

Background: Two flexible hinges may control electron transfer and catalysis in NOS enzymes.
Results: Shortening or extending the FMN-FAD/NADPH hinge lowered NO synthesis, altered electron transfer, and uncoupled NADPH consumption.
Conclusion: Native hinge length achieves a best compromise between NADPH oxidation, electron transfer, and NO synthesis.
Significance: Determining how hinge length impacts catalysis helps reveal enzyme structure-activity relationships in NOS.
Keywords: Electron Transfer, Enzyme Catalysis, Flavoproteins, Nitric Oxide, Nitric-oxide Synthase, Conformational Equilibrium, Flavin Reduction, Hemeprotein, Hinge, Pre-steady-state Kinetics
Abstract
In nitric-oxide synthases (NOSs), two flexible hinges connect the FMN domain to the rest of the enzyme and may guide its interactions with partner domains for electron transfer and catalysis. We investigated the role of the FMN-FAD/NADPH hinge in rat neuronal NOS (nNOS) by constructing mutants that either shortened or lengthened this hinge by 2, 4, and 6 residues. Shortening the hinge progressively inhibited electron flux through the calmodulin (CaM)-free and CaM-bound nNOS to cytochrome c, whereas hinge lengthening relieved repression of electron flux in CaM-free nNOS and had no impact or slowed electron flux through CaM-bound nNOS to cytochrome c. How hinge length influenced heme reduction depended on whether enzyme flavins were pre-reduced with NADPH prior to triggering heme reduction. Without pre-reduction, changing the hinge length was deleterious; with pre-reduction, the hinge shortening was deleterious, and hinge lengthening increased heme reduction rates beyond wild type. Flavin fluorescence and stopped-flow kinetic studies on CaM-bound enzymes suggested hinge lengthening slowed the domain-domain interaction needed for FMN reduction. All hinge length changes lowered NO synthesis activity and increased uncoupled NADPH consumption. We conclude that several aspects of catalysis are sensitive to FMN-FAD/NADPH hinge length and that the native hinge allows a best compromise among the FMN domain interactions and associated electron transfer events to maximize NO synthesis and minimize uncoupled NADPH consumption.
Introduction
Nitric oxide (NO) is a biological mediator and is produced in animals by three NO synthase (NOS)2 isozymes, (EC 1.14.13.39): inducible NOS (iNOS), neuronal NOS (nNOS), and endothelial NOS (eNOS) (1–4). All NOS are modular enzymes comprised of an N-terminal oxygenase domain (NOSoxy) and a C-terminal flavoprotein domain, with a calmodulin (CaM)-binding site connecting the two domains (5, 6). The NOS enzymes have different gene expression patterns, protein interactions, post-translational modifications, and catalytic behaviors that enable specific roles in biology (7–11).
During catalysis, the NOS flavoprotein domain transfers NADPH-derived electrons to a heme located in the NOSoxy domain. This enables heme-dependent oxygen activation and a stepwise conversion of l-Arg to NO and citrulline (12, 13). CaM binding triggers heme reduction in eNOS and nNOS (14–18) and also increases electron flux through the flavoprotein domain as demonstrated by its increasing the cytochrome c reductase activity of either enzyme (18, 19). The NOS flavoprotein domain is itself comprised of two separate domains, one that binds NADPH and FAD (FAD/NADPH domain) and another that binds FMN (FMN domain). The FMN domain accepts electrons from the FAD/NADPH domain and can then transfer the electrons to the NOSoxy domain or to an external acceptor like cytochrome c. Electron shuttling by the FMN domain involves dynamic conformational changes in which the FMN domain switches between an electron-accepting state (FMN and FAD/NADPH domains in complex, FMN-shielded) and an electron-donating state (FMN domain free from the FAD/NADPH domain, FMN-deshielded) (Fig. 1) (20–23). CaM binding shifts the conformational equilibrium toward the FMN-deshielded state (21, 24), and this change is associated with CaM increasing electron flux through the NOS flavoprotein domain. Electron transfer through the related dual-flavin enzymes cytochrome P450 reductase (CPR), methionine synthase reductase, and novel reductase 1 (NR1) also likely involves similar conformational motions of their FMN domains (25–28).
FIGURE 1.

