of memorization, boring at times, but I'll say one thing: put me in any environment on Earth, if an invertebrate should come walking, crawling, flying, or swimming my way, I can make a pretty good stab at telling you class, order, family, genus. In the second year I took physical chemistry, microbiology, math, and physiology. Microbiology was engrossing, and when the Molecular Biology Revolution later engulfed us all, it proved fundamentally valuable. The final year consisted entirely of physiology, with a research project and thesis required. The American reader will note the absence of liberal arts courses. In truth, we would have regarded them as a waste of precious time. I enjoyed my first exposure to research. The department was strong in gastroenterology. The head, A. A. Harper, was the discoverer of pancreozymin (now cholecystokinin) and had recruited several first-rate experimentalists. My project, under the direction of J. David Reed, entailed a study of components of cat gastric secretion and was quite biochemical.
H. S. A. (“Stan”) Sherratt had been recruited into the physiology department to beef up biochemical aspects, and he described to me a project that would make an ideal Ph.D. thesis study. I graduated with honors in physiology in the summer of 1964 and agreed to start work under Stan in October. There were two other important events that year. First, I met my future wife, Glenda. She subsequently graduated from the botany honors degree program at Newcastle. (So, together, we have both the fauna and the flora nailed down.) Second, I received an invitation from Gordon W. Searle, professor of physiology at the University of Iowa, to spend ten weeks in his laboratory doing research on glucose transport in rat small intestine. I had written to physiology departments in the United States asking if they could offer a summer research experience for a just-graduated young chap, and when Gordon replied positively, I was more than happy. It turned out to be a life-changing experience. I thoroughly enjoyed my ten weeks in Iowa City, particularly in the laboratory. I could see that American laboratories were better equipped than UK laboratories and that, in the post-Sputnik era, there was money for young researchers. It planted in my mind the idea that living and working in America might be a future option. As it turned out, this is what happened, and I am deeply grateful to Gordon for affording me the opportunity to work in his laboratory. An interesting aside is that the flight from London to New York was my first experience of flying, at age 21, and that it took exactly the same time in 1964 as it does today.
Back in Newcastle, I began Ph.D. studies. I had a three-year Science Research Council scholarship, so the goal was to finish in three years. My project was to study the mechanism of action of pent-4-enoic acid, a hypoglycemic agent. The ackee fruit Blighia sapida was brought originally to Jamaica by Captain Bligh (the “Bounty” man) and had become a staple of Jamaican diet. However, eating unripe fruit could bring on “Jamaican vomiting sickness,” which killed ∼5000 people in the first half of the 20th century. Victims evinced profound hypoglycemia due to the presence in unripe fruit of hypoglycin (l-2-amino-3-methylenecyclopropylpropionic acid), which is metabolized to methylenecyclopropylacetic acid, the toxic agent. Drug companies were interested because there was good evidence that hypoglycemia was hormone-independent, and so a route to oral hypoglycemic agents might emerge. It was apparent that the backbone configuration (C=C-C-C-COOH) endowed hypoglycemic potency by an unknown mechanism. Pent-4-enoic acid was the simplest compound to show the effect, and there were several nonhypoglycemic controls (pentanoic acid, pent-2-enoic acid, and cyclopropanecarboxylic acid) that I could use. At the time, elucidation of metabolic pathways was “hot,” and every laboratory had the latest Boehringer metabolic pathways chart on the wall, so I would be working in an evolving area.
I conducted a series of pharmacological and biochemical experiments and, to summarize, I determined that hypoglycemia was caused by inhibition of fatty acid oxidation in mitochondria and impaired gluconeogenesis. Reduced fatty acid oxidation leads to enhanced dependence on glucose as fuel; once glycogen stores are exhausted and because gluconeogenesis is insufficient, hypoglycemia results. I had used Hans Krebs' technique for gluconeogenesis measurements, and when I presented my results at the Biochemical Society meeting in 1967, I met and discussed them with him, a memorable moment. My thesis was submitted in October 1967; my external examiner was Peter Garland, who gave me a thorough but positive grilling, and I passed. On January 1, 1968, my family and I, with green cards in our pockets, were en route to a frigid Madison, Wisconsin, where I was to start a postdoctoral fellowship funded by the National Institutes of Health (NIH) under David E. Green at the Institute for Enzyme Research.
It later transpired that pent-4-enoic acid is metabolized by mitochondria to 3-oxopent-4-enoyl-CoA, which potently inhibits 3-oxoacyl-CoA thiolase. Unfortunately, side effects precluded any clinical use of the compounds. As I showed, pent-4-enoic acid is strongly ketogenic; hypoglycin metabolites also affect amino acid metabolism through inhibition of isovaleryl-CoA dehydrogenase (for reviews of this field, see Refs. 1 and 2).
At the Enzyme Institute
Green's laboratory had 16 postdoctoral fellows when I arrived, with a strong supporting technical staff. By 1968, Green had accumulated an exceptional list of accomplishments, including the discovery of the four respiratory complexes I–IV, which constitute the foundation of oxidative metabolism in eukaryotes. I was to spend four years at the Enzyme Institute and to learn a huge amount about proteins, membranes, and bioenergetics. Not least, I was to interact closely and form lasting friendships with a number of fellow postdocs who went on to distinguished careers. I had landed therefore in an exceptional situation. There was just one fly in the ointment: the project that I was assigned was a complete dud. The idea was that “structural proteins” formed the backbone of biological membranes and bound the membrane enzymes and phospholipids. Insoluble preparations thought to represent such proteins had been reported by several groups. My project was to characterize the membrane-forming, protein-binding, and phospholipid-binding properties of mitochondrial structural protein. Which I did exhaustively, concluding that mitochondrial structural protein was not a structural component of mitochondrial membranes; rather, it was a heterogeneous mixture containing denatured mitochondrial ATPase as one major component. Its protein- and lipid-binding properties were physiologically insignificant. There were redeeming features, to be sure. I published my first JBC paper, with David MacLennan (3). After my work, structural protein as a concept vanished, so I saved the nation some research dollars. And I now knew a lot about mitochondrial ATPase.
Soluble mitochondrial ATPase had been purified by Maynard Pullman, Harvey Penefsky, Anima Datta, and Efraim Racker in 1960. They named it “F1.” When bound back to mitochondrial inner membrane from which it had previously been stripped, it restored oxidative phosphorylation. It was therefore evident that F1 contained catalytic sites for ATP hydrolysis and synthesis and that detailed study of it would get us to the heart of oxidative phosphorylation. I decided to pursue this avenue, and Green was supportive. I was joined in late 1969 by a talented postdoctoral fellow, Jack Brooks. The ensuing years have shown that F1 is one part of the ATP synthase enzyme, also called F1F0, which consists of a membrane sector (F0) and extramembranous F1, working together to catalyze proton gradient-driven ATP synthesis or ATP hydrolysis-driven proton pumping. ATP synthase occurs widely in archaea, eubacteria, and eukaryotes, and F1 is remarkably similar across species. We did not know at the time that we were dealing with such a truly fundamental enzyme.
The original purification procedure did not provide sufficient material, so the first requirement was a better one. This we accomplished by introduction of ion exchange and gel filtration, producing hundreds of milligrams of homogeneous protein (4). The protocol that we introduced was followed in principle for the next twenty-five years by laboratories studying F1 in bacteria, chloroplasts, and mitochondria. We showed the molecular size to be 360 kDa and that F1 contained five subunits, ranging from 7.5 to 53 kDa, plus a sixth reversibly dissociable subunit (5, 6), the latter corresponding to the “specific inhibitor protein” described originally by Pullman and Gladys Monroy. The likely ratio of the five core subunits was 3:3:1:1:1. Each subunit was purified, and amino acid analysis verified that they were unique entities (7). In further work, I purified subunit OSCP (oligomycin sensitivity-conferring protein) to homogeneity in large amount and characterized it in detail (8). OSCP originally had been identified by David MacLennan and Alex Tzagoloff as a component of one of Racker's “coupling factors,” which facilitated binding of F1 to membranes. I showed that it was a component of several coupling factors. We now know that OSCP is a component of the peripheral stalk that anchors F1 to F0.
