

Background: Regulation of group I p21-activated kinases is based on dimeric conformation.
Results: PAK3a forms regulatory heterodimers with PAK1 in vitro and in vivo.
Conclusion: Heterodimerization of PAK3a and PAK1 links PAK pathways in brain.
Significance: Analyzing the molecular mechanisms of PAKs regulation could help to understand the pathophysiology of some cancers and mental disabilities.
Keywords: Brain, Cancer, Mutant, Neurological Diseases, Signaling, Dimerization, Intellectual Disability, Kinase, Regulation, p21-activated Kinase
Abstract
p21-activated kinase 1 (PAK1) and PAK3 belong to group I of the PAK family and control cell movement and division. They also regulate dendritic spine formation and maturation in the brain, and play a role in synaptic transmission and synaptic plasticity. PAK3, in particular, is known for its implication in X-linked intellectual disability. The pak3 gene is expressed in neurons as a GTPase-regulated PAK3a protein and also as three splice variants which display constitutive kinase activity. PAK1 regulation is based on its homodimerization, forming an inactive complex. Here, we analyze the PAK3 capacity to dimerize and show that although PAK3a is able to homodimerize, it is more likely to form heterodimeric complexes with PAK1. We further show that two intellectual disability mutations impair dimerization with PAK1. The b and c inserts present in the regulatory domain of PAK3 splice variants decrease the dimerization but retain the capacity to form heterodimers with PAK1. PAK1 and PAK3 are co-expressed in neurons, are colocalized within dendritic spines, co-purify with post-synaptic densities, and co-immunoprecipitate in brain lysates. Using kinase assays, we demonstrate that PAK1 inhibits the activity of PAK3a but not of the splice variant PAK3b in a trans-regulatory manner. Altogether, these results show that PAK3 and PAK1 signaling may be coordinated by heterodimerization.
Introduction
The p21-activated kinases (PAK)4 are effectors of Rac and Cdc42 GTPases and control cell proliferation and movement (1). The mammalian group I of PAK includes PAK1, PAK2, and PAK3 which all display a very high degree of sequence identity and share structural and functional similarities. Their N-terminal domain interacts with regulators such as GTPases through the p21-binding domain (PBD), with Nck adaptors through the first proline-rich sequence and with PIX guanine exchange factors through an atypical proline-rich sequence (2–5). The PBD partially overlaps with the auto-inhibitory domain (AID). This latter domain can be subdivided into three domains: the dimerization domain, the inhibitory switch domain and the kinase inhibitory segment, which directly interacts with the kinase domain to inhibit its activity (6, 7). Mechanisms of PAK regulation have been mainly studied for PAK1 and were shown to be driven through the formation of dimers. Interaction between the regulatory domain and the catalytic domain is determinant for kinase regulation (6–11). Dimeric assembly positions the AID of one monomer inside the catalytic cleft of the kinase domain of the other monomer, forcing the T-loop to adopt an inactive conformation (6, 11). Membrane recruitment and GTPase-binding to the PBD induce conformational changes of the inhibitory switch domain, leading to the dissociation of the dimer and cis- and trans-phosphorylations allowing the activation of each partner (7, 8, 11–13). The dynamics of the different steps appears to be very complex and may proceed through different reaction pathways (11). This model of regulation is thought to be more generally applicable to the whole group I PAK proteins on the basis of their strong sequence identity.
PAK3 has specific functions in neuronal physiology and pak3 mutations lead to intellectual disability (ID) (14–16). PAK3 is the only group I member whose expression is relatively restricted to neurons whereas PAK1 is highly expressed in the nervous system but also in other tissues (17, 18). The mammalian pak3 gene contains two alternatively spliced exons called b and c (17, 19) and encodes PAK3a (without any spliced exon) and three splice variants referred to as PAK3b, PAK3c, and PAK3cb. The inserts are present in the AID, and their sequences are highly conserved during evolution, suggesting that these splice variants support unique functions for PAK3 (17). Unlike PAK3a, these splice variants are constitutively active and their interaction with active GTPases of the Rac/Cdc42 family are profoundly diminished compared with PAK3a. Moreover, their AID cannot inhibit the kinase activity of a PAK3a kinase domain (19). The molecular basis of the specific functions of PAK3 and its splice variants are largely unknown (20), and their mechanisms of regulation deserve to be extensively studied. Mechanisms of regulation of PAK1 were largely studied in regard to its role in cancer (21, 22) and the basis of PAK1 regulation is dependent upon its capacity to form dimeric complexes. In an attempt to characterize the mechanisms of regulation of PAK3a and of the three PAK3 splice variants, we analyzed, in vitro, the ability of PAK3 proteins to form dimers and to interact with the other neuronal PAK protein, PAK1. In transfected cells, we have demonstrated the existence of PAK3 homodimers and also heterodimeric complexes between PAK1 and the four PAK3 proteins. However, we observed a preference for heterodimeric complexes with PAK1 rather than PAK3 homodimers. In vivo, we showed that the different pak3 mRNAs are expressed together with pak1 in single pyramidal neurons and that PAK1 and PAK3 proteins colocalized in dendritic spines and are both present in the post-synaptic density (PSD) fraction. Moreover, we report that PAK1 and PAK3 co-immunoprecipitate with each other in mouse brain lysates. Finally, we demonstrated that PAK1/PAK3 heterodimers allow a trans-inhibition of the catalytic activity of PAK3a but not of the PAK3b splice variant. Altogether, our data show that PAK3 proteins form heterodimers with PAK1 and suggest that PAKs heterodimerization can coordinate PAK signaling and help to synchronize actin polymerization and spine stabilization.
EXPERIMENTAL PROCEDURES
Antibodies
Immunoblot and immunochemistry analyses were performed using anti-HA (rabbit: Santa Cruz Biotechnologies and rat: Roche), anti-FLAG (rabbit and mouse: Sigma-Aldrich), anti-PSD95 K28/43 (NeuroMab), anti-paxillin (Upstate-Millipore), HRP-conjugated secondary antibodies (Bio-Rad), Rabbit TrueBlot antibody (eBioscience), and anti-rabbit-FITC-, anti-rat-TRITC-, anti-mouse-Alexa-568-, and anti-mouse-Alexa-633-conjugated secondary antibodies (Jackson ImmunoResearch, Sigma, and Invitrogen). The anti-synaptophysin rabbit serum was previously described (23). For PAK analysis, several commercial or homemade sera were used and their characteristics were described in supplemental Fig. S1. N19-PAK3 directed against the 19-N-terminal amino acids of PAK3 and the N20-PAK1 directed against the 20 N-terminal amino acids of PAK1 were from Santa Cruz Biotechnologies, and the monoclonal antibody, named 3A12-PAK3 was from Sigma-Aldrich. A new polyclonal antiserum directed against the PAK3 protein was generated in rabbit (Sigma Genosys). The ASPAAPNKEDIPPSAENA (residues 211–228) synthetic peptide, common to all the mouse PAK3 proteins, was conjugated to keyhole limpet hemocyanin before immunization. The serum obtained was named rb-211-PAK3. The rabbit polyclonal antibody raised against the PAK3 exon c-encoded peptide, named Ec, was previously described (17). A rabbit polyclonal serum, directed against the GEFTPDLY (residues 94–101) peptide, only present in the mouse PAK3b protein, was similarly processed (Neosystem) and named rb-svs-PAK3b. Two new polyclonal antisera were obtained after immunization with synthetic peptides, unique to the mouse PAK1 protein (GenScript): a serum named ch-209-PAK1 was prepared from chicken, using the LTVTPTRDVAT (residues 209–219) peptide, and another one from rabbit, named rb-211-PAK1, using the VTPTRDVATSPISPTEN (residues 211–227) peptide. PAK antibodies were affinity-purified by Ultralink column chromatography (Perbio Science).