Model of FMN domain function in electron transfer and heme reduction. Two hinge elements (H1 and H2) connect the FMN domain in NOS enzymes. The interactions required for electron transfer can be described by a two-equilibrium, three-state model. In equilibrium A, nNOS fluctuates between a closed conformation competent for FMN-FAD/NADPH electron transfer (FMN-shielded, left) and an open conformation where the FMN domain can interact with electron acceptors such as cytochrome c (FMN-deshielded, center). In equilibrium B, nNOS fluctuates between the FMN-deshielded conformation and a conformation competent for FMN to heme electron transfer. Complementary surface charge interactions (gray, negative, and black, positive) are involved in the domain interactions. See text for details.
In NOS enzymes, two flexible hinges connect the FMN domain to the rest of the enzyme (Fig. 2A). During catalysis, these hinge elements may guide FMN domain interactions with its FAD/NADPH domain and NOSoxy domain partners (20, 22, 29–32) and in this way help determine electron flux through the flavoprotein, the rate of heme reduction (kr), and the NO synthesis activity. The hinge connecting the FMN and FAD/NADPH domains in NOS enzymes (FMN-FAD/NADPH hinge; Fig. 2A) is also present in related dual-flavin reductases. The FMN-FAD/NADPH hinge has a varied sequence composition among NOS enzymes (Fig. 2B) and is two residues longer in nNOS than in eNOS or iNOS. In our previous study, we found that incorporating the FMN-FAD/NADPH hinge of nNOS into eNOS increased its NO synthesis activity 4-fold to a level that is two-thirds that of nNOS. It also decreased uncoupled NADPH oxidation and supported faster heme reduction (19), establishing that the composition and/or length of the FMN-FAD/NADPH hinge is important for determining electron transfer and catalysis in NOS. To more systematically investigate the role of the FMN-FAD/NADPH hinge, we constructed a series of nNOS mutants that either shorten or lengthen its 25-residue FMN-FAD/NADPH hinge by 2, 4, and 6 residues (Fig. 2C). The deletions or insertions ((Ala-Ser) × 1, 2, or 3) were done near the middle of the FMN-FAD/NADPH hinge and designed to change hinge length while having a minimal impact on hinge flexibility. Our results provide a comprehensive picture of how FMN-FAD/NADPH hinge length impacts the electron transfer, protein conformational, and catalytic properties of nNOS.
FIGURE 2.

Hinge elements connecting the FMN domain in NOS. A, diagram of NOS subunit composition emphasizing the two connecting hinges (Oxy-FMN and FMN-FAD/NADPH). AI, autoinhibitory insert; CT, C-terminal tail. B, sequence alignment of the FMN-FAD/NADPH hinges of various NOS enzymes. C, composition of the FMN-FAD/NADPH hinge mutants of nNOS. The top line lists the amino acid sequence of the FMN-FAD/NADPH hinge in wild-type rat nNOS. The H+2, H+4, and H+6 mutants have the indicated two, four, and six amino acids inserted at the indicated position in hinge. The HΔ2, HΔ4, and HΔ6 deletion mutants are missing amino acids indicated by gaps in the sequence.
EXPERIMENTAL PROCEDURES
General Methods and Materials
All reagents and materials were obtained from Sigma, Amersham Biosciences, or other sources as reported previously (19, 33–35). Absorption spectra and steady-state kinetic data were obtained using a Shimadzu UV-2401PC spectrophotometer. All plots and some additional curve-fitting were done using Origin® 8.0 (OriginLab, Northampton, MA). All experiments were repeated two or more times with at least two independently prepared batches of proteins to ensure consistent reproducibility of the results. Data were analyzed and are expressed as mean ± S.D.