By introducing modern protein purification and analytical techniques, Jack and I moved the field forward significantly. I summarized our work in a review, which was well received (9). We were among the first to use the new technique of SDS-gel electrophoresis (10), which became so important in biochemistry. There was one event that I still regret. Jack and I had the SDS gels running well, so we obtained samples of purified respiratory complexes I–IV from adjoining laboratories and ran them. Each contained so many components that we doubted the purity of the preparations! We wrote up a manuscript but decided not to submit. In hindsight, we had it right: they do contain multiple components, as many as 40 in the case of complex I, and we would have been the first to show it.
Rochester
In 1971, Glenda and I had to make the decision whether to return to the United Kingdom or to stay in the United States. Fortuitously, the choice was made for us. Out of the blue, Elmer Stotz, Chairman of the Department of Biochemistry at the University of Rochester Medical Center, contacted me and asked me if I was interested in the job of assistant professor. Rochester had just expanded its medical school class size, and the Department of Biochemistry had moved into a new building, so they needed more faculty. As Elmer explained it to me, as well as establishing a research program, teaching medical students would be important. This suited me fine because I had enjoyed my teaching experiences in Newcastle while a Ph.D. student. I interviewed in Rochester in June 1971, and on the way back to the airport, Elmer told me that I had made a good impression on the faculty and would hear from him next week. A job offer duly appeared in the mail, and we accepted with a start date of November 1. When I tell this story to younger faculty or postdoctoral fellows currently hunting for a job, their eyes tell me they do not believe a word of it. Times have changed.
Rochester is a midsize metropolitan area, notable for heavy winter snowfalls. I often say that if snow removal were an Olympic event, Rochester would win the gold more often than not. Kodak and Xerox were established there; when we arrived and for many years later, money was flowing into the city from all corners of the planet, so it was a wealthy place. The University then and now is small but punches well above its weight. It helped greatly that Glenda got a good job teaching biology to deaf students at the National Technical Institute for the Deaf, a college of the Rochester Institute of Technology.
I sent off a grant to NIH; it was approved for funding in 1972. Then President Nixon imposed a halt in disbursements. The nation was engaged in Vietnam, and the financial costs were becoming clear. Only one year later did the NIH money come through. My plan was to characterize the catalytic sites on mitochondrial F1 by chemical modification, nucleotide binding measurements, and divalent metal cofactor manipulations. To cut a long story short, the work led to publication of several solid papers, none of which opened up significant new insights. Chemical modification proved difficult when applied to the large complex F1. Measurements conducted by Ph.D. student Richard Leimgruber uncovered a complicated set of nucleotide-binding sites, some catalytic and some not. Divalent cation (Mg2+)-binding sites of both catalytic and structural nature were demonstrated, but we failed to incorporate useful metal reporter probes. It would have been frustrating except that one collaboration had flourished and promised a bright future.
Australian Collaboration
I met Frank Gibson at a Gordon Conference in 1974. He gave a talk describing his Australian group's work on E. coli membrane ATPase, the bacterial equivalent of mitochondrial F1F0. It was clear that genetic techniques provided a powerful tool with which to manipulate the enzyme. The point mutants that Frank described were unequivocally specific, which, given the frustrations that chemical modification of mitochondrial enzyme entailed, was attractive. I wrote to Frank suggesting I spend a sabbatical in his laboratory, and he agreed. The Josiah Macy Jr. Foundation awarded me a grant, and I flew to Canberra in late 1977 to start work. The E. coli system had numerous advantages. It was then and remains today the only system in which mutagenesis of F1F0 can be combined conveniently with assays of growth and oxidative phosphorylation in vivo and with assays of ATPase, ATP-driven proton pumping, and ATP synthesis in vitro. ATPase activity, which is technically much easier to characterize than ATP synthesis, is a true physiological role of F1F0 in E. coli. The mutant strains were stable; membrane preparations could be kept frozen for years; and as the Molecular Biology Revolution took hold, the possible genetic manipulations far exceeded what I or indeed anyone else had imagined. Charles Yanofsky and Allan Downie had succeeded in cloning the genes encoding E. coli F1F0 on a multicopy plasmid. Downie and Graeme Cox, a long-time colleague of Gibson, were excited about future possibilities, and their infectious enthusiasm fired me up for what was to be a productive visit to Australia. We quickly derived a method for large-scale production of pure F1 and prepared mutant enzymes with impaired catalysis. Using two-dimensional electrophoresis, we showed that the uncA gene encoded the α-subunit and confirmed that uncD encoded the β-subunit (11, 12). We devised an assay to quantitatively assess binding of soluble F1 to F0 and demonstrated that p-aminobenzamidine increased binding affinity by a mechanism unrelated to its anti-proteolysis property, making it valuable for preparing membranes enriched in F1. This work opened up the area of studies of F1 catalytic mechanism using mutant enzymes. In 1982, with a grant from the National Science Foundation, I made a second visit to Canberra. In this visit, with Lyndall (Langman) Hatch, we identified and characterized a new group of β-subunit mutants with altered catalytic properties (13). On my return, I shifted all my efforts to E. coli enzyme and continued to collaborate with Gibson and Cox for many years. In those early days, we all imagined that mutations would be defined by protein sequencing; as it turned out, DNA sequencing was the method used. Like so many facets of the Molecular Biology Revolution, this was totally unpredicted.
A Cyclical Catalytic Mechanism
By 1982, there was growing evidence that the α- and β-subunits of F1F0 existed in an alternating hexagonal arrangement with the catalytic sites probably on β, and speculative cyclical mechanisms had been proposed. In 1983, Ph.D. students John Wise and Tom Duncan showed that there were six nucleotide-binding sites on E. coli F1, three that were nonexchangeable and thus noncatalytic and three potential catalytic sites that exchanged with medium nucleotide (14). Tom went on to establish that the catalytic sites were on β by characterization of mutants that he sequenced with postdoctoral fellow Derek Parsonage (15, 16); they also formulated a structural model of the catalytic site (17). In 1987, Derek achieved the first site-directed mutagenesis of F1 (18), mutagenizing the predicted catalytic site and thereby supporting the model. Ph.D. student Rajini Rao and postdoctoral fellow Marwan Al-Shawi showed that isolated β-subunit contained a single nucleotide site (19), but, interestingly, it has no catalytic activity (20). Meanwhile, John, focusing on four “dead” α-subunit mutants, found that they interrupted α/β-subunit interaction without affecting catalytic site properties (21, 22). Penefsky's laboratory had shown that ATP hydrolysis at one catalytic site in bovine F1 was greatly accelerated upon binding of ATP at a second or third catalytic site. This acceleration was blocked in the α-subunit mutants. The conclusion was that normal catalysis involved signal transmission from one β-subunit to another through an intervening α-subunit, strongly implying a three-site cyclical catalytic mechanism. In 1987, Rajini used an ingenious approach to prove this by generating mixtures of hybrid enzymes containing zero to three α-subunit mutants with the corresponding complement of wild-type α (23). Sequencing of the α-subunit mutants by M.S. student Mary Beth Maggio and postdoctoral fellow Janet Pagan showed that the four dead α-subunit mutants clustered between α-subunit residues 345 and 375, which we designated as “α/β signal transmission region” (reviewed in Refs. 24 and 25). Later, after John Walker, Andrew Leslie, and colleagues published their enormously valuable x-ray structures of bovine F1, it transpired that the α/β signal transmission region lines the α/β-subunit interface proximate to the catalytic site (see Fig. 9 of Ref. 26), which fit nicely with our conclusions. In 1997, Noji and colleagues published their famous experiment visually demonstrating rotation of the γ-subunit within the α/β-subunit hexagon, leaving no doubt whatsoever that the catalytic mechanism is cyclical.
Optical Probes
Postdoctoral fellow Joachim (Achim) Weber came to the laboratory in 1991. He was familiar with the fluorescent nucleotides lin-benzo-ATP and lin-benzo-ADP and, with postdoctoral fellow Rita Lee, was able to apply them profitably to E. coli F1, showing that β-Tyr-331 lay adjacent to the adenine ring of bound substrate MgATP (27). They put a Trp at that position (28), and it yielded a strong fluorescent signal, totally quenched by bound MgATP, MgADP, or other nucleotides and of equal strength at each of the three catalytic sites. Thus, we had a real-time, quantitative, and specific probe of catalytic site occupancy under true equilibrium conditions. Subsequently, with x-ray structures as guides, Achim put Trp probes in multiple locations, allowing us to monitor a wide variety of properties. The ability to generate Trp-free F1 that mirrored the wild type in properties was helpful (Ref. 29). This work was summarized in Refs. 30 and 31. Fig. 1 shows Trp probes that were developed for studies of the catalytic sites (32).