Plasmid Constructs
The following pcDNA3-HA-based plasmids were previously described: pcDNA3-HA-PAK3a-WT, -PAK3a-K297L (kinase-dead), -PAK3b-WT, -PAK3b-K297L (19), -PAK3a-A365E, -PAK3a-R419X, -PAK3a-R67C (24), -PAK3c-WT, -PAK3cb-WT (17). The following FLAG-based constructs were previously described (5): pFLAG-PAK3a-WT, pFLAG-PAK3a-K297L, pFLAG-PAK1-K299R (kinase-dead and named pFLAG-PAK1-KD). The plasmid encoding HA-PAK2 protein was provided by Herma Renkema (Tampere, Finland) and the HA-tagged wild-type PAK1 plasmid was provided by M. C. Parrini (7). Mutagenesis was performed using procedures based upon the QuickChange protocol (Stratagene) with the oligonucleotides listed in supplemental Table S2. PCR on pcDNA3-HA-PAK3a-K297L with the oligonucleotides set 1 gave rise to the pcDNA3-HA-PAK3a-L102F-K297L plasmid. The KpnI/XbaI digested fragment from the pcDNA3-HA-PAK3a-L102F-K297L plasmid was introduced in the pFLAG3S2X vector (Sigma), to obtain the pFLAG-PAK3a-L102F-K297L plasmid. Mutagenesis on the pFLAG-PAK3a-WT and the pFLAG-PAK3a-K297L plasmids using the oligonucleotides set 2 led to the pFLAG-PAK3a-R436E-K437D and the pFLAG-PAK3a-R436E-K437D-K297L plasmids, respectively. A fragment obtained after PfmlI digestion of the pFLAG-PAK3a-R436E-K437D-K297L construct, was introduced in the PfmlI digested pFLAG-PAK3a-L102F-K297L plasmid, giving rise to the pFLAG-PAK3a-L102F-R436E-K437D-K297L plasmid. The pFLAG-PAK3a-R436E-K437D-K297L was digested by KpnI/XbaI and the fragment obtained was cloned in the pcDNA3-HA vector, leading to the pcDNA3-HA-PAK3a-R436E-K437D-K297L plasmid. PCR amplifications were performed from the pFLAG-PAK3a-K297L and the pFLAG-PAK3a-R436E-K437D using the oligonucleotides set 3. pLex and pGAD plasmids previously were previously described (5). PCR products were digested by BamHI/SalI and the fragments obtained were inserted in the pLex vector to obtain the pLex-PAK3-(232–544) plasmid (named pLex-PAK3-Cter) and the pLex-PAK3-R436E-K437D-(232–544) plasmid (named pLex-PAK3-Cter-R436E-K437D), respectively. PCR amplification was also performed on pcDNA3-HA-PAK1-K299R with the oligonucleotide set 4. PCR products were digested by BamHI/XbaI then inserted into pLex or pGAD vectors to obtain pLex-PAK1-(235–545) named pLex-PAK1-Cter and pGAD-PAK1-(235–545) named pGAD-PAK1-Cter, respectively. The BamHI/SalI fragment PAK3a-(2–270) from the pcDNA3-HA-PAK3a-WT was cloned in the pLex vector, giving rise to the pLex-PAK3a-(2–270) plasmid, named pLex-PAK3-Nter. To obtain the pGAD-PAK3-(232–544) plasmid, named pGAD-PAK3-Cter, a PCR was performed on pcDNA3-HA-PAK3a-K297L with the oligonucleotide set 5. PCR products were digested by BamHI/XbaI and the insert obtained was cloned into the GAD vector. PCR amplifications were performed from the plasmids pcDNA3-HA-PAK3a-L102F-K297L, pcDNA3-HA-PAK3b, pcDNA3-HA-PAK3c, and pcDNA3-HA-PAK3cb with the oligonucleotide set 7. PCR products digested by BamHI/XbaI were cloned into the pGAD vector to obtain pGAD-PAK3a-(2–270)-L102F, pGAD-PAK3b-(2–270), pGAD-PAK3c-(2–270), and pGAD-PAK3cb-(2–270) constructs (all named Nter in place of 2–270). The BamHI/XbaI insert obtained after digestion of the pFLAG-PAK1-K299R plasmid (5) was inserted in the pcDNA3-HA vector (Invitrogen) in order to obtain the pcDNA3-HA-PAK1-K299R plasmid. The pGAD-PAK1-(2–270) plasmid, named pGAD-PAK1-Nter was obtained by amplifying pcDNA3-HA-PAK1 with set 6. The insert was then cloned in the GAD vector. The GST-Cdc42-V12 prokaryotic expression plasmid was previously described (19). All plasmid coding sequences obtained were confirmed by sequencing.
Cell Line and Neuron Culture, Immunocytochemistry, and Imaging
HeLa cell cultures, primary cultures of dissociated hippocampal neurons and transfections were previously described (5). For HeLa cells, PAK-transfected cells were fixed after 24 h and triple immunolabeled using FLAG, HA, and paxillin antibodies, followed by anti-rabbit-FITC-, anti-rat-TRITC- and anti-mouse-Alexa-633-conjugated secondary antibodies, respectively. At 21 days-in vitro (DIV), neurons were fixed and labeled for native PAK proteins, using specific PAK1 and PAK3 antibodies, followed by anti-rabbit-FITC- and anti-mouse-Alexa-568-conjugated secondary antibodies, respectively, in the presence of Alexa-633-phalloidin to label actin cytoskeleton. Images were acquired in a sequential mode, as previously described (5), using a Zeiss LSM 700 confocal microscope, equipped with 488, 555, and 639 nm lasers. Bleed-through was checked by imaging cells labeled with a single fluorophore and by acquiring dual channel images with the same setup used for the triple-labeled cells, indicating the total absence of overlap between the different fluorophores used. Analysis of the PAK1/PAK3 co-localized pixels was performed from the same focal plane acquired sequentially in the two channels, using the RG2B co-localization plug-in of NIH ImageJ software.
Immunoprecipitation and Western Blot
24–30 h after transfection, HeLa cells were lysed in KLB buffer as described by Ref. 7. Cell lysates were immunoprecipitated overnight with HA-agarose conjugate or with protein G-agarose (Sigma-Aldrich) in the presence of the FLAG antibody. For brain immunoprecipitation, tissues were cut in KLB buffer (10% weight/volume) then homogenized in a Potter-Elvehjem homogenizer. After a centrifugation step, tissue supernatants were then incubated a few hours in the presence of protein G-agarose as a clearing step. Immunoprecipitation was then performed overnight at 4 °C by adding protein G-agarose and appropriate PAK antibodies. Samples were resolved by electrophoresis and Western blotting as previously described (5). Expression of transfected proteins was checked in each experiment by Western blotting of Total Cell Lysate (TCL) samples.
Kinase Assay
The immunoprecipitates obtained from transfected cells were resuspended in the kinase buffer (25 mm HEPES, pH 7.4, 25 mm MgCl2, 25 mm-glycerophosphate, 2 mm dithiothreitol, 0.1 mm orthovanadate) and incubated in the absence or the presence of the active GTPase GST-Cdc42V12, to activate the kinase activity, as previously described in Thévenot et al., (5). Samples were then incubated in the presence of 5 μCi of [γ-32P]ATP (Amersham Biosciences) for 20 min at 30 °C under agitation, in the presence of 3 μg of Myelin Basic Protein (Invitrogen) as a substrate. Reaction was stopped by adding SDS-Laemmli sample buffer. Boiled samples were resolved by SDS-PAGE and incorporation of 32P was quantified using a PhosphorImager (Amersham Biosciences).
Yeast Two-hybrid Assay, Histidine Test, and β-Galactosidase Activity
The two-hybrid assay was applied according to standard procedures. L40 yeasts strain (L40 Mata, Trp-1, Leu-2, His-3, lys::(lexAop)4-HIS3, ura3::(LexAop)8-LacZ), were co-transfected with pLex and pGAD constructs by the lithium acetate method and transformants were grown on appropriate selective medium. Following overnight growth on YPDA medium, patches were replicated on plates lacking His, Trp, and Leu for selection of diploids. Liquid β-galactosidase assay was performed following the method of Crouin et al. (25). In each experiment, the quantification of the interaction between the PAK3a-Nterminal domain and the PAK3-Cterminal domain constitutes the 100% reference for all other tested interactions.
Single Cell RT-PCR on Cortical Neurons
Experiments were performed as previously described (26). Briefly, layer V visual cortex neurons of 3-week-old rats were patched and following whole cell recording to identify neurons, as much cytoplasm as possible was aspirated into the patch pipette. The cytoplasm was added to a reverse-transcription mix and processed for first strand elongation for three hours at 48 °C. The RT products were then amplified by two successive rounds of PCR. First, a multiplex PCR was performed directly in the same tubes by adding 90 μl of the PCR mix. This mix contained pairs of primers that hybridize with similar Tm. A second specific amplification was then performed in several parallel PCRs, using 1 μl of the PCR1 products (in 25 μl final PCR volume) and one primer set. PCR products were analyzed after electrophoresis in ethidium bromide containing agarose gels. Primer sequences are described in supplemental Table S2 and in Ref. 17.