Molecular Biology
Wild-type and mutant nNOS proteins containing a His6 tag attached to their N termini were overexpressed in Escherichia coli strain BL21(DE3) using a modified pCWori vector as described (35, 36). Restriction digestions, cloning, and bacterial growth were performed using standard procedures. Transformations were performed using a TransformAid bacterial transformation kit (Fermentas, Hanover, MD). Oligonucleotides used to construct mutants in nNOS were obtained from Integrated DNA Technologies (Coralville, IA). To delete/extend amino acids in the nNOS hinge region, we used PCR oligonucleotides containing an AatII site (5′-oligonucleotides are listed in supplemental Table S1) and a 3′-oligonucleotide, CGT GGG CGG CGT GGT GAT, to amplify nNOS between bases corresponding to amino acids 942 and 1105. The PCR products (∼450 bp) and the pCW nNOS vector were digested with AatII and BsmI restriction enzymes. The digested PCR fragment (∼200 bp) and vector were gel-purified and ligated into the vector replacing the wild-type hinge region. All mutated constructs were confirmed by DNA sequencing at the Cleveland Clinic Genomics Core.
Expression and Purification of Wild-type and Mutant Proteins
All proteins were purified in the presence of H4B and l-Arg as described previously. The ferrous heme-CO adduct absorbing at 444 nm was used to measure hemeprotein content with an extinction coefficient of ϵ444 = 74 mm−1 cm−1 (A444 − A500) (37). Purity of each protein was assessed by SDS-PAGE and spectral analysis.
NO Synthesis, NADPH Oxidation, and Cytochrome c Reduction
Steady-state activities of wild-type and mutant proteins were determined separately at 25 °C by using spectrophotometric assays that were described in detail previously (33, 35). For the electron flux measurement through the NOS heme during steady-state catalysis, we measured the rate of NADPH oxidation by each CaM-bound enzyme in the absence of arginine substrate or in the presence of 2 mm agmatine (19, 38).
Anaerobic Heme Reduction Measurements
The kinetics of heme reduction in CaM-free or CaM-bound enzymes were analyzed at 10 °C as described previously (33, 35, 37–39) using a stopped-flow apparatus and diode array detector (TGK Scientific KinetAsyst SF-61DX2) equipped for anaerobic analysis. Ferric heme reduction was followed by formation of the ferrous heme-CO complex at 444 nm. For proteins that exhibited slower flavin reduction kinetics than the wild-type nNOS (hinge 2, 4, and 6 extension mutants), their apparent heme reduction rates were adjusted to take into account the concurrent loss of 444 nm absorbance due to ongoing flavin reduction.
Anaerobic Flavin Reduction Measurements
The kinetics of flavin reduction were monitored at 485 nm at 10 °C by using a stopped-flow apparatus and diode array detector (TGK Scientific KinetAsyst SF-61DX2). Reactions were initiated by rapidly mixing an anaerobic buffered solution containing 100 μm NADPH with an anaerobic buffered solution containing wild-type or mutant nNOS (∼10 μm), 100 mm EPPS (pH 7.6), 100 mm NaCl, 20 μm (6R)-tetrahydrobiopterin, 0.3 mm DTT, 30 μm CaM, 2 mm CaCl2, and 1 mm l-N-nitro-arginine methyl ester (l-NAME; added to block any concurrent heme reduction). Signal-to-noise ratios were improved by averaging 8–10 individual mixing experiments. The time course of the absorbance change was fit to double exponential equations by using a nonlinear least squares method provided by Hi-Tech Scientific, under the constraint where the 1st and 2nd phases each represented 50% of the total absorbance change at 485 nm.
Steady-state Flavin Reduction
Wild-type and mutant nNOS enzymes were diluted to 4 μm in a cuvette that contained oxygen-saturated buffer, H4B, Ca2+, CaM, and other additives as used for the NO synthesis assay, except that here oxyhemoglobin and NADPH were omitted and 2 mm l-agmatine replaced l-Arg (34). A spectrum was collected, and then a solution containing NADPH, 5 units of glucose-6-phosphate dehydrogenase, and 4 mm glucose 6-phosphate (enzymatic NADPH-regenerating system) was added to maintain 100 μm NADPH, and a second spectrum was collected during NADPH and O2 consumption. A final spectrum was collected after O2 depletion. Subtraction of these spectra provided the percentage of reduced flavins during the steady state.
Fluorescence Spectroscopy
Flavin fluorescence measurements were done using a Hitachi model F-2500 spectrofluorometer as described previously (40). A 1-ml cuvette with a path length of 1 cm was used. Dilution effects were less than 0.5%, and the samples were maintained at 25 °C during measurement. The wild-type and mutant nNOS proteins were diluted to 2 μm in 40 mm EPPS (pH 7.6), containing 0.6 mm EDTA, 1 mm DTT, and 12 μm CaM. Proteins were irradiated with 457 nm light, and flavin fluorescence emission was monitored at 520 nm versus time before and after addition of 2 mm Ca2+ followed by 4 mm EDTA.