FIGURE 1.

Trp probes in the F1 catalytic site. Residues located around MgAMPPNP (an analog of MgATP; shown in green) in the catalytic site of F1 were substituted with Trp. Residues indicated in yellow gave the same fluorescence response to MgADP and MgATP, valuable for measuring binding affinity and occupancy. Residues indicated in red responded differently to MgADP versus MgATP. β-Trp-297 and α-Trp-291 responded specifically to site one (highest affinity), yielding proof that this site persists in steady-state hydrolysis. The residue shown in gray did not respond to nucleotide. This figure was reprinted from Ref. 32 with permission.
Molecular Mechanism of Catalysis
Using the β-Trp-331 probe, we found that the F1 catalytic sites showed strongly heterogeneous behavior. The three Kd values for MgATP were 1 nm, 1 μm, and 100 μm, respectively (26, 28, 33). Kd3 corresponded closely to Km(ATP), so it was clear that binding to the third site is required to attain the Vmax for ATP hydrolysis. With MgATP bound at only two sites, hydrolysis rates are negligible and certainly nonphysiological. Like ATPase activity, binding heterogeneity was dependent on Mg2+. In its absence, all three sites showed a Kd of ∼100 μm. These data provide the strongest evidence available that the three F1 catalytic sites exist in different conformations and that conformational heterogeneity is required for activity. Comparing MgADP versus MgATP, the Kd at site 1 was not of such high affinity, but Kd values at sites 2 and 3 were similar. Postdoctoral fellow Sabine Löbau extended these findings to intact F1F0 containing β-Trp-331, obtaining the same results (34).
The introduction of the β-Trp-148 probe in 1996 (26, 35) enabled us to confidently formulate a detailed cyclical MgATP hydrolysis mechanism for the first time. Located close to the catalytic site (Fig. 1), this probe responds differently to MgADP versus MgAMPPNP (5′-adenylyl imidodiphosphate) or MgADP-BeFx (two analogs of MgATP). It indicated that, during steady-state hydrolysis, the enzyme contains, in time average, one MgATP and two MgADP molecules in catalytic sites. Putting all the data together, it was evident that, in a first step, MgATP binds to an empty site; it then lingers on the enzyme until a second MgATP is bound, at which time it is hydrolyzed; and then the resultant product MgADP lingers until a third MgATP is bound, after which it is released. The mechanism is shown in Fig. 2 (36). Subsequent single-molecule experiments in the laboratories of M. Yoshida/K. Kinosita and H. Noji have visually monitored MgATP binding and product release. There have been some tweaks, e.g. their timing of product MgADP and Pi release differs in detail from Fig. 2, but the main framework is confirmed.
FIGURE 2.

Mechanism of ATP hydrolysis by F1. From Ref. 36. In 2004, we combined our original mechanism (26, 35) with rotational information from single-molecule experiments of M. Yoshida and K. Kinosita. Catalytic sites (circles) are designated O for open and H, M, or L for high, medium, or low affinity for MgATP. In a series of enzyme states (ABCDA), one MgATP is consumed, and the γ-subunit rotates 120°. Panel a, binding of red ATP generates rotation of γ, hydrolysis of already-bound green ATP, and a switch in site conformations (“binding change”). Already-bound blue ADP is then released. Note that red ATP binding, green ATP hydrolysis, and blue ADP release occur at three different catalytic sites. Panel b, a new (black) ATP binds and brings about the same set of events, with this time prebound red ATP being hydrolyzed and green ADP being released. Panel c, finally, in a third series, the red ADP product is released.
Postdoctoral fellow Marwan Al-Shawi had compared the thermodynamic parameters of E. coli and mitochondrial F1 catalysis, concluding that the latter was a more evolved enzyme (37), and had extended these studies to a group of mutants, thereby making a start on attribution of specific roles to individual residues (38). Next, applying optical probes in combination with directed mutagenesis, we were able to designate the roles of residues responsible for binding MgATP and MgADP. For example, we showed that the Walker A Lys residue has its major interaction with the γ-phosphate of MgATP, that the Walker B Asp residue interacts with Mg2+ in MgATP, and that the critical β-Glu-181 binds neither nucleotide nor Mg2+; rather, its role is in reaction chemistry per se (39). X-ray structures of bovine F1 were invaluable in suggesting candidate residues, but of course, one cannot deduce quantitative binding parameters from structures. In the case of β-Glu-181, x-ray structures do indicate that it acts as a catalytic base because it is coordinated to a suitable attacking water molecule. Fig. 3 shows our deduced octahedral coordination of Mg2+ in substrate MgATP (40). Residues from both Walker A and B motifs and elsewhere in the β-subunit engage the Mg2+, in some cases through intervening waters. Removal of any one ligand abolishes Mg2+ binding, i.e. it is highly cooperative. Fig. 4 shows our proposed chemical transition state structure for ATP hydrolysis, deduced by determination of quantitative binding parameters for substrate MgATP, product MgADP, and transition state analogs MgADP-fluoroaluminate and MgADP-fluoroscandium combined with mutagenesis of candidate side chains by postdoctoral fellow Sashi Nadanaciva (41, 42). One residue of particular interest is α-Arg-376, which we named the “arginine finger” by analogy with G-proteins (41). It plays no role in binding MgATP or MgADP but is crucial for formation of the transition state. Thus, its role is to accelerate catalysis by inserting into the catalytic site to generate the transition state. It lies at the end of the α/β signal transmission region, which we had previously identified in the α-subunit (see above), providing a nice explanation of the earlier results. The need for the arginine finger coming from the α-subunit also explained why the isolated β-subunit has no activity. We summarized five factors responsible for catalytic rate acceleration in ATP hydrolysis in Ref. 43.
FIGURE 3.

Mg2+ coordination in the catalytic site of F1. As deduced in Ref. 40. Green sphere, Mg2+; cyan spheres, water molecules in the first coordination shell. Residues involved in Mg2+ coordination are β-Thr-156, β-Glu-185, and β-Asp-242. The catalytic carboxylate β-Glu-181 is not involved, although it is in close proximity. This figure was reprinted from Ref. 45 with permission.
FIGURE 4.

The F1 catalytic transition state. As deduced in Ref. 41. Ligands to the pentacovalent phosphorus are shown, with the reactant water molecule in cyan, the β-subunit in gray, and the α-subunit in green. α-Arg-376 is the arginine finger. This figure was reprinted from Ref. 45 with permission.
Studying the molecular mechanism of ATP synthesis proved a greater challenge. We were not able to obtain Trp-free F1F0, and the requirement for a lipid bilayer added high optical background. In several reviews (44–46), we discussed the difficulties and proposed possible mechanisms. It is clear that ATP synthesis cannot be the exact reverse of ATP hydrolysis. The synthesis of ATP is one of the most important reactions in biology, yet we know little about it in molecular terms. Hopefully, the ideas in those reviews will prove helpful for future workers.
Our conclusion that significant ATPase activity occurs only when all three catalytic sites become occupied by MgATP generated a steady stream of criticism from one influential quarter. It had previously been assumed that a two-site (“bisite”) mechanism pertained, as formulated in an oft-cited article from 1981. The 1997 chemistry Nobel Prize announcement pictured this mechanism as if it were an established fact. We were therefore at pains to justify our experimental protocols. Then one morning, Achim Weber walked into my office and said, “All these extra controls are unnecessary; the bisite mechanism has a fundamental flaw.” And, indeed it did. In ATP hydrolysis, at the step of substrate binding, MgATP had the choice of binding to either of two empty catalytic sites, one of lower affinity than the other, and at every turnover, it was required to bind only to the site of lower affinity. Yet this formulation had survived for nearly twenty years! Further analysis revealed additional flaws (47), so that particular discussion had a surprise ending.