Synaptosome Extraction and PSD Purification
Mice brains were cut in small pieces and resuspended in 5% (weight/volume) of resuspension solution A (5 mm Hepes, 320 mm sucrose, 150 mm NaCl, 2 mm EDTA, pH 7.4, and protease and phosphatase inhibitors), as described in Ref. 27 (see supplemental Fig. S3). Homogenization of tissues was performed by a Precellys tissue homogenizer. The TBL was centrifuged to give the P2 fraction, according to the protocol of Ref. 28. P2 was then resuspended in a small volume of solution B (320 mm sucrose, 1 mm NaHCO3 with protease and phosphatase inhibitors), which was then mixed with 15 ml of 14% Ficoll (GE Healthcare). This preparation was loaded below a 7.5% Ficoll solution. Both Ficoll solutions were diluted in solution A. Gradients were centrifuged during 1 h at 80,000 × g. Synaptosomes migrated at the interface between both the two Ficoll solutions, so this cream-colored band was collected. Synaptosomes were diluted in B solution, and the obtained volume was half diluted with solution C (12 mm Tris pH 8.0, 1% Triton X-100 with protease and phosphatase inhibitors). The preparation was incubated 15 min in ice then centrifuged 20 min at 32,800 × g. The pellet containing the PSD fraction was resuspended in a small volume of Kreb's buffer (20 mm Hepes, 10 mm glucose, 1.2 mm NaH2PO4, 1.3 mm MgCl2, 1.2 mm CaCl2, 5 mm KCl, 145 mm NaCl, 2 mm EDTA, with protease, and phosphatase inhibitors). All the collected fractions were then analyzed by Western blotting using appropriate antibodies.
Animal Experimentation
All experiments were conducted under appropriate biological containment in accordance with European Communities Council Directive (CEE 86/609) for animal care and experimentation, and were conducted following the guidelines of the animal facility in Orsay (France) approved by the national direction of veterinary services (Direction des Services Vétérinaires, France, agreement no. B91-471-104).
RESULTS
PAK3a Forms Homodimers
Crystallographic analysis and co-immunoprecipitation assays showed that PAK1 forms homodimers (6, 7). Since PAK1 and PAK3 possess a high sequence identity and share an overall similar structure (Fig. 1A), we first asked whether PAK3 may form homodimers like PAK1 does. The ability of PAK3a to form dimers was tested by a co-immunoprecipitation assay. HeLa cells were co-transfected with HA- and FLAG-tagged PAK3a constructs. As a control, cells were transfected by HA-or FLAG- PAK3 plasmids alone or with empty vectors. To bypass the problem of dissociation of the dimers following PAK activation, we chose to work with kinase-dead (KD) constructs (PAK3-K297L and PAK1-K299R) as previously performed for PAK1 (7). The mutation of the PAK3 lysine 297 residue that is homolog to the lysine 299 in the PAK1 protein, totally inactivates the kinase activity of PAK3 (19). Co-immunoprecipitated proteins were revealed by Western blot analysis with anti-FLAG antibodies on HA-precipitates. As shown in Fig. 1B, the FLAG-PAK3 protein is present in the HA-immunoprecipitated complex, demonstrating that PAK3a is able to form homodimers in co-transfected cells. Specificity of the signal was verified with the absence of co-immunoprecipitated protein in experiments from single-transfected cells, and from cells co-transfected with an HA or FLAG empty vector and a corresponding tagged PAK3 plasmid (Fig. 1B, fourth and fifth lanes). It was previously shown that in PAK1 the Leu-107 residue in the Inhibitory Switch domain and the Arg-Lys-438–439 residues in the large lobe of the kinase domain are involved in the molecular interaction between the N-terminal regulatory domain and the catalytic C-terminal domain and their mutation disrupts the dimeric complex (6, 7). We introduced the homolog mutations L102F and R436E-K437D, alone or together, in the pak3 coding sequence (see Fig. 1A) and tested the capacity of the mutated proteins to dimerize (Fig. 1C). The Western blot analysis indicates that mutation L102F present on only one monomer was sufficient to strongly reduce the interaction (4.84 ± 2.00% of PAK3a homodimer) (Fig. 1D). Present in both partners, L102F totally suppressed the interaction (0.68 ± 0.39% of PAK3a homodimer). The double mutation R436E-K437D in the catalytic domain of one partner also decreased the co-immunoprecipitation (51.08 ± 2.34% of PAK3a homodimer) but to a lesser extent than the L102F mutation. The presence of the mutation R436E-K437D in both partners strengthened the effect of a single mutation (25.36 ± 5.88% of PAK3a homodimer). Moreover, the presence of the mutations L102F and R436E-K437D in one PAK3 protein totally abrogates dimer formation (1.39 ± 0.8% of PAK3a homodimer). These results show that L102 and R436-K437 residues are strongly involved in PAK3 dimer formation. To define the precise domains involved in the complex formation, a two-hybrid assay was performed with the N-terminal moiety and the C-terminal moiety of PAK3 fused to the Lex binding domain or the GAD transactivation domain. Protein interactions were observed on selective medium devoid of histidine (histidine test) and quantified by measure of β-galactosidase activity. Double transfected yeasts that co-expressed N-terminal and C-terminal moieties displayed a significant growth on selective media and a high β-galactosidase activity, indicating a strong interaction between the N-terminal and the C-terminal domains of PAK3a (Fig. 1E). In contrast, interactions between two N-terminal moieties of PAK3a and between two C-terminal moieties of PAK3 were very weak referred to the interaction between the N-terminal and C-terminal parts of the protein: 4.19 ± 1.39% for PAK3a-Nter/PAK3a-Nter interaction and 3.45 ± 1.15% for PAK3-Cter/PAK3-Cter interaction. The L102F or the R436E-K437D mutations both strongly decrease the N-terminal/C-terminal interaction (L102F: 17.06 ± 5.68%; R436E-K437D: 1.22 ± 0.40%). Taken together, these data show that PAK3a forms homodimers via a strong N-terminal/C-terminal interaction, leucine 102, arginine 436, and lysine 437 being critical residues for this complex.
FIGURE 1.

PAK3a forms homodimers. A, structure of the PAK3 protein indicating location of interaction mutations. B, PAK3a co-immunoprecipitates with itself. Lysates of HeLa cells transfected with empty HA- or FLAG vectors (empty v.) or with plasmids encoding HA-PAK3a-KD, FLAG-PAK3a-KD, or both, were HA-precipitated (IP HA). Total cell lysates (TCL) were controlled using the corresponding antibodies as indicated (third and fourth panels). HA-precipitated proteins were revealed by anti-HA Western blotting (second panel) and the co-immunoprecipitated PAK3a proteins were revealed by anti-FLAG Western blotting (first panel). Images shown are representative of three independent experiments. C, characterization of PAK3a mutations that disrupt the dimer. All PAK3a constructs carry the K297L (KD) mutation that inactivates the kinase domain. HA or FLAG-tagged PAK3a-WT, -L102F or -R436E-K437D plasmids were co-transfected in HeLa cells before HA-precipitation (IP HA) and Western blotting using FLAG antibody (co-immunoprecipitates, first panel) or HA antibody (immunoprecipitates, second panel) as indicated. Levels of FLAG-PAK3a proteins in TCL were controlled by anti-FLAG immunoblotting (third panel). Images shown are representative of three independent experiments. D, amount of FLAG-PAK3a proteins co-immunoprecipitated compared with the amount of HA-immunoprecipitated PAK3a proteins in three experiments. Results are expressed relative to the PAK3a/PAK3a interaction. Comparison with Student's t test: ***, p < 0.001, n = 3. E, PAK3a interaction involves a strong Nter/Cter interaction as demonstrated with the two-hybrid assay. Yeast growth on histidine minus media (histidine test) indicates a protein-protein interaction (+). β-Galactosidase activity was expressed relative to the PAK3a-Nter/PAK3a-Cter interaction. Comparison with Student's t test: ***, p < 0.001, n = 3.
PAK3a Forms Heterodimers with PAK1
Given that PAK1 and PAK3 share a very high level of sequence identity, we wondered whether PAK3a can form heterodimers with PAK1. Because PAK1 and PAK3 proteins can form heterodimers only if they localize in the same subcellular regions, we analyzed their co-localization, in particular in focal adhesions whose structure and reorganization are highly regulated by PAKs (4). HeLa cells were co-transfected with FLAG-tagged PAK3a-KD and HA-tagged PAK1-KD expressing plasmids (Fig. 2A). Immunolabeling was performed using FLAG and HA antibodies to detect PAK3 and PAK1 proteins, respectively, and paxillin antibody to detect focal adhesions. We observed that the PAK3a and PAK1 proteins colocalize (Fig. 2A, right panel), and colocalize also with paxillin in the focal adhesions at the edge of the cell.
FIGURE 2.