RESULTS
General Properties of the FMN-FAD/NADPH Hinge Mutants
All the hinge mutants had normal protein expression level and a normal content of bound flavins and heme as judged by their UV-visible spectral features (supplemental Fig. S1), indicating they were suitable to evaluate the role of FMN-FAD/NADPH hinge length in nNOS function.
Cytochrome c Reductase Activity
Electron flux through the nNOS flavoprotein domain can be measured by the NADPH-dependent cytochrome c reductase activity. The electron flux through nNOS is repressed in the CaM-free state, and the repression is relieved by CaM binding (41). We compared the steady-state cytochrome c reductase activities of each mutant in the absence or presence of bound CaM (Fig. 3, upper and lower panels). All assays contained superoxide dismutase to ensure that we measured only the direct electron transfer from the FMN hydroquinone (FMNH2 or FMNhq) in nNOS to cytochrome c (20, 42). For the CaM-free enzyme, shortening the FMN-FAD/NADPH hinge by 2, 4, and 6 residues resulted in ∼50–75% lower reductase activities relative to wild-type nNOS. In contrast, extending the hinge length by 2 residues had no effect, and longer extensions of 4 and 6 residues caused a 2-fold increase in the cytochrome c reductase activity.
FIGURE 3.

Rate of steady-state cytochrome c reduction versus FMN-FAD/NADPH hinge length in nNOS. Rates were measured at 25 °C either in the absence (upper panel) or presence (lower panel) of bound CaM. Assay conditions are described under “Experimental Procedures.” The activities of wild-type nNOS are boxed and represented by the dashed lines. Values represent the mean and standard deviations of at least five independent measurements.
In the CaM-bound nNOS, shortening the hinge by 4 or 6 residues caused an approximate 50% drop in the reductase activity, whereas lengthening the hinge had no effect except with the longest extension (6 residues), which caused an ∼2.5-fold drop in activity. The data establish that the FMN-FAD/NADPH hinge length helps control electron flux through the nNOS flavoprotein. Shortening the hinge inhibits electron flux through both the CaM-free and CaM-bound enzymes, whereas lengthening the hinge relieves the repression on electron flux that is present in the CaM-free enzyme and has either no impact or is deleterious for electron flux through the CaM-bound enzyme.
Rates of Heme Reduction
We next compared heme reduction rates in the hinge mutants and wild-type enzyme (35, 43). We used CO binding to detect heme reduction under anaerobic conditions at 10 °C and used two different protocols to initiate heme reduction in our stopped-flow spectrophotometer. In one case, we initiated the reaction by mixing excess NADPH with each CaM-bound ferric enzyme. Under this condition, NADPH reduction of the FAD and FMN groups, which is normally fast, must take place before the electron transfer from FMNhq to the heme can occur. In the second case, we pre-reduced each CaM-free enzyme with NADPH just prior to our initiating heme reduction by mixing the NADPH-pre-reduced enzyme with excess Ca2+/CaM to trigger CaM binding. Under this condition, the flavins are already reduced by the NADPH pre-treatment, and electron transfer from FMNhq to the heme may begin as soon as CaM binds to the enzyme. The measured rates are compared in Fig. 4 (kinetic traces and rate values are shown in supplemental Fig. S2 and supplemental Table S2). The heme reduction rate was almost unaffected by shortening the FMN-FAD/NADPH hinge by 2 or 4 residues or by increasing the hinge length by 2 residues. Shortening the hinge by 6 residues caused a modest (20–30%) inhibition according to both heme reduction protocols. Interestingly, lengthening the hinge had opposite effects on the heme reduction rate depending on which experimental protocol was used. When heme reduction was initiated by mixing CaM-bound enzyme with NADPH, lengthening the hinge progressively inhibited the heme reduction rate. However, when heme reduction was initiated by triggering CaM binding to the NADPH pre-reduced enzyme, the hinge extension mutants had increased rates of heme reduction compared with wild type. Conceivably, such results could result if the NADPH pre-treatment had caused a significant extent of heme reduction to occur in the anaerobic enzyme samples prior to their being mixed with CaM to trigger heme reduction. To test this, we directly examined the rate and extent of heme reduction in the CaM-free wild-type enzyme and in the 2-, 4-, and 6-residue hinge extension mutants when they were mixed with NADPH under the anaerobic condition. As shown in supplemental Fig. S3, all four proteins exhibited a delayed build up of reduced heme under this condition, but the process was slow enough to preclude build up of a significant amount of ferrous enzyme within the time frame of our mixing protocol when we measured CaM-triggered heme reduction. Together, our results show that heme reduction in nNOS is sensitive to changes in FMN-FAD/NADPH hinge length. Moreover, increasing the hinge length is only detrimental when heme reduction is initiated with NADPH, and it is beneficial when heme reduction is triggered by CaM binding to NADPH pre-reduced enzymes.