Noncatalytic Sites
As mentioned above, E. coli F1 has three nonexchangeable, noncatalytic nucleotide sites. They lie on α-subunits, and apart from confusing researchers, they appeared to have no function. To probe possible functions, we placed a Trp probe at α-Arg-365, equivalent to β-Tyr-331 in sequence. It gave a strong fluorescent signal in an empty site that was totally quenched upon binding of nucleotide. This allowed real-time specific monitoring of occupancy of noncatalytic sites, and we found that, in wild-type enzyme devoid of noncatalytic site nucleotide (48) or in mutants unable to bind nucleotide in these sites (49), both ATP hydrolysis and synthesis proceeded normally. A variety of nucleotides, PPi, and PPPi, were shown to bind to the sites, and quantitative binding parameters could be obtained. For each ligand, all three sites bound with equal affinity in strongly Mg2+-dependent fashion. The nonexchangeable nature of the sites toward adenine nucleotide is because of a conformational change after binding that sequesters the nucleotide, which is not seen with guanine or inosine nucleotide (26). However, we could detect no regulatory function, so the noncatalytic sites remain a puzzle.
The Phosphate-binding Pocket
Our earlier work with E. coli F1 indicated that H2PO4− is the species that binds to the catalytic sites in ATP synthesis. Binding is normally weak but is strengthened by a proton gradient (50, 51). This was corroborated by single-molecule experiments showing that Pi binding is linked to a substep of subunit rotation (reviewed in Ref. 52). Expedited capture of Pi into an empty catalytic site is a key requirement in ATP synthesis because Kd(MgADP) and Kd(MgATP) at the lowest affinity catalytic site are essentially the same (∼100 μm). The MgATP/MgADP concentration ratio in cells is usually 10:1 or more, so MgADP binding would be infrequent. We proposed that binding of Pi into a pocket within the catalytic site sterically prevents access to MgATP but not MgADP, so we were interested in characterizing the Pi-binding pocket.
A 2001 x-ray structure from John Walker and colleagues enabled detailed studies. In one catalytic site, it contained a sulfate ion, possibly mimicking phosphate, guiding the choice of residues for mutagenesis. Using an assay (developed by Stuart Ferguson's laboratory) that fortuitously measures binding only in the empty site to which Pi normally binds for ATP synthesis, postdoctoral fellow Zulfiqar Ahmad defined a triangular Pi-binding pocket at one end of the catalytic site, involving four positively charged residues plus Ser-OH (53–55). Modulation of positive charge density within this pocket by deletion or introduction of Arg residues markedly affected Pi binding (56). Not all candidate residues were “hits”; for example, from the x-ray structure, β-Asn-243 seemed a likely Pi-binding residue, but mutagenesis showed that it was not involved (57).
The Peripheral or Stator Stalk
Stanley Dunn had shown in the early 1980s using proteolysis that binding of F1 to the membrane F0 required the δ-subunit, which attached to the N-terminal region of the α-subunit. In agreement, in our laboratory, Ph.D. student Andrea Hazard and M.S. student Mary Beth Maggio found mutants in δ and α that were functionally deleterious, consistent with impaired F1 binding to F0 (58, 59). In addition, using proteolysis and antibody approaches, postdoctoral fellow David Perlin showed that subunit b was required for F1 to bind to F0 (60). Up until 1994, the only connection (“stalk”) that had been seen by electron microscopy was one joining the base of F1 to F0, so that is where we assumed δ and b to be located. However, the 1994 x-ray structure demolished this idea by showing that this stalk (the “rotor” stalk) contained only γ- and ϵ-subunits. Therefore, δ and b had to be on the outside. Using electron microscopy, Roderick Capaldi's laboratory found them on the periphery, in the “stator” stalk, so named because it serves to prevent co-rotation of the α/β-subunit hexagon with γ/ϵ. The α-subunit N-terminal region carrying the binding site for δ is, counterintuitively, at the very top of the molecule, most distant from the membrane (Fig. 5). Beginning in 2002, Achim Weber applied optical probes to determine quantitative binding parameters in the stator stalk.
FIGURE 5.

E. coli F1F0. This structure (based on Ref. 87) is a composite from x-ray structures and molecular modeling. The stator stalk consists of subunits b2 and δ, interacting with subunits a and α, respectively.
Stephan Wilkens' NMR structure of δ allowed informed insertion of Trp residues, providing two good probes of binding, but the best probe was the naturally occurring δ-Trp-28. We found that, in absence of subunit b, δ bound to F1 with a Kd of ∼1 nm; in presence of subunit b, this value decreased to ∼3 pm (61, 62). Clearly, the stator stalk is “overengineered”: it is more than capable of resisting the rotor strain. The stoichiometry of δ/F1 binding never exceeded 1:1, which was mysterious because there are three α-subunits in F1. Mg2+ ions strongly promote binding, although their location is not yet known. Mutagenesis of conserved residues in δ enabled us to formulate a likely hydrophobic binding site on δ for the α-subunit N terminus, located between two parallel helices, and to estimate contributions to the binding energy of individual δ-subunit residues (62). Next, we took a synthetic 22-mer peptide corresponding to α-subunit residues 1–22 and found that it bound to δ with a Kd of ∼100 nm and 1:1 stoichiometry (63). By inserting truncations, deletions, and “mutations” in the peptide, we established that it is helical, with a hydrophobic surface that binds to δ, and we identified key hydrophobic residues responsible for binding (64). This enabled us to formulate a model in which the helical peptide nestles between the two helices on δ. Finally, in collaboration with Wilkens, we obtained an NMR structure of the α-subunit peptide bound to δ, shown in Fig. 6 (65). We also showed that subunit b itself binds directly to F1 with a Kd of ∼100 nm (66). Its binding site(s) has not yet been determined.
FIGURE 6.

Binding of the δ-subunit to the N terminus of the α-subunit. Shown is the binding of a 22-residue peptide corresponding to the N-terminal region of the α-subunit to the δ-subunit as deduced by NMR (65). The helical peptide (gray) nestles between two helices (blue and orange) on δ. The naturally occurring δ-Trp-28 provided an excellent optical probe.
How and why the stoichiometry of binding of δ to F1 is controlled at 1/1 was solved in Ref. 67. We found that δ would not bind to isolated α-subunit; rather, it would bind only to α that was pre-incorporated into F1. The N-terminal region of α that binds δ was susceptible to proteolysis in F1 but not in isolated α-subunit, showing that it is sequestered until α is incorporated into F1, when it becomes available to δ. The N termini of the three α-subunits lie in close proximity. Presumably, binding of one δ-subunit to one α-subunit N-terminal region in F1 sterically prevents binding of additional δ-subunits to either of the other two α-subunits. Conversely, if isolated δ- and α-subunits could freely form α-δ complexes, this would prevent assembly of the α/β-subunit hexagon.
P-glycoprotein, a Second ATPase
I had been aware of P-glycoprotein (Pgp) for some years before the human and mouse genes were sequenced in the late 1980s. Pgp had been recognized by Victor Ling and colleagues as a plasma membrane protein that endowed multidrug resistance upon cells, notably to anticancer agents. The “P” in the name stood for “permeability” because Ling originally thought that membrane permeability to drugs was reduced. However, after the sequences came out, it seemed that the “P” might more realistically stand for “pump” because the protein contained two sets of Walker A and B motifs, denoting two nucleotide-binding domains (NBDs) that clearly might have ATPase function to drive drug export. If so, this would be a two-cylinder ATPase, in contrast to the three-cylinder F1F0, different enough to make it interesting but similar enough that my previous experience could be expeditious. As it turned out, Pgp is a member of the ATP-binding cassette (ABC) transporter superfamily, members of which have common sequence motifs located in two NBDs, so the work we were about to embark on became of wide interest. In 1990, I submitted a grant application to the American Cancer Society, and the grant funded me for three years. I had no preliminary data on Pgp, but I think I can say with pride that we justified the investment.