PAK3a forms heterodimers with PAK1. A, PAK1 and PAK3 colocalize at focal adhesions of HeLa cells. FLAG-PAK3a-KD and HA-PAK1-KD plasmids were transfected in HeLa cells. Immunolabeling was performed using FLAG and HA antibodies to detect PAK proteins and paxillin antibodies to identify focal adhesions. The PAK1/PAK3 colocalized pixels are shown (right). B, PAK3a co-immunoprecipitates with PAK1. HA- and FLAG-tagged PAK3a-KD and PAK1-KD plasmids were co-transfected in HeLa cells then cell lysates were HA-precipitated (IP HA). The co-immunoprecipitated PAK proteins were revealed by anti-FLAG Western blotting (first panel). HA-precipitated proteins and TCL were controlled using the corresponding antibodies as indicated (second and third panels, respectively). Images shown are representative of three independent experiments. C, amount of FLAG-PAK proteins co-immunoprecipitated compared with the amount of HA-precipitated PAK proteins. Results are expressed relative to the PAK3a/PAK3a interaction. Comparison with Student's t test: NS, p > 0.05; ***, p < 0.001, n = 3. D, wild-type PAK isoforms interact together. Cells were co-transfected with HA-PAK1-KD and FLAG-PAK3a-KD (third lane), HA-PAK1-KD, and FLAG-PAK3a-WT (fourth lane), and HA-PAK1-WT and FLAG-PAK3a-WT (fifth lane). HA-proteins were immunoprecipitated (second panel) and co-immunoprecipitated FLAG proteins were visualized by anti-FLAG Western blotting (first panel). Co-transfection of PAK1 with empty vector (empty v.) was done as control (first two lanes). Images shown are representative of three independent experiments. E, analysis of the PAK1/PAK3a interaction using the two-hybrid assay. Yeasts growth on histidine minus media indicated a protein-protein interaction (+). β-Galactosidase activity was expressed relative to the PAK3a-Nter/PAK3a-Cter interaction. Comparison with Student's t test: NS, p > 0.05; *, p < 0.05; ***, p < 0.001; n = 3.
Thus, since these proteins were localized in the same cellular structures, we examined whether they could form heterodimers by a co-immunoprecipitation assay. As mentioned above, since PAK activation may interfere with the capacity to form dimers (7), we tested interaction with kinase-dead proteins. HeLa cells were co-transfected with plasmids encoding differentially tagged PAK3a-KD or PAK1-KD (Fig. 2B) and a co-immunoprecipitation assay revealed that PAK3a could form homodimers. Interestingly, significantly higher levels of heterodimers were formed with PAK1 (Fig. 2B). Comparison of the ratio co-immunoprecipitate/precipitate intensity reveals that PAK3a interacts about 7-fold more with PAK1 than with itself (674.71% of PAK3a homodimer ± 224.90) (Fig. 2C). In contrast, PAK1 forms similar amounts of homodimers (431.22% of PAK3a homodimer ± 143.74) than heterodimers.
We then compared co-immunoprecipitation between wild-type and kinase-dead proteins (Fig. 2D). Controls with empty vectors verified the specificity of the assay (first two lanes). We observed that the interaction between PAK1 and PAK3a is maintained for wild-type partners, demonstrating that this interaction is not abrogated by phosphorylation. On the contrary, we observed that PAK3a-WT co-immunoprecipitates more with PAK1-KD than the PAK3a-KD does. This suggests that, in contrast to that what previously reported for PAK1 (7), the wild-type PAK3a protein more easily forms heterodimers than the inactive protein, and that the regulation of dimer stability by autophosphorylation may differ for the different PAK isoforms.
Using a two-hybrid assay, we observed that the PAK3 C-terminal domain strongly interacts with the PAK1 N-terminal domain of its partner, and reciprocally, contributing to heterodimer formation (Fig. 2E). The L102F and R436E-K437D mutations also disrupted heteromeric interaction between PAK3a and PAK1, as analyzed by the two-hybrid assay. Note that the quantitative difference observed between PAK3a/PAK3a homodimers and PAK3a/PAK1 heterodimers in the co-immunoprecipitation assay was not observed in the two-hybrid assay, suggesting that preferential heteromeric conformation is supported by properties dependent on full length structure. Altogether, our results indicate that, in transfected cells, the two proteins PAK1 and PAK3 localized and that PAK3a preferentially forms heterodimers with PAK1.
Intellectual Disability Mutations Differentially Affect PAK3 Dimerization
To date, five mutations in the pak3 gene coding sequence have been identified in patients with a non syndromic form of ID (reviewed in Ref. 20). Because we demonstrated that PAK3 preferentially forms heterodimers with PAK1, we wondered if these ID mutations could affect dimerization. We tested the R67C mutation previously described to decrease both the Cdc42 binding and the induced PAK3 activity, and two other mutations located in the C-terminal moiety, one missense mutation (A365E) and one nonsense mutation (R419X), that were both shown to abrogate PAK3 kinase activity (Fig. 3A) (24). HeLa cells were co-transfected with FLAG-PAK1-KD in the presence of either HA-tagged PAK3 plasmids PAK3a-WT, -KD, -A365E, -R419X, and -R67C. Cell lysates were HA-immunoprecipitated and the co-immunoprecipitated PAK1 proteins were revealed using FLAG antibodies (Fig. 3B). Concerning ID mutations, the R67C mutation did not affect the PAK1/PAK3 interaction. In contrast, the A365E and R419X PAK3 mutations dramatically decreased PAK1/PAK3 complex formation. Thus, PAK3 ID mutations of the Ala-365 residue and C-terminal deletion from Arg-419 residue are directly responsible for the loss of dimerization with PAK1.
FIGURE 3.
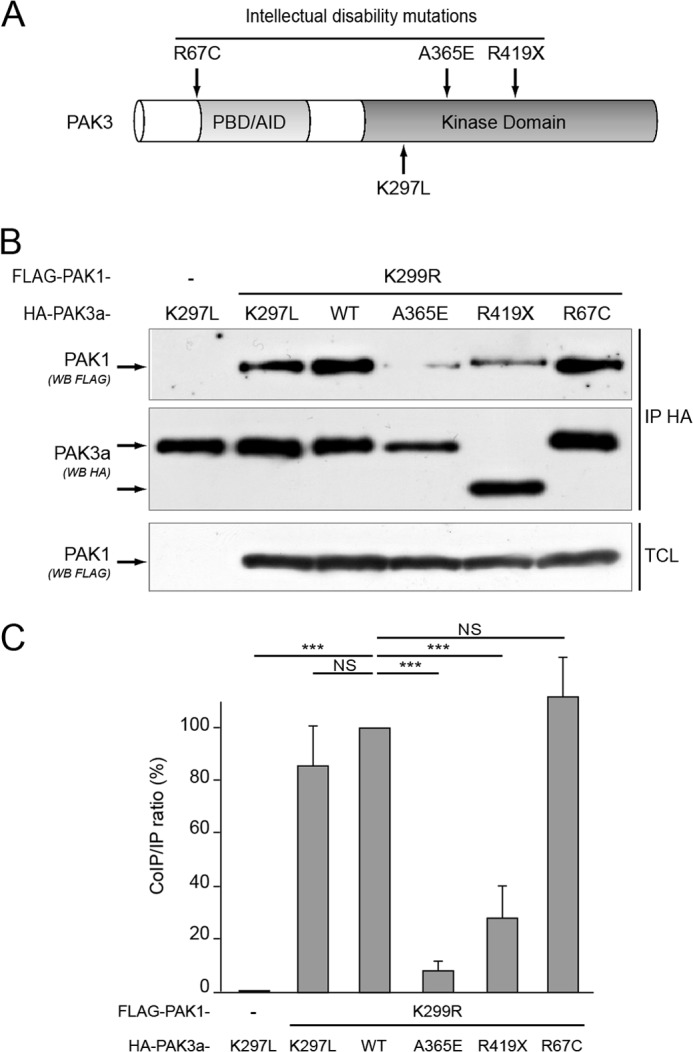
Two intellectual disability mutations of PAK3 affect their dimerization properties. A, location of three mutations responsible for intellectual disability. B, the A365E and R419X mutations affect the capacity to form heterodimers. HeLa cells were co-transfected with FLAG-PAK1-KD and HA-tagged PAK3a-KD, -WT, -A365E, -R419X, or -R67C. Co-immunoprecipitated proteins were revealed using FLAG antibodies (first panel) and HA-immunoprecipitated proteins (IP HA) were controlled by anti-HA labeling (second panel). Expression of FLAG PAK1 protein was controlled on TCL by an anti-FLAG Western blotting (third panel). Image shown is representative of three independent experiments. C, amount of FLAG-PAK1 proteins co-immunoprecipitated compared with the amount of HA-precipitated PAK proteins, in the experiment illustrated in B. Results are expressed relative to the PAK1/PAK3-WT interaction. Comparison with Student's t test: NS, p > 0.05; ***, p < 0.001, n = 3.