FIGURE 4.

Rate of ferric heme reduction versus FMN-FAD/NADPH hinge length. All reactions were performed at 10 °C in a stopped-flow spectrophotometer as described under “Experimental Procedures.” Heme reduction was triggered by either NADPH or CaCl2 addition. The rates observed for wild-type nNOS are boxed. Values are the means ± S.D. of 7–10 individual reactions and are representative of experiments done with two enzyme preparations for each.
Kinetics of Flavin Reduction by NADPH
We next determined if changes in hinge length were impacting flavin reduction kinetics. The CaM-bound enzymes were rapidly mixed with excess NADPH under anaerobic conditions in a stopped-flow spectrophotometer. Fig. 5 (A and B) compares the 485 nm absorbance traces derived from the diode array data sets for each hinge-shortened (Fig. 5A) and each hinge-lengthened (Fig. 5B) mutant relative to wild-type. The traces could be described as biphasic in all cases. We fit the data under the added constraint that each phase was associated with ∼50% of the total absorbance change. The resulting rate constants are listed in Table 1. Hinge deletions had relatively little impact on flavin reduction kinetics relative to wild type (Fig. 5, A and C). In contrast, all the hinge extension mutants had similar initial rates of flavin reduction but had progressively slower second phase rates relative to wild type (Fig. 5, B and C). This suggests that the hinge-lengthened mutants developed a kinetic barrier to further reduction after their FAD received two electrons from the first NADPH hydride transfer.
FIGURE 5.
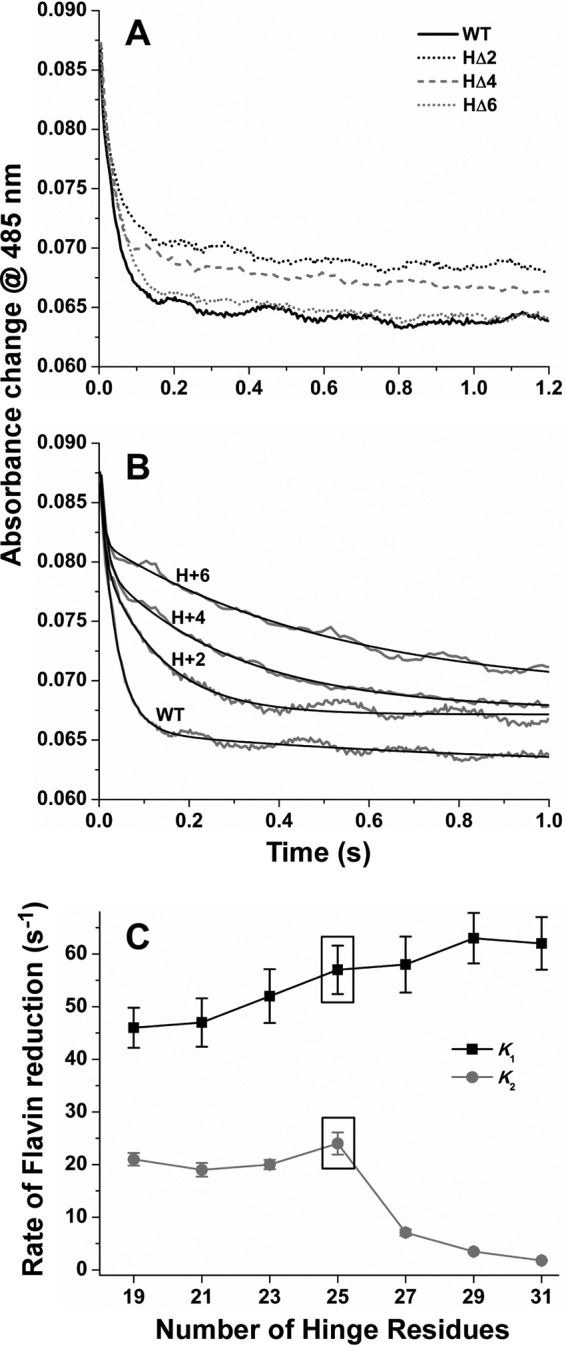
Kinetics of flavin reduction in wild-type nNOS and FMN-FAD/NADPH hinge mutants. nNOS enzymes were rapidly mixed with excess NADPH at 10 °C under anaerobic conditions in a stopped-flow spectrophotometer to initiate the reaction. Traces show time-dependent absorbance loss at 485 nm associated with flavin reduction in CaM-bound enzymes (A and B) and solid calculated lines of best fit (B). Results are representative of two separate trials. C shows the two rates (k1 and k2) of flavin reduction versus FMN-FAD/NADPH hinge length.
TABLE 1.
Kinetics of nNOS flavin reduction by NADPH and levels of reduced flavins maintained during the steady state