Getting Enough Protein
I realized that, just as with F1 twenty years earlier, we would need a good source of protein and an efficacious purification scheme. Ling sent us a Chinese hamster ovary cell line that was enriched for Pgp, and by successive rounds of selection in colchicine, Marwan Al-Shawi was able to bump up the content of Pgp in plasma membranes to ∼20% (68). Postdoctoral fellow Ina Urbatsch purified fully active protein by detergent solubilization, column chromatography, and reconstitution into proteoliposomes (69). One nice outcome was that pure reconstituted Pgp behaved very much the same as Pgp in plasma membranes, so for many experiments, we could use the latter. These materials sufficed for early characterization, but one could not make mutants, and later, when structural biochemists with insatiable appetites became involved, enhanced production became imperative. Philippe Gros and colleagues pioneered application of the Pichia pastoris expression system to Pgp and introduced a purification procedure using the now ubiquitous His tag method. We were able to greatly improve purification of wild-type and mutant mouse and human Pgp from Pichia (70), and this material has since supported biochemical and structural studies.
A Different Kind of ATPase
We found quickly that our Pgp had easily measurable, drug-stimulated ATPase and that there was no phosphorylated intermediate and no high-affinity catalytic site, distinguishing it from P-type ATPases, F1F0-ATPases, and myosin. There was just one Km(MgATP) (approximately millimolar) and low specificity for nucleotide. Several inhibitors were identified, notably N-ethylmaleimide and 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole, which react covalently in the Walker A motif and are ATP-protectable, and it was interesting that the latter gave total inhibition of ATPase at a stoichiometry of 1:1 (71). It appeared that there was either only one active NBD or very strong cooperativity between two equivalent NBDs, the latter being consistent with early mutation studies in the Gros laboratory. We also found that the lipid environment affected both ATPase turnover and drug stimulation profiles (72).
One inhibitor was vanadate (Vi). Postdoctoral fellows Ina Urbatsch and Banu Sankaran found that, as in myosin, it inhibited Pgp by forming an MgADP-Vi complex, which, mimicking the ATP hydrolysis transition state, became tenaciously trapped in the catalytic site. Interestingly, trapping of one MgADP-Vi complex per Pgp gave complete inhibition of ATPase, yet the MgADP-Vi complex was distributed evenly between the two NBDs, showing that both were catalytically active and equivalent but not capable of acting independently (73, 74). Thus, there was strong cooperativity, a pattern that we were later to see repeated with catalytic site mutants and with MgADP-BeFx, a tight-binding analog of MgATP. It seemed that when one NBD entered the transition state, the other NBD was prohibited from doing so.
Alternating Sites Mechanism
The above work led to our proposal of the alternating sites mechanism (75), shown in Fig. 7. This was the first catalytic mechanism proposed for Pgp (and indeed for any ABC transporter), so it was an important step. It provided a framework for later experiments and retains strong support as the likely mechanism in Pgp. As the study of ABC transporters has progressed, it has become evident that there are nuances that differentiate them; thus, it appears that no universal mechanism will apply to the superfamily, and new proposals have emerged, often based on similar experimentation to ours.
FIGURE 7.
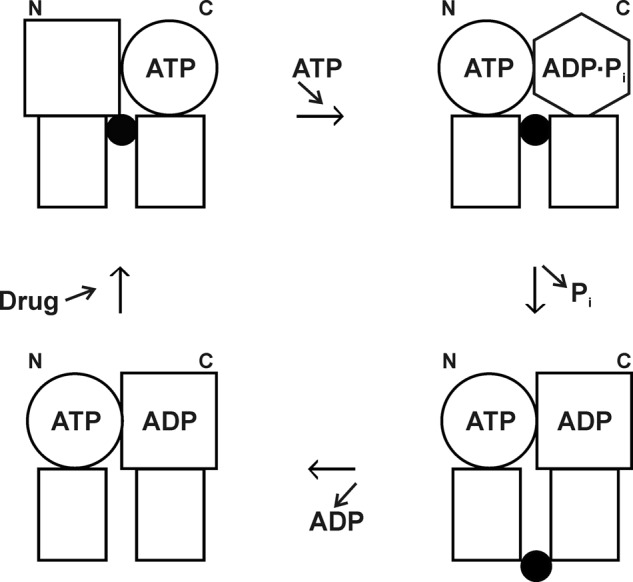
P-glycoprotein alternating sites mechanism. Rectangles, transmembrane domains; circles, squares, and hexagon, different conformations of the N- and C-terminal catalytic sites (NBDs); black circles, drug molecule. Binding of ATP to an empty N-terminal site (upper left) brings about hydrolysis of ATP in the C-terminal site (upper right). Relaxation of the C-terminal site drives drug from inward-facing higher affinity to outside-facing lower affinity (lower right) as Pi is released. Drug and ADP dissociate (lower left), and in the next cycle, it will be the C-terminal site that binds ATP and the N-terminal site that hydrolyzes it. This figure was reprinted from Ref. 75 with permission.
Postdoctoral fellow Greg Tombline looked into events occurring at a catalytic site as MgATP binds and progresses through the catalytic cycle. Each NBD contains a conserved Glu residue, recently proved to act as a catalytic base in hydrolysis (reviewed in Ref. 76). Evidence in the literature suggested that, upon mutagenesis of these residues, catalysis was halted and substrate became bound tightly. Tombline mutated both Glu residues and confirmed that MgATP did become “occluded” but, importantly, in only one NBD (77). Following up with a series of experiments using mutants, drugs, and inhibitors, he was able to show that the “occluded nucleotide” lies on the pathway of hydrolysis and is the nucleotide that is committed to hydrolysis (78), as shown in Fig. 8. The data fit well with structural data (in other ABC transporters) showing an NBD dimer sandwich, and with the alternating sites mechanism (79).
FIGURE 8.

The occluded nucleotide conformation in the P-glycoprotein catalytic pathway. From Ref. 78. Each half-circle represents an NBD. The occluded nucleotide conformation occurs as a closed NBD dimer at step III, with the ATP that is tightly bound and committed to hydrolysis shown in boldface. Steps inhibited by mutations and covalent reagents or accelerated by drug are shown. NBD-Cl, 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole; NEM, N-ethylmaleimide.
Structural Characterization
As work progressed, we were able to draw several inferences regarding the structure of the NBDs. For example, Banu Sankaran showed that the widely used photolabel 8-azido-ATP reacted with a specific conserved Tyr residue in each NBD, which very likely stacks against the adenine ring when MgATP binds (80). Mutagenesis of conserved Gln residues in the Q-loop motif by Ina Urbatsch indicated that the Q-loop was responsible for communication between membrane-located drug sites and NBDs (81). Other mutagenesis studies and further teasing of the MgADP-Vi-inhibited state suggested that residues from one NBD interdigitated with residues from the other NBD (82, 83). All of these inferences were consistent with structural analyses in various ABC transporters, but clearly structures of Pgp itself were needed.
For structural analysis of Pgp, we generated mutants that abolished N-glycosylation and removed all Cys residues. This has now begun to show benefits. In a collaboration with Eric Lee and Stephan Wilkens, we published low-resolution electron microscopy structures of both nucleotide-free and nucleotide-bound Pgp embedded in lipid bilayer (84, 85). The first x-ray structure of Pgp recently appeared (86), and it was gratifying that our mutants and purification protocols contributed significantly. Watching this field develop is going to be a pleasure.
Teaching
From day one in Rochester, I was involved in medical and graduate student teaching in metabolism and bioenergetics. Teaching provided a change of pace from research and often enhanced it. The publication in 1970 of Biochemistry by Albert Lehninger made life easier for all of us. Until that point, there was no satisfactory textbook. Lehninger's book started where organic chemistry textbooks ended and finished where physiological chemistry textbooks started. It described the burgeoning body of biochemical information in between in lucid prose with great diagrams. In a very real sense, Lehninger's textbook defined for the first time the boundaries of the modern discipline, marking it as a truly singular accomplishment.