PAK3 Splice Variants Still Form Heterodimers with PAK1
The b and c inserts are located between amino acids 92 and 93 at the N-terminal part of the inhibitory switch domain (Fig. 4A), which is proximal to the dimerization segment and the Leu-102 residue involved in dimer stability (6, 7, 17). We thus wondered whether PAK3 splice variants could also form dimers and tested this using a co-immunoprecipitation assay (Fig. 4B). Cells were transfected with plasmids expressing FLAG-tagged PAK3a-KD or PAK1-KD in the presence of HA-tagged PAK3 splice variants in their kinase-dead forms. Interestingly, we observed that the presence of the b and c inserts dramatically decreased their co-immunoprecipitation with PAK3a compared with that of PAK3a with itself (Fig. 4, B and C). We further observed that the three splice variants were able to co-immunoprecipitate with PAK1 about 5-fold less than PAK3a does with PAK1. However, the level of splice variant dimerization with PAK1 is equivalent to that of PAK3a dimerization with itself (Fig. 4, B and C). Two-hybrid analysis confirmed that the presence of the insert in the N-terminal part of PAK3 protein fused to GAD bait decreases the interaction with the C-terminal moieties of PAK1 and PAK3 fused to Lex, suggesting that the inserts strongly modify structures involved in dimer formation (supplemental Fig. S4). In transfected mammalian cells, PAK3 splice variants (PAK3sv) were relatively impaired in their capacity to form dimers with PAK3a, but they retained their capacity to form heterodimers with PAK1. Interestingly, in both cases, relative to PAK3a-PAK3a, the decrease of dimer formation was more pronounced for PAK3sv-PAK3a (more than 10-fold) than for PAK3sv-PAK1 (3–5-fold), suggesting that in vivo, PAK3 splice variants heterodimerize with PAK1.
FIGURE 4.
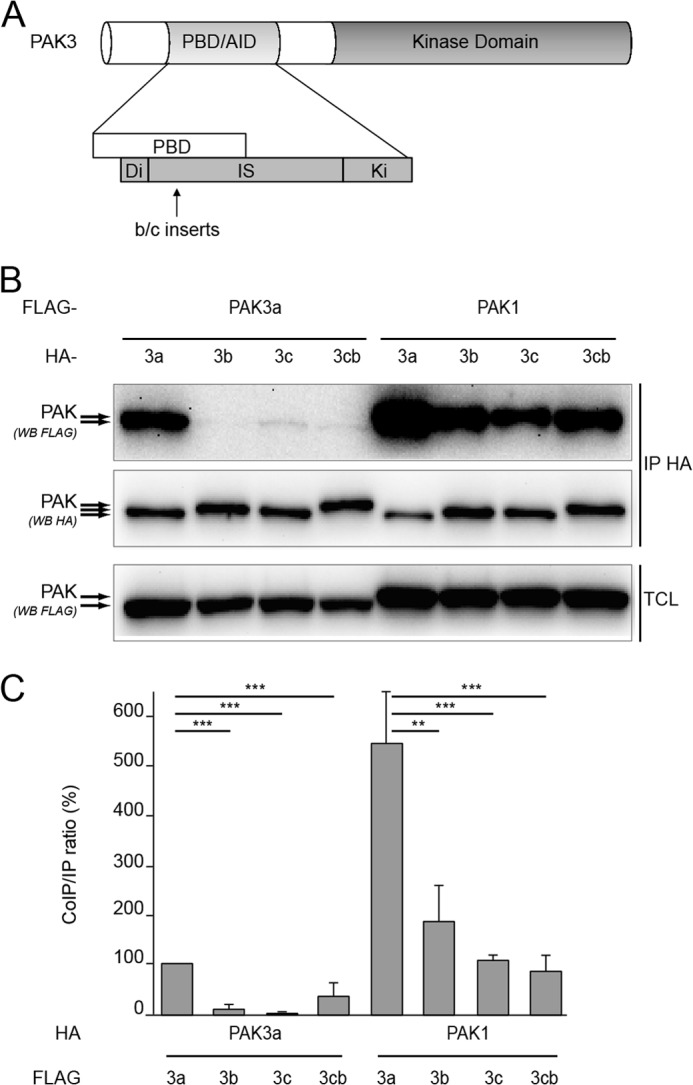
PAK3 splice variants form heterodimers with PAK1. A, schematic representation of the PAK3 protein indicating the location of the b and c inserts. B, presence of inserts decreases dimer formation. All the PAK1 and PAK3 expressed proteins carry the kinase dead mutation (KD). FLAG-tagged PAK3a-KD or PAK1-KD plasmids were transfected in HeLa cells in the presence of HA-tagged PAK3-KD-encoding variants. Cell lysates were HA-precipitated (IP HA). The co-immunoprecipitated PAK proteins were revealed by anti-FLAG Western blotting (first panel). HA-precipitated proteins and TCL were controlled using the corresponding antibodies as indicated (second and third panels, respectively). Images shown are representative of three independent experiments. C, quantification of the amount of FLAG-PAK3a-KD or FLAG-PAK1-KD proteins co-immunoprecipitated in the experiment illustrated in B, relative to the amount of HA-immunoprecipitated PAK3 proteins. Comparison with Student's t test: **, p < 0.01; ***, p < 0.001, n = 3.
PAK3 and PAK1 Form Complexes in Brain
One required condition for heterodimer formation is the expression of transcripts coding for the two partners in the same cell. To test this, we investigated whether pak1 mRNA and the different pak3 transcripts are co-expressed in a neuron by single cell RT-PCR. Pyramidal neurons of the visual cortex layer V of juvenile rat were patched and the cytoplasm was aspirated to be processed for RT-PCR with specific primers that amplified pak1 transcripts, all the pak3 transcripts (indicated as PAK3s) or each pak3 splice variant, i.e. a, b, c, cb variants as previously described (17, 26) (Fig. 5A). Actin was used as control, to attest the ability of the cDNA amplification from each sample. Among the actin-positive neurons (n = 37), 26 were positive for pak1 and 11 for pak3. Importantly, all of the pak3 positive cells were also positive for pak1 transcripts. Among pak3 positive neurons, 5 expressed pak3a, 4 expressed pak3b and 2 expressed pak3a and pak3cb. As an illustration, we show here (Fig. 5B) three neurons, positive for at least one pak3 transcript, indicating that a single neuron could express different pak3 transcripts. These results show that in a single neuron, both pak1 and pak3 genes are expressed and also that several pak3 splice variants can be co-expressed.
FIGURE 5.

PAK3 and PAK1 are coexpressed in single neurons, colocalize in dendritic spines, co-purified with the postsynaptic density, and co-immunoprecipitate in brain extracts. A, schematic representation of the location of the different PAK3 oligonucleotides used for single-cell RT-PCR. Forward and reverse oligonucleotides are indicated by arrows. Primers were chosen near or within the exons 2–3 portion to detect all the PAK3 cDNAs (1), and at exon/exon junction to specifically detect PAK3a (2), PAK3b (3), PAK3c (4), and PAK3cb (5) cDNAs. B, PAK3 and PAK1 are co-expressed in single neuron. RNAs from single pyramidal neurons were extracted and RT-PCR was performed using the different primer sets. Actin amplification was used as control (panel 7). Specific primer sets were used for PAK1 (first panel), all PAK3s (second panel), PAK3a, b, c, cb (third to sixth panel). C, PAK3 and PAK1 partially colocalize in dendritic spines of differentiated hippocampal neurons. Endogenous PAK proteins were immunolabeled with rb-211-PAK1 antibodies (first panel) and monoclonal 3A12 PAK3 antibodies (second panel). Dendritic spines were visualized by Alexa-633-phalloidin labeling (third panel) on dissociated hippocampal neurons at DIV21. On the PAK1/PAK3 colocalization image (fourth panel), white arrows indicate some spines where PAK1 and PAK3 proteins are co-expressed. Scale bar, 2 μm. D, PAK3 and PAK1 partially co-purify with some synaptosomal fractions as shown by Western blots of the different fractions purified from brain lysates. TBL from adult mice brain were fractionated to obtain the second pellet (P2) fraction, the synaptosomal fraction (Syn) and the postsynaptic densities (PSD) fraction. The same amount of protein was loaded for each fraction. The different fractions were probed with either the N20-PAK1 antibodies specific for PAK1 (first panel), the rb-211-PAK3 antibodies that recognized the PAK3s proteins (second panel), the rb-svs-PAK3b antibodies specific for PAK3b variant (third panel), the Ec antibodies that recognized the PAK3c and PAK3cb variants (fourth panel). Western blots with the synaptophysin antibodies as a presynaptic marker (fifth panel), and the PSD-95 antibodies as a postsynaptic density marker (sixth panel) confirm quality of tissue fractionation. Images shown are representative of three experiments. E, PAK1 co-immunoprecipitates with PAK3. TBL were prepared from adult wild-type mice (WT) and as a negative control, from pak3− mice (KO). In TBL, PAK1, and PAK3 proteins were detected by immunoblotting using the N20-PAK1 antibodies (third panel) and the N19-PAK3 (fourth panel), respectively. PAK3 proteins were immunoprecipitated with the N19-PAK3 sera (second panel) and co-immunoprecipitated PAK1 proteins were detected with ch-209-PAK1 (first panel). F, PAK3 co-immunoprecipitates with PAK1. TBL were processed as for E, to analyze PAK3 (third panel) and PAK1 (fourth panel) expression. PAK1 proteins were immunoprecipitated using the N20-PAK1 antibodies (second panel) and co-immunoprecipitated PAK3 proteins were then detected with the rb-211-PAK3 sera (first panel). Images shown are representative of two experiments.