Rates of flavin reduction in CaM-bound nNOS proteins were determined at 10 °C under anaerobic conditions in a stopped-flow spectrophotometer as described under “Experimental Procedures”. The absorbance traces at 485 nm were fit to a double exponential equation (k1 and k2 refer to the rates for biphasic reactions) under the constraint where the 1st and 2nd phases each represented 50% of the total absorbance change at 485 nm. The flavin reduction values are the mean ± S.D. of 7–10 individual reactions. The percentage of reduced flavins in CaM-bound enzymes during their steady-state NADPH consumption was determined spectroscopically as described under “Experimental Procedures.” The percentages are representative of three independent trials. HΔ2 indicates 2-residue deletion in hinge; HΔ4, indicates 4-residue deletion in hinge; HΔ6, indicates 6-residue deletion in hinge; H+2, indicates 2-residue insertion in hinge; H+4, indicates 4-residue insertion in hinge; H+6, indicates 6-residue insertion in hinge.
| Protein | Flavin reduction rate | Reduced flavins during the steady state |
|---|---|---|
| s−1 | % | |
| WT nNOS | k1 = 57 ± 4.6 (50%) | 33 ± 3.0 |
| k2 = 24 ± 2.1 (50%) | ||
| HΔ2 nNOS Fl | k1 = 52 ± 5.1 (50%) | 46 ± 3.9 |
| k2 = 20 ± 0.90 (50%) | ||
| HΔ4 nNOS Fl | k1 = 47 ± 4.6 (50%) | 38 ± 3.8 |
| k2 = 19 ± 1.3 (50%) | ||
| HΔ6 nNOS Fl | k1 = 46 ± 3.8 (50%) | 37 ± 2.1 |
| k2 = 21 ± 1.2 (50%) | ||
| H+2 nNOS Fl | k1 = 58 ± 5.3 (50%) | 72 ± 6.6 |
| k2 = 7.1 ± 0.6 (50%) | ||
| H+4 nNOS Fl | k1 = 63 ± 4.8 (50%) | 60 ± 5.9 |
| k2 = 3.5 ± 0.21 (50%) | ||
| H+6 nNOS Fl | k1 = 62 ± 5 (50%) | 66 ± 4.3 |
| k2 = 1.8 ± 0.1 (50%) |
Flavin Fluorescence Measures to Examine the nNOS Conformational Equilibrium
The flavin fluorescence emission in nNOS is indicative of the position of its reductase domain conformational equilibrium, with greater fluorescence emission indicating a more open structure (Fig. 1) (40). The flavin fluorescence emission profiles of wild-type and mutant nNOS proteins are compared in supplemental Fig. S4. In these traces, the basal flavin fluorescence for each CaM-free protein is first recorded; then Ca2+ is added to allow CaM binding, and finally excess EDTA is added to cause CaM dissociation. In wild-type nNOS, CaM binding caused flavin fluorescence to increase, matching previous reports (40), implying that CaM shifts the nNOS conformational equilibrium to a more open form. The basal and CaM-bound fluorescence values of most of the hinge mutants were similar to wild type. However, the 6-residue hinge extension mutant had a higher basal flavin fluorescence emission that was near the level achieved with CaM binding. The results imply that in the flavin-oxidized state, most of the hinge mutants maintain a conformational set point and CaM response that is close to that of wild-type nNOS, but the 6-residue extension mutant has an altered conformational set point that favors a more open state.