Breakthrough experiments, so incisive that they speak for themselves, provide strong teaching tools. One, of course, was the demonstration of rotation of subunits in F1, mentioned above. I would impress on students that, as they sat in lecture, billions of F1 nanomotors were spinning around in their mitochondria at 100 revolutions/s, just as in the video. Two other examples come to mind. Peter Mitchell's chemiosmotic mechanism of oxidative phosphorylation initially met resistance, but two standout experiments came along to vindicate him. One was the “pH jump” experiment of André Jagendorf and Ivan Ryrie, beautiful in its simplicity. The other was the experiment of Efraim Racker and Walther Stoeckenius in which they co-reconstituted the light-driven proton pump bacteriorhodopsin from Halobacterium with bovine heart ATP synthase, and lo and behold, when they illuminated the proteoliposomes, ATP was synthesized. Gratifyingly, former students now well into their careers will regularly tell me how much they have appreciated my lectures. Invariably, toward the end of the conversation, they lean forward with a smile and tell me they remember one very important fact: “It's dark inside a cow.”
Acknowledgments
Biochemistry is a team exercise. While the principal investigator is away at conferences and study sections or teaching lectures, it is critical that the folks back in the laboratory keep things moving. I would like to thank each and every person who worked with me for their contribution to keeping things moving. Remembering my experience in Iowa City, I made it a point to provide each year a paid summer research experience to one or two undergraduates, for a total of 50 students. They repaid us in spades with their energy and vitality, and I thank them all. Throughout my career, I was fortunate to work with a series of superior technicians, namely Graeme Chambers (Newcastle); Sandra Winder and Pauline Cuff (Madison); Barry and Diane Webb (Canberra); and Christine Broadhurst Whitman, Lisa Richardson Latchney, Anne Ferguson, David Cox, Cheryl Bowman, Sean Hammond, Sumedha Bhagat, Khursheed Gimi, Lori Bartholomew White, and Alma Muharemagić (Rochester). Special thanks go to Susan Wilke-Mounts, my laboratory manager for twenty years. I noted the contributions of my five Ph.D. students in the text; to these should be added Steven Martin, M.D./Ph.D. student, who, together with postdoctoral fellow John Hamlyn found and characterized a membrane ecto-ATPase in the pancreas. Lack of resources prevented follow-up on that topic. Russell Hilf has been a valued mentor, colleague, and friend in Rochester since 1971. We collaborated and published together on mammary gland mitochondria and prolactin receptors. Thank you, Russ. Finally, I must surely acknowledge my strongest supporter, for forty-seven years, Glenda Senior; and pay the utmost respect to Eric and Kath Senior, who, in the dark days of 1941, had the courage and optimism to marry and plan a family.
REFERENCES
- 1. Sherratt H. S. A. (1986) Hypoglycin, the famous toxin of the unripe Jamaican ackee fruit. Trends Pharmacol. Sci. 7, 186–191 [Google Scholar]
- 2. Schulz H., Fong J. C. (1981) 4-Pentenoic acid. Methods Enzymol. 72, 604–610 [DOI] [PubMed] [Google Scholar]
- 3. Senior A. E., MacLennan D. H. (1970) Mitochondrial “structural protein.” A reassessment. J. Biol. Chem. 245, 5086–5095 [PubMed] [Google Scholar]
- 4. Senior A. E., Brooks J. C. (1970) Studies on the mitochondrial oligomycin-insensitive ATPase. I. An improved method of purification and the behavior of the enzyme in solutions of various depolymerizing agents. Arch. Biochem. Biophys. 140, 257–266 [DOI] [PubMed] [Google Scholar]
- 5. Senior A. E., Brooks J. C. (1971) The subunit composition of the mitochondrial oligomycin-insensitive ATPase. FEBS Lett. 17, 327–329 [DOI] [PubMed] [Google Scholar]
- 6. Brooks J. C., Senior A. E. (1971) Studies on the mitochondrial oligomycin-insensitive ATPase. II. The relationship of the specific protein inhibitor to the ATPase. Arch. Biochem. Biophys. 147, 467–470 [DOI] [PubMed] [Google Scholar]
- 7. Brooks J. C., Senior A. E. (1972) Methods for purification of each subunit of the mitochondrial oligomycin-insensitive adenosine triphosphatase. Biochemistry 11, 4675–4678 [DOI] [PubMed] [Google Scholar]
- 8. Senior A. E. (1971) On the relationship between the oligomycin-sensitivity conferring protein and other mitochondrial coupling factors. J. Bioenerg. 2, 141–150 [DOI] [PubMed] [Google Scholar]
- 9. Senior A. E. (1973) The structure of mitochondrial ATPase. Biochim. Biophys. Acta 301, 249–277 [DOI] [PubMed] [Google Scholar]
- 10. Shapiro A. L., Viñuela E., Maizel J. V. (1967) Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacrylamide gels. Biochem. Biophys. Res. Commun. 28, 815–820 [DOI] [PubMed] [Google Scholar]
- 11. Senior A. E., Downie J. A., Cox G. B., Gibson F., Langman L., Fayle D. R. (1979) The uncA gene codes for the α-subunit of the adenosine triphosphatase of Escherichia coli. Electrophoretic analysis of uncA mutant strains. Biochem. J. 180, 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Senior A. E., Fayle D. R., Downie J. A., Gibson F., Cox G. B. (1979) Properties of membranes from mutant strains of Escherichia coli in which the β-subunit of the adenosine triphosphatase is abnormal. Biochem. J. 180, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Senior A. E., Langman L., Cox G. B., Gibson F. (1983) Oxidative phosphorylation in Escherichia coli. Characterization of mutant strains in which F1-ATPase contains abnormal β-subunits. Biochem. J. 210, 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wise J. G., Duncan T. M., Latchney L. R., Cox D. N., Senior A. E. (1983) Properties of F1-ATPase from the uncD412 mutant of Escherichia coli. Biochem. J. 215, 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duncan T. M., Senior A. E. (1985) The defective proton-ATPase of uncD mutants of Escherichia coli. Two mutations which affect the catalytic mechanism. J. Biol. Chem. 260, 4901–4907 [PubMed] [Google Scholar]
- 16. Parsonage D., Duncan T. M., Wilke-Mounts S., Kironde F. A., Hatch L., Senior A. E. (1987) The defective proton-ATPase of uncD mutants of Escherichia coli. Identification by DNA sequencing of residues in the β-subunit which are essential for catalysis or normal assembly. J. Biol. Chem. 262, 6301–6307 [PubMed] [Google Scholar]
- 17. Duncan T. M., Parsonage D., Senior A. E. (1986) Structure of the nucleotide-binding domain in the β-subunit of Escherichia coli F1-ATPase. FEBS Lett. 208, 1–6 [DOI] [PubMed] [Google Scholar]
- 18. Parsonage D., Wilke-Mounts S., Senior A. E. (1987) Directed mutagenesis of the β-subunit of F1-ATPase from Escherichia coli. J. Biol. Chem. 262, 8022–8026 [PubMed] [Google Scholar]
- 19. Rao R., Al-Shawi M. K., Senior A. E. (1988) Trinitrophenyl-ATP and -ADP bind to a single nucleotide site on isolated β-subunit of Escherichia coli F1-ATPase. In vitro assembly of F1-subunits requires occupancy of the nucleotide-binding site on β-subunit by nucleoside triphosphate. J. Biol. Chem. 263, 5569–5573 [PubMed] [Google Scholar]