To go further, we analyzed PAK1 and PAK3 localization by immunofluorescence and biochemical fractionation. PAKs immunofluorescence was performed on hippocampal neurons after 21 days of in vitro differentiation using specific antibodies as described in supplemental Fig. S1. PAK3 labeling shows an enrichment of PAK3 proteins in dendritic spines which are enriched in actin, as observed by Phalloïdin labeling (Fig. 5C, second and third panels), a result previously described (29–33). PAK1 labeling displays a more widespread distribution in the dendritic shaft, but is also detectable in certain dendritic spines. However, we observed clear colocalization of PAK1 and PAK3 in some spines (Fig. 5C, fourth panel, white arrows), indicating that in some functional domains of neurons, PAK1 and PAK3 proteins colocalize.
We then asked whether PAK1 and PAK3 proteins were enriched in the same subcellular domain in particular in the post-synaptic densities where PAK1 and PAK3 were described to localize (32). Synaptosomes were isolated from mice brain lysates on Ficoll-sucrose gradient and the postsynaptic densities (PSD) fractions were purified by Triton-X100 treatment (supplemental Fig. S3) as described (27). The different fractions obtained were analyzed by Western blot using the PAK antibodies characterized in supplemental Fig. S1, and as controls, the pre-synaptic marker synaptophysin and the post-synaptic marker PSD-95 (Fig. 5B, two lower panels). Results showed that synaptophysin was highly enriched in the synaptosomal fraction and completely absent from the PSD fraction. By contrast and as expected, PSD-95 is almost completely recovered in the PSD fraction. We observed that PAK1 was enriched in the synaptosomal fraction and was also present in the PSD fraction whereas PAK3 is present at a low level in the synaptosomal fraction and is highly enriched in the PSD fraction. These data are in agreement with the immunolabeling results indicating a more restricted localization of PAK3 in dendritic spines. The different PAK3 splice variants segregate differentially in postsynaptic fractions, the PAK3b splice variant being particularly enriched in PSD whereas the splice variants containing exon c-encoded insert are enriched in synaptosomes. Thus PAK1 and the different PAK3 proteins partially co-purified in the PSD fraction from adult mouse brain.
We next investigated whether dimers exist in the mouse brain between the two endogenous proteins by co-immunoprecipitation using specific immunoprecipitating antibodies (supplemental Fig. S1) and pak3 knock-out mouse brains as control. PAK1 protein was equally detected in both models (Fig. 5E, third panel, and 5F, fourth panel) and equal amount of immunoprecipitated PAK1 proteins were detected in brain lysates from wild-type mice and pak3 KO mice (Fig. 5F, second panel). On the other hand, PAK3 protein was only detectable in WT animals (Fig. 5E, fourth panel, and 5F, third panel), as expected (30). We detected PAK1 protein in the PAK3 immunoprecipitate from wild-type mice and not from pak3 KO mice (Fig. 5E, first panel). In the reverse experiment, we detected PAK3 protein in the PAK1 immunoprecipitated from wild-type mice, while no signal was detected from pak3 knock-out mice (Fig. 5F, first panel). Thus, using pak3 knock-down mice as a negative control and specific immunoprecipitating antibodies, we demonstrated by co-immunoprecipitation assays that endogenous PAK1 and PAK3 proteins form heterodimers in mouse brain. To resume, PAK3 and PAK1 are co-expressed in single neurons, and to some extent, colocalize in dendritic spines, co-purify in post-synaptic densities and co-immunoprecipitate in mouse brain demonstrating that endogenous PAK1 and PAK3 proteins form heterodimers in neurons.
PAK3a Is Regulated by Heterodimerization
Since PAK3 can dimerize, and PAK1 is regulated by its homodimerization, we wondered whether PAK3 heterodimerization regulates its own kinase activity. We developed a multi-step assay in which we first immunoprecipitated a PAK1/PAK3 complex. The co-immunoprecipitated material was divided into two equal aliquots, the first one being directly tested for its kinase activity while the second being incubated with active recombinant Cdc42 GTPase, before testing for its kinase activity. In both cases only the activity of the co-immunoprecipitated protein was measured since the precipitated protein was a kinase-dead mutant. We verified for the presence of immunoprecipitated and co-immunoprecipitated proteins by Western blot (Fig. 6, A and B) and the dissociation of the dimers following activation by Cdc42 (data not shown), as previously described (7). Kinase activity was analyzed by measuring the incorporation of γ-32P in myelin basic protein (MBP) as a substrate. The comparison between the kinase activities of the two fractions with or without the GTPase reflects the level of inhibition of the co-immunoprecipitated partner in the non-dissociated complex. To do this, HeLa cells were co-transfected with FLAG-PAK1 kinase-dead and HA-PAK3a or -PAK3b wild type plasmids. Immunoprecipitation and Western blot analysis were performed with appropriate tag antibodies as indicated in Fig. 6B. A control was performed to confirm that PAK3a-WT has a catalytic activity, only in presence of Cdc42-V12, whereas PAK3b is constitutively activated as previously published (17, 19) (Fig. 6C, first panel). Kinase assay with the inactive kinase-dead PAK1 protein indicated the background of the test likely due to other kinases co-immunoprecipitated with the PAK complex (Fig. 6C, second panel). When PAK3a is associated with PAK1, its activity is inhibited and this inhibition is relieved after activation by Cdc42 (Fig. 6D). On the contrary, we did not observed a Cdc42-dependent increase of MBP phosphorylation upon dissociation of PAK1-KD/PAK3b dimer suggesting that PAK3b is not inhibited in dimers (Fig. 6E). These results strongly suggest that heterodimerization with PAK1 regulates PAK3a activity but not activities of splice variants.
FIGURE 6.

PAK1 regulates PAK3 activity through heterodimerization. A, control of PAK expression. HA-tagged PAK3a-WT and PAK3b-WT plasmids were transfected in HeLa cells, and cell lysates were HA-immunoprecipitated (IP HA), then the amount of PAK3 proteins was determined using HA antibodies. B, control of co-immunoprecipitation of PAK3s proteins with PAK1. FLAG-PAK1-KD plasmid was transfected in HeLa cells alone or in the presence of HA-PAK3a-WT or HA-PAK3b-WT. Cell lysates were FLAG-immunoprecipitated (IP FLAG) then the amount of PAK1 proteins was determined using FLAG antibodies (second panel). The co-immunoprecipitated PAK3a-WT and PAK3b-WT proteins were revealed by HA blotting (WB HA, first panel). TCLs were controlled using the HA antibody (third panel). C, PAK3a displays an inducible kinase activity whereas PAK3b possesses a constitutive activity. Catalytic activity of HA-PAK3a-WT; HA-PAK3b-WT and FLAG-PAK1-KD previously obtained by immunoprecipitation was tested in the presence or in the absence of recombinant Cdc42-V12. D and E, catalytic activity of complexed or dissociated PAK3 proteins. HA-PAK3a-WT (D) or HA-PAK3b-WT (E) obtained by co- immunoprecipitation (B, first panel) was analyzed after incubation with or without recombinant Cdc42-V12 protein. Images shown are representative of three experiments
DISCUSSION
Molecular Mechanisms of Regulation of PAK3
Kinase dimerization is a key regulatory mechanism in the kinome. A large range of scenarios exist in life and the importance of kinase dimerization was recently highlighted in relation to pathophysiology (34). Dimerization is a canonical step in receptor tyrosine kinase activation (35) but plays more diverse roles in serine/threonine kinases, being responsible for inactivation, activation, subcellular compartment retention or localization and substrate specification (36–38). PAK dimerization was first described ten years ago (6) and since then the PAK1 regulation mechanisms have been extensively studied (6, 7, 11, 12). Data demonstrated that, under cellular resting conditions, PAK1 exists in an autoinhibited inactive dimeric conformation (6, 7). The activation process of PAK1 requires a succession of steps: membrane recruitment, modification of domain folding, and phosphorylation of key residues that modify the conformation of the catalytic cleft and other regulatory sites leading to changes in the intermolecular assembly (1, 13, 39). The PAK1 trans-inhibition regulatory mechanism is thought to be valid also for the other kinases of group I since they share high sequence identities and functional similarities. We demonstrated here for the first time, that PAK3 is able to form homodimers, in vitro, as PAK1 does. However, another interesting question concerns the potentiality for PAK kinases to form heterodimers. It was previously mentioned (7) that PAK1 can form heterodimers with PAK2. Here we present the first evidence that PAK3 can also form heterodimers with PAK1, through interactions mainly between the N-ter domain and the C-ter domain. The L102F mutation or the R436E-K437D mutation introduced in the IS domain and the catalytic domain respectively, strongly or totally abrogate homomeric interaction of PAK3a and also the heteromeric interaction between PAK3a and PAK1. This suggests that PAK3a may form homodimers as PAK1 does, and also that heterodimeric complexes have similar structures compared with the homodimeric ones described for PAK1. Moreover, we demonstrated that PAK1 regulates PAK3a kinase activity within heterodimers, showing that PAK heterodimerization also allows trans-inhibition.