- 20. Al-Shawi M. K., Parsonage D., Senior A. E. (1990) Adenosine triphosphatase and nucleotide binding activity of isolated β-subunit preparations from Escherichia coli F1F0-ATP synthase. J. Biol. Chem. 265, 5595–5601 [PubMed] [Google Scholar]
- 21. Wise J. G., Latchney L. R., Senior A. E. (1981) The defective proton-ATPase of uncA mutants of Escherichia coli. Studies of nucleotide-binding sites, bound aurovertin fluorescence, and labeling of essential residues of the purified F1-ATPase. J. Biol. Chem. 256, 10383–10389 [PubMed] [Google Scholar]
- 22. Wise J. G., Latchney L. R., Ferguson A. M., Senior A. E. (1984) Defective proton ATPase of uncA mutants of Escherichia coli. 5′-Adenylyl imidodiphosphate binding and ATP hydrolysis. Biochemistry 23, 1426–1432 [DOI] [PubMed] [Google Scholar]
- 23. Rao R., Senior A. E. (1987) The properties of hybrid F1-ATPase enzymes suggest that a cyclical catalytic mechanism involving three catalytic sites occurs. J. Biol. Chem. 262, 17450–17454 [PubMed] [Google Scholar]
- 24. Senior A. E. (1985) The proton-ATPase of Escherichia coli. Curr. Top. Membr. Transp. 23, 135–151 [Google Scholar]
- 25. Senior A. E. (1990) The proton-translocating ATPase of Escherichia coli. Annu. Rev. Biophysics. Biophys. Chem. 19, 7–41 [DOI] [PubMed] [Google Scholar]
- 26. Weber J., Senior A. E. (1997) Catalytic mechanism of F1-ATPase. Biochim. Biophys. Acta 1319, 19–58 [DOI] [PubMed] [Google Scholar]
- 27. Weber J., Lee R. S., Grell E., Wise J. G., Senior A. E. (1992) On the location and function of tyrosine β331 in the catalytic site of Escherichia coli F1-ATPase. J. Biol. Chem. 267, 1712–1718 [PubMed] [Google Scholar]
- 28. Weber J., Wilke-Mounts S., Lee R. S., Grell E., Senior A. E. (1993) Specific placement of tryptophan in the catalytic sites of Escherichia coli F1-ATPase provides a direct probe of nucleotide binding. Maximal ATP hydrolysis occurs with three sites occupied. J. Biol. Chem. 268, 20126–20133 [PubMed] [Google Scholar]
- 29. Wilke-Mounts S., Weber J., Grell E., Senior A. E. (1994) Tryptophan-free Escherichia coli F1-ATPase. Arch. Biochem. Biophys. 309, 363–368 [DOI] [PubMed] [Google Scholar]
- 30. Weber J., Senior A. E. (1996) F1F0-ATP synthase: development of direct optical probes of the catalytic mechanism. Biochim. Biophys. Acta 1275, 101–104 [DOI] [PubMed] [Google Scholar]
- 31. Weber J., Senior A. E. (2004) Fluorescent probes applied to catalytic cooperativity in ATP synthase. Methods Enzymol. 380, 132–152 [DOI] [PubMed] [Google Scholar]
- 32. Weber J., Wilke-Mounts S., Hammond S. T., Senior A. E. (1998) Tryptophan substitutions surrounding the nucleotide in catalytic sites of F1-ATPase. Biochemistry 37, 12042–12050 [DOI] [PubMed] [Google Scholar]
- 33. Weber J., Wilke-Mounts S., Senior A. E. (1994) Cooperativity and stoichiometry of substrate binding to the catalytic sites of Escherichia coli F1-ATPase. Effects of magnesium, inhibitors, and mutation. J. Biol. Chem. 269, 20462–20467 [PubMed] [Google Scholar]
- 34. Löbau S., Weber J., Senior A. E. (1998) Catalytic site nucleotide binding and hydrolysis in F1F0-ATP synthase. Biochemistry 37, 10846–10853 [DOI] [PubMed] [Google Scholar]
- 35. Weber J., Bowman C., Senior A. E. (1996) Specific tryptophan substitution in catalytic sites of Escherichia coli F1-ATPase allows differentiation between bound substrate ATP and product ADP in steady-state catalysis. J. Biol. Chem. 271, 18711–18718 [DOI] [PubMed] [Google Scholar]
- 36. Senior A. E., Weber J. (2004) Happy motoring with ATP synthase. Nat. Struct. Mol. Biol. 11, 110–112 [DOI] [PubMed] [Google Scholar]
- 37. Al-Shawi M. K., Senior A. E. (1988) Complete kinetic and thermodynamic characterization of the unisite catalytic pathway of Escherichia coli F1-ATPase. Comparison with mitochondrial F1-ATPase and application to the study of mutant enzymes. J. Biol. Chem. 263, 19640–19648 [PubMed] [Google Scholar]
- 38. Senior A. E., Al-Shawi M. K. (1992) Further examination of seventeen mutations in Escherichia coli F1-ATPase β-subunit. J. Biol. Chem. 267, 21471–21478 [PubMed] [Google Scholar]
- 39. Löbau S., Weber J., Wilke-Mounts S., Senior A. E. (1997) F1-ATPase, roles of three catalytic site residues. J. Biol. Chem. 272, 3648–3656 [DOI] [PubMed] [Google Scholar]
- 40. Weber J., Hammond S. T., Wilke-Mounts S., Senior A. E. (1998) Mg2+ coordination in catalytic sites of F1-ATPase. Biochemistry 37, 608–614 [DOI] [PubMed] [Google Scholar]
- 41. Nadanaciva S., Weber J., Wilke-Mounts S., Senior A. E. (1999) Importance of F1-ATPase residue α-Arg-376 for catalytic transition state stabilization. Biochemistry 38, 15493–15499 [DOI] [PubMed] [Google Scholar]
- 42. Senior A. E., Weber J., Nadanaciva S. (2000) The catalytic transition state in ATP synthase. J. Bioenerg. Biomembr. 32, 523–529 [DOI] [PubMed] [Google Scholar]
- 43. Senior A. E., Nadanaciva S., Weber J. (2000) Rate acceleration of ATP hydrolysis by F1F0-ATP synthase. J. Exp. Biol. 203, 35–40 [DOI] [PubMed] [Google Scholar]
- 44. Weber J., Senior A. E. (2000) ATP synthase: what we know about ATP hydrolysis and what we do not know about ATP synthesis. Biochim. Biophys. Acta 1458, 300–309 [DOI] [PubMed] [Google Scholar]
- 45. Senior A. E., Nadanaciva S., Weber J. (2002) The molecular mechanism of ATP synthesis by F1F0-ATP synthase. Biochim. Biophys. Acta 1553, 188–211 [DOI] [PubMed] [Google Scholar]
- 46. Weber J., Senior A. E. (2003) ATP synthesis driven by proton transport in F1F0-ATP synthase. FEBS Lett. 545, 61–70 [DOI] [PubMed] [Google Scholar]
- 47. Weber J., Senior A. E. (2001) Bisite catalysis in F1-ATPase: does it exist? J. Biol. Chem. 276, 35422–35428 [DOI] [PubMed] [Google Scholar]
- 48. Weber J., Wilke-Mounts S., Grell E., Senior A. E. (1994) Tryptophan fluorescence provides a direct probe of nucleotide binding in the noncatalytic sites of Escherichia coli F1-ATPase. J. Biol. Chem. 269, 11261–11268 [PubMed] [Google Scholar]
- 49. Weber J., Bowman C., Wilke-Mounts S., Senior A. E. (1995) α-Aspartate 261 is a key residue in noncatalytic sites of Escherichia coli F1-ATPase. J. Biol. Chem. 270, 21045–21049 [DOI] [PubMed] [Google Scholar]
- 50. Al-Shawi M. K., Senior A. E. (1992) Catalytic sites of Escherichia coli F1-ATPase. Characterization of unisite catalysis at varied pH. Biochemistry 31, 878–885 [DOI] [PubMed] [Google Scholar]
- 51. Al-Shawi M. K., Senior A. E. (1992) Effects of dimethyl sulfoxide on catalysis in Escherichia coli F1-ATPase. Biochemistry 31, 886–891 [DOI] [PubMed] [Google Scholar]
- 52. Senior A. E. (2007) ATP synthase: motoring to the finish line. Cell 130, 220–221 [DOI] [PubMed] [Google Scholar]
- 53. Ahmad Z., Senior A. E. (2004) Mutagenesis of residue β-Arg-246 in the phosphate-binding subdomain of catalytic sites of Escherichia coli F1-ATPase. J. Biol. Chem. 279, 31505–31513 [DOI] [PubMed] [Google Scholar]
- 54. Ahmad Z., Senior A. E. (2005) Involvement of ATP synthase residues α-Arg-376, β-Arg-182, and β-Lys-155 in Pi binding. FEBS Lett. 579, 523–528 [DOI] [PubMed] [Google Scholar]