In vivo, we demonstrated that PAK1 and PAK3a are co-expressed in the same cells, they colocalize in the same subcellular area, and they also biochemically partially co-purify in the same structure, three properties necessary for their interaction. Interestingly, we observed a higher level of PAK3a/PAK1 heterodimers than PAK3a/PAK3a homodimers in vitro whereas PAK1 interacts with itself or with PAK3a with the same efficiency. Moreover, the relative amount of endogenous proteins in neurons is much more in favor of PAK3a/PAK1 heterodimer formation than PAK3a homodimer formation. Actually, all these results strongly reinforce the idea that PAK3 forms complexes with PAK1 in vivo, a hypothesis that we demonstrated by detecting PAK1/PAK3 complexes in brain immunoprecipitates.
Moreover, we report here that PAK3 splice variants can form heterodimers with PAK1. Insertions of 15, 21, and 36 amino acids encoded by the two alternatively spliced exons b and c, and present in the splice variants b, c, and cb, respectively, are localized in the β-sheet of the IS domain, and so are predicted to strongly modify the structure of the regulatory region. We previously demonstrated that the AID of PAK3b does not inhibit a PAK3a wild-type kinase domain (19). PAK3 splice variants show a relatively decreased capacity to form complexes with PAK3a and heterodimers with PAK1, but heterodimers are more efficiently formed than dimers with PAK3 suggesting that splice variants exist mainly as complexes with PAK1. In addition, we demonstrated that whereas PAK1 could regulate PAK3a kinase activity by dimerization, dimerization with PAK3 splice variants does not inhibit their kinase activity. This observation suggests that dimer formation is not sufficient to permit a trans-inhibition of one monomer by another one. It is also possible that the residual activity of PAK3b in dimers is sufficient to phosphorylate crucial residues such as Thr-421 or Ser-139 (144 for PAK1) which play important role in kinase regulation (12). Moreover this suggests that the kinase activity of splice variants is regulated by other unknown mechanisms, such as post-transcriptional modulation of splicing or protein degradation. Finally, recent data demonstrated an asymmetry in the PAK dimer complex, suggesting that the two monomers have distinct roles in the molecular mechanisms of kinase inhibition (11). Since we report that PAK dimers contain one PAK1 and one PAK3 monomers, this questions whether the two isoforms have distinct roles in the asymmetric complex, or there is a random repartition of the two distinct roles.
Biological Consequence of Dimerization
Our data suggest that PAK3 and PAK1 signaling are linked together, at least until the moment of their activation. Interestingly, PAK1 protein is also involved in synaptic plasticity via regulation of cofilin phosphorylation and actin cytoskeleton modeling (40) whereas PAK3 probably acts through other pathways involving CREB and Nck2 (5, 30). A recent study that characterized the pak1/pak3 double KO mice pointed out new neuronal defects due to an impaired post-natal brain growth (41). Altogether, these data suggest that in addition to unique functions and to shared functions, some PAK functions may depend on a crosstalk between PAK1 and PAK3 during the activation process. As a consequence, PAK1-dependent PAK3 regulation could coordinate their specific signaling pathway and help to synchronize actin polymerization and spine stabilization.
In addition, a consequence of the link between PAK1 and PAK3 signaling concerns the relationship between intelligence disability PAK3 linked ID mutations and the alteration of PAK1 signaling. The five ID mutations which have been shown to modify different properties of PAK3 such as the kinase activity, the binding to the Rac-Cdc42 GTPases (24), or the binding to Nck2/Grb4 (5), and which are associated with moderate to severe cognitive defects (reviewed in (20)), differently affect dimer formation. We observed that the R67C mutated protein does not alter dimerization with PAK1 in contrast with the two kinase-dead ID mutated proteins. Since the PAK3-R67C protein displays a defect in GTPase binding and a consequential defect in the activation of the kinase (24), the PAK3-R67C protein in complex with PAK1 will probably maintain PAK1 inactive. Thus, the more severe phenotype associated with the R67C mutation could be due to a defect of PAK1 activation, in addition to partial PAK3 impairment, supporting the idea that some mutated PAK3 proteins may act as dominant-negative proteins with respect to PAK1. In contrast, PAK3-A365E and PAK3-R419X mutations that alter the PAK3 ability to dimerize with PAK1 could favor PAK1 to form homodimers and thus these mutations would not impact PAK1 regulation. So, some phenotypic characteristics associated with pak3 mutations may be due to anomalies in PAK1 regulation in addition to the PAK3 signaling defect.
To conclude, it appears that PAK regulation is a complex process that involves homodimer and heterodimer formation dependent upon intra and inter molecular interactions, in a face-to-face conformation, allowing a dynamic succession of trans- and cis- phosphorylation events associated with spatial rearrangements of several domains of the protein, also dependent on activators such as active GTPases, or to a lesser extent on the guanine exchange factor of the PIX/Cool family, on membrane interaction, and on lipid association. We report here a novel level of complexity in one of these parameters, demonstrating that a combinatory of dimer assembly exists, that a hierarchy in complex formation exists, and that these heteromeric interactions may have a functional role. We show that PAK isoforms do not have the same propensity to form homodimers versus heterodimers. Thus, in addition to the concept that heterodimers support a signaling network of related kinases (6), we propose that heterodimers may link together PAK1 and PAK3 signaling pathways in physiological as well as in pathophysiological conditions.
Supplementary Material
Acknowledgments
We thank Zhengping Jia for providing us with the pak3 KO strain, Maria-Carla Parrini for the PAK1 WT plasmid, Herma Renkema for the PAK2 plasmid, and Nicolas Morel for the synaptophysin antibody. We also thank Seana O'Regan for helpful comments on the manuscript. We are grateful to Nathalie Samson and Pascale Veyrac (CNRS, France) for animal care, Sandrine Poëa-Guyon for help in confocal imaging, and Bernadette Wiszniowski for technical assistance.
This work was supported in part by a grant from the Agence Nationale de la Recherche (ANR-MNP-2009) and the Fondation Jérôme Lejeune.

This article contains three supplemental figures and one supplemental table.
- PAK
- p21-activated kinase
- PBD
- p21-binding domain
- AID
- auto-inhibitory domain
- PSD
- post-synaptic density
- DIV
- days in vitro
- TCL
- total cell lysate
- TBL
- total brain lysate
- KD
- kinase-dead
- PAK3sv
- PAK3 splice variant
- KD
- kinase dead.
REFERENCES
- 1. Bokoch G. M. (2003) Biology of the p21-activated kinases. Annu. Rev. Biochem. 72, 743–781 [DOI] [PubMed] [Google Scholar]
- 2. Burbelo P. D., Drechsel D., Hall A. (1995) A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J. Biol. Chem. 270, 29071–29074 [DOI] [PubMed] [Google Scholar]
- 3. Bagrodia S., Taylor S. J., Jordon K. A., Van Aelst L., Cerione R. A. (1998) A novel regulator of p21-activated kinases. J. Biol. Chem. 273, 23633–23636 [DOI] [PubMed] [Google Scholar]
- 4. Manser E., Huang H. Y., Loo T. H., Chen X. Q., Dong J. M., Leung T., Lim L. (1997) Expression of constitutively active α-PAK reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol. 17, 1129–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thévenot E., Moreau A. W., Rousseau V., Combeau G., Domenichini F., Jacquet C., Goupille O., Amar M., Kreis P., Fossier P., Barnier J. V. (2011) p21-Activated kinase 3 (PAK3) protein regulates synaptic transmission through its interaction with the Nck2/Grb4 protein adaptor. J. Biol. Chem. 286, 40044–40059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lei M., Lu W., Meng W., Parrini M. C., Eck M. J., Mayer B. J., Harrison S. C. (2000) Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell 102, 387–397 [DOI] [PubMed] [Google Scholar]
- 7. Parrini M. C., Lei M., Harrison S. C., Mayer B. J. (2002) Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol. Cell 9, 73–83 [DOI] [PubMed] [Google Scholar]
- 8. Zenke F. T., King C. C., Bohl B. P., Bokoch G. M. (1999) Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J. Biol. Chem. 274, 32565–32573 [DOI] [PubMed] [Google Scholar]
- 9. Buchwald G., Hostinova E., Rudolph M. G., Kraemer A., Sickmann A., Meyer H. E., Scheffzek K., Wittinghofer A. (2001) Conformational switch and role of phosphorylation in pak activation. Mol. Cell. Biol. 21, 5179–5189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pirruccello M., Sondermann H., Pelton J. G., Pellicena P., Hoelz A., Chernoff J., Wemmer D. E., Kuriyan J. (2006) A dimeric kinase assembly underlying autophosphorylation in the p21 activated kinases. J. Mol. Biol. 361, 312–326 [DOI] [PubMed] [Google Scholar]
- 11. Wang J., Wu J. W., Wang Z. X. (2011) Structural insights into the autoactivation mechanism of p21-activated protein kinase. Structure 19, 1752–1761 [DOI] [PubMed] [Google Scholar]
- 12. Chong C., Tan L., Lim L., Manser E. (2001) The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J. Biol. Chem. 276, 17347–17353 [DOI] [PubMed] [Google Scholar]
- 13. Parrini M. C., Camonis J., Matsuda M., de Gunzburg J. (2009) Dissecting activation of the PAK1 kinase at protrusions in living cells. J. Biol. Chem. 284, 24133–24143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Allen K. M., Gleeson J. G., Bagrodia S., Partington M. W., MacMillan J. C., Cerione R. A., Mulley J. C., Walsh C. A. (1998) PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 20, 25–30 [DOI] [PubMed] [Google Scholar]
- 15. Bienvenu T., des Portes V., McDonell N., Carrié A., Zemni R., Couvert P., Ropers H. H., Moraine C., van Bokhoven H., Fryns J. P., Allen K., Walsh C. A., Boué J., Kahn A., Chelly J., Beldjord C. (2000) Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am. J. Med. Genet. 93, 294–298 [DOI] [PubMed] [Google Scholar]
- 16. Gedeon A. K., Nelson J., Gecz J., Mulley J. C. (2003) X-linked mild non-syndromic mental retardation with neuropsychiatric problems and the missense mutation A365E in PAK3. Am. J. Med. Genet. 120, 509–517 [DOI] [PubMed] [Google Scholar]
- 17. Kreis P., Rousseau V., Thévenot E., Combeau G., Barnier J. V. (2008) The four mammalian splice variants encoded by the p21-activated kinase 3 gene have different biological properties. J. Neurochem. 106, 1184–1197 [DOI] [PubMed] [Google Scholar]
- 18. Arias-Romero L. E., Chernoff J. (2008) A tale of two Paks. Biol. Cell 100, 97–108 [DOI] [PubMed] [Google Scholar]
- 19. Rousseau V., Goupille O., Morin N., Barnier J. V. (2003) A new constitutively active brain PAK3 isoform displays modified specificities toward Rac and Cdc42 GTPases. J. Biol. Chem. 278, 3912–3920 [DOI] [PubMed] [Google Scholar]
- 20. Kreis P., Barnier J. V. (2009) PAK signaling in neuronal physiology. Cell. Signal. 21, 384–393 [DOI] [PubMed] [Google Scholar]
- 21. Molli P. R., Li D. Q., Murray B. W., Rayala S. K., Kumar R. (2009) PAK signaling in oncogenesis. Oncogene 28, 2545–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dummler B., Ohshiro K., Kumar R., Field J. (2009) Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 28, 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morel N., Dedieu J. C., Philippe J. M. (2003) Specific sorting of the a1 isoform of the V-H+ATPase a subunit to nerve terminals where it associates with both synaptic vesicles and the presynaptic plasma membrane. J. Cell Sci. 116, 4751–4762 [DOI] [PubMed] [Google Scholar]
- 24. Kreis P., Thévenot E., Rousseau V., Boda B., Muller D., Barnier J. V. (2007) The p21-activated kinase 3 implicated in mental retardation regulates spine morphogenesis through a Cdc42-dependent pathway. J. Biol. Chem. 282, 21497–21506 [DOI] [PubMed] [Google Scholar]
- 25. Crouin C., Arnaud M., Gesbert F., Camonis J., Bertoglio J. (2001) A yeast two-hybrid study of human p97/Gab2 interactions with its SH2 domain-containing binding partners. FEBS Lett. 495, 148–153 [DOI] [PubMed] [Google Scholar]
- 26. Poëa-Guyon S., Amar M., Fossier P., Morel N. (2006) Alternative splicing controls neuronal expression of v-ATPase subunit a1 and sorting to nerve terminals. J. Biol. Chem. 281, 17164–17172 [DOI] [PubMed] [Google Scholar]
- 27. Sans N., Racca C., Petralia R. S., Wang Y. X., McCallum J., Wenthold R. J. (2001) Synapse-associated protein 97 selectively associates with a subset of AMPA receptors early in their biosynthetic pathway. J. Neurosci. 21, 7506–7516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Standley S., Roche K. W., McCallum J., Sans N., Wenthold R. J. (2000) PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 28, 887–898 [DOI] [PubMed] [Google Scholar]
- 29. Boda B., Alberi S., Nikonenko I., Node-Langlois R., Jourdain P., Moosmayer M., Parisi-Jourdain L., Muller D. (2004) The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus. J. Neurosci. 24, 10816–10825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meng J., Meng Y., Hanna A., Janus C., Jia Z. (2005) Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J. Neurosci. 25, 6641–6650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rex C. S., Lin C. Y., Kramár E. A., Chen L. Y., Gall C. M., Lynch G. (2007) Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J. Neurosci. 27, 3017–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hayashi M. L., Choi S. Y., Rao B. S., Jung H. Y., Lee H. K., Zhang D., Chattarji S., Kirkwood A., Tonegawa S. (2004) Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron 42, 773–787 [DOI] [PubMed] [Google Scholar]
- 33. Dubos A., Combeau G., Bernardinelli Y., Barnier J. V., Hartley O., Gaertner H., Boda B., Muller D. (2012) Alteration of synaptic network dynamics by the intellectual disability protein PAK3. J. Neurosci. 32, 519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wan P. T., Garnett M. J., Roe S. M., Lee S., Niculescu-Duvaz D., Good V. M., Jones C. M., Marshall C. J., Springer C. J., Barford D., Marais R. (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867 [DOI] [PubMed] [Google Scholar]
- 35. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yatime L., Hein K. L., Nilsson J., Nissen P. (2011) Structure of the RACK1 dimer from Saccharomyces cerevisiae. J. Mol. Biol. 411:486–498 [DOI] [PubMed] [Google Scholar]
- 37. Wimmer R., Baccarini M. (2010) Partner exchange: protein-protein interactions in the Raf pathway. Trends Biochem. Sci. 35, 660–668 [DOI] [PubMed] [Google Scholar]
- 38. Hatzivassiliou G., Song K., Yen I., Brandhuber B. J., Anderson D. J., Alvarado R., Ludlam M. J., Stokoe D., Gloor S. L., Vigers G., Morales T., Aliagas I., Liu B., Sideris S., Hoeflich K. P., Jaiswal B. S., Seshagiri S., Koeppen H., Belvin M., Friedman L. S., Malek S. (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435 [DOI] [PubMed] [Google Scholar]
- 39. Eswaran J., Soundararajan M., Kumar R., Knapp S. (2008) UnPAKing the class differences among p21-activated kinases. Trends Biochem. Sci. 33, 394–403 [DOI] [PubMed] [Google Scholar]
- 40. Asrar S., Meng Y., Zhou Z., Todorovski Z., Huang W. W., Jia Z. (2009) Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 56, 73–80 [DOI] [PubMed] [Google Scholar]
- 41. Huang W., Zhou Z., Asrar S., Henkelman M., Xie W., Jia Z. (2011) p21-Activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol. Cell. Biol. 31, 388–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.