- 55. Li W., Brudecki L. E., Senior A. E., Ahmad Z. (2009) Role of α-subunit VISIT-DG sequence residues Ser-347 and Gly-351 in the catalytic sites of Escherichia coli ATP synthase. J. Biol. Chem. 284, 10747–10754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ahmad Z., Senior A. E. (2005) Modulation of charge in the phosphate-binding site of Escherichia coli ATP synthase. J. Biol. Chem. 280, 27981–27989 [DOI] [PubMed] [Google Scholar]
- 57. Ahmad Z., Senior A. E. (2004) Role of β-Asn-243 in the phosphate-binding subdomain of catalytic sites of Escherichia coli F1-ATPase. J. Biol. Chem. 279, 46057–46064 [DOI] [PubMed] [Google Scholar]
- 58. Hazard A. L., Senior A. E. (1994) Defective energy coupling in δ-subunit mutants of Escherichia coli F1F0-ATP synthase. J. Biol. Chem. 269, 427–432 [PubMed] [Google Scholar]
- 59. Maggio M. B., Parsonage D., Senior A. E. (1988) A mutation in the α-subunit of F1-ATPase from Escherichia coli affects the binding of F1 to the membrane. J. Biol. Chem. 263, 4619–4623 [PubMed] [Google Scholar]
- 60. Perlin D. S., Cox D. N., Senior A. E. (1983) Integration of F1 and the membrane sector of the proton-ATPase of Escherichia coli. Role of subunit “b” (UncF protein). J. Biol. Chem. 258, 9793–9800 [PubMed] [Google Scholar]
- 61. Weber J., Wilke-Mounts S., Senior A. E. (2002) Quantitative determination of binding affinity of δ-subunit in Escherichia coli F1-ATPase. Effects of mutation, Mg2+, and pH on Kd. J. Biol. Chem. 277, 18390–18396 [DOI] [PubMed] [Google Scholar]
- 62. Weber J., Wilke-Mounts S., Senior A. E. (2003) Identification of the F1-binding surface on the δ-subunit of ATP synthase. J. Biol. Chem. 278, 13409–13416 [DOI] [PubMed] [Google Scholar]
- 63. Weber J., Muharemagić A., Wilke-Mounts S., Senior A. E. (2003) F1F0-ATP synthase. Binding of δ-subunit to a 22-residue peptide mimicking the N-terminal region of α-subunit. J. Biol. Chem. 278, 13623–13626 [DOI] [PubMed] [Google Scholar]
- 64. Weber J., Muharemagić A., Wilke-Mounts S., Senior A. E. (2004) Analysis of sequence determinants of F1F0-ATP synthase in the N-terminal region of α-subunit for binding of δ-subunit. J. Biol. Chem. 279, 25673–25679 [DOI] [PubMed] [Google Scholar]
- 65. Wilkens S., Borchardt D., Weber J., Senior A. E. (2005) Structural characterization of the interaction of the δ- and α-subunits of the Escherichia coli F1F0-ATP synthase by NMR spectroscopy. Biochemistry 44, 11786–11794 [DOI] [PubMed] [Google Scholar]
- 66. Weber J., Wilke-Mounts S., Nadanaciva S., Senior A. E. (2004) Quantitative determination of direct binding of b subunit to F1 in Escherichia coli F1F0-ATP synthase. J. Biol. Chem. 279, 11253–11258 [DOI] [PubMed] [Google Scholar]
- 67. Senior A. E., Muharemagić A., Wilke-Mounts S. (2006) Assembly of the stator in Escherichia coli ATP synthase. Complexation of α-subunit with other F1 subunits is prerequisite for δ-subunit binding to the N-terminal region of α. Biochemistry 45, 15893–15902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Al-Shawi M. K., Senior A. E. (1993) Characterization of the adenosine triphosphatase activity of Chinese hamster P-glycoprotein. J. Biol. Chem. 268, 4197–4206 [PubMed] [Google Scholar]
- 69. Urbatsch I. L., Al-Shawi M. K., Senior A. E. (1994) Characterization of the ATPase activity of purified Chinese hamster P-glycoprotein. Biochemistry 33, 7069–7076 [DOI] [PubMed] [Google Scholar]
- 70. Lerner-Marmarosh N., Gimi K., Urbatsch I. L., Gros P., Senior A. E. (1999) Large-scale purification of detergent-soluble P-glycoprotein from Pichia pastoris cells and characterization of nucleotide-binding properties of wild-type, Walker A, and Walker B mutant proteins. J. Biol. Chem. 274, 34711–34718 [DOI] [PubMed] [Google Scholar]
- 71. Al-Shawi M. K., Urbatsch I. L., Senior A. E. (1994) Covalent inhibitors of P-glycoprotein ATPase activity. J. Biol. Chem. 269, 8986–8992 [PubMed] [Google Scholar]
- 72. Urbatsch I. L., Senior A. E. (1995) Effects of lipids on ATPase activity of purified Chinese hamster P-glycoprotein. Arch. Biochem. Biophys. 316, 135–140 [DOI] [PubMed] [Google Scholar]
- 73. Urbatsch I. L., Sankaran B., Weber J., Senior A. E. (1995) P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 270, 19383–19390 [DOI] [PubMed] [Google Scholar]
- 74. Urbatsch I. L., Sankaran B., Bhagat S., Senior A. E. (1995) Both P-glycoprotein nucleotide-binding sites are catalytically active. J. Biol. Chem. 270, 26956–26961 [DOI] [PubMed] [Google Scholar]
- 75. Senior A. E., Al-Shawi M. K., Urbatsch I. L. (1995) The catalytic cycle of P-glycoprotein. FEBS Lett. 377, 285–289 [DOI] [PubMed] [Google Scholar]
- 76. Senior A. E. (2011) Reaction chemistry ABC-style. Proc. Natl. Acad. Sci. U.S.A. 108, 15015–15016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tombline G., Bartholomew L. A., Tyndall G. A., Gimi K., Urbatsch I. L., Senior A. E. (2004) Properties of P-glycoprotein with mutations in the “catalytic carboxylate” glutamate residues. J. Biol. Chem. 279, 46518–46526 [DOI] [PubMed] [Google Scholar]
- 78. Tombline G., Muharemagić A., White L. B., Senior A. E. (2005) Involvement of the “occluded nucleotide conformation” of P-glycoprotein in the catalytic pathway. Biochemistry 44, 12879–12886 [DOI] [PubMed] [Google Scholar]
- 79. Tombline G., Senior A. E. (2005) The occluded nucleotide conformation of P-glycoprotein. J. Bioenerg. Biomembr. 37, 497–500 [DOI] [PubMed] [Google Scholar]
- 80. Sankaran B., Bhagat S., Senior A. E. (1997) Photoaffinity labeling of P-glycoprotein catalytic sites. FEBS Lett. 417, 119–122 [DOI] [PubMed] [Google Scholar]
- 81. Urbatsch I. L., Gimi K., Wilke-Mounts S., Senior A. E. (2000) Investigation of the role of glutamine 471 and glutamine 1114 in the two catalytic sites of P-glycoprotein. Biochemistry 39, 11921–11927 [DOI] [PubMed] [Google Scholar]
- 82. Urbatsch I. L., Gimi K., Wilke-Mounts S., Senior A. E. (2000) Conserved Walker A Ser residues in the catalytic sites of P-glycoprotein are critical for catalysis and involved primarily at the transition state step. J. Biol. Chem. 275, 25031–25038 [DOI] [PubMed] [Google Scholar]
- 83. Urbatsch I. L., Tyndall G. A., Tombline G., Senior A. E. (2003) P-glycoprotein catalytic mechanism: studies of the ADP-vanadate-inhibited state. J. Biol. Chem. 278, 23171–23179 [DOI] [PubMed] [Google Scholar]
- 84. Lee J. Y., Urbatsch I. L., Senior A. E., Wilkens S. (2002) Projection structure of P-glycoprotein by electron microscopy. Evidence for a closed conformation of the nucleotide-binding domains. J. Biol. Chem. 277, 40125–40131 [DOI] [PubMed] [Google Scholar]
- 85. Lee J. Y., Urbatsch I. L., Senior A. E., Wilkens S. (2008) Nucleotide-induced structural changes in P-glycoprotein observed by electron microscopy. J. Biol. Chem. 283, 5769–5779 [DOI] [PubMed] [Google Scholar]
- 86. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Weber J. (2010) Structural biology: toward the ATP synthase mechanism. Nat. Chem. Biol. 6, 794–795 [DOI] [PubMed] [Google Scholar]