Steady-state Electron Flux through Heme
We next examined how hinge length would impact electron flux through the nNOS heme, under conditions where O2 acts as the electron acceptor in the absence of NO synthesis. To achieve this, we measured the rate of NADPH oxidation by each CaM-bound enzyme in the presence of l-agmatine, which binds in the l-Arg-binding site without serving as a substrate for NO synthesis. This eliminates NO feedback inhibition of the electron flux that normally occurs when nNOS catalyzes NO synthesis from l-arginine (19, 38, 44). Thus, the NADPH oxidation rate in the presence of l-agmatine reports on the steady-state electron flux through the nNOS heme to O2 in the CaM-bound enzyme, which should be proportional to the rate and extent of heme reduction under the same condition. The NADPH consumption rates we obtained in the presence of l-agmatine are listed in supplemental Table S3. Fig. 6 plots these steady-state rates against the corresponding rates of heme reduction that we obtained for each protein under the NADPH-triggered protocol (see supplemental Table S2 and Fig. 4).3 A very good correlation was observed (r2 = 0.984) across the series. This implies that the FMN-FAD/NADPH hinge deletions or extensions alter the steady-state electron flux through nNOS primarily by altering the heme reduction rate.
FIGURE 6.

Correlation between the heme reduction rate and the NADPH oxidation rate for nNOS and hinge mutants. Points were calculated using the rates of NADPH-triggered heme reduction and the rates of steady-state NADPH oxidation in the presence of l-agmatine that were measured at 10 and 25 °C, respectively, as described under “Experimental Procedures.” The slope and fit of the line are indicated.
Corresponding Levels of Flavin Reduction during Steady-state Electron Flux
We next recorded UV-visible scans from 350 to 700 nm before and after initiating the steady-state NADPH oxidation by each CaM-bound enzyme in the presence of l-agmatine. From the traces we created difference spectra that allowed us to measure the percent of flavin reduction maintained by each CaM-bound nNOS protein during steady-state electron flux through the heme. Spectra obtained for the wild type and the 2-, 4-, and 6-residue hinge extension mutants are shown in Fig. 7, and the rest are shown in supplemental Fig. S5; Table 1 reports the calculated percentages of reduced flavins present during the steady state in each case. The spectra taken with wild-type nNOS show that it contained partially reduced flavins and ferric heme during the steady-state reaction. This is consistent with heme reduction being the slow step for electron flux through the enzyme under these conditions. Reduced flavin is indicated in the difference spectra by negative absorbances centered near 393 and 475 nm and the broad positive absorbance centered near 600 nm, which indicated that a significant amount of flavin semiquinone is present. The spectrum recorded after the reaction consumed all the O2 shows that flavin reduction became complete and was accompanied by the reduction of nNOS ferric heme to ferrous. This is clearly indicated in the second difference spectra, which shows ferric to ferrous heme absorbance changes at 397, 429, 550, and 650 nm. A comparison of the absorbance change at 485 nm in the two difference spectra, which is a specific wavelength for monitoring the change in the flavin reduction state alone (485 nm is an isosbestic point for the ferric to ferrous heme spectral transition in NOS) (45), indicates that the flavins were 33% reduced in wild-type nNOS during the steady state. Through an identical analysis, we found that the three hinge deletion mutants maintained 37–46% reduced flavin levels during the steady state, somewhat higher than but similar to wild type, and had lower levels of flavin semiquinone build up (supplemental Fig. S5 and Table 1). In comparison, the three hinge extension mutants (Fig. 7) maintained twice the level of reduced flavins in the steady state (60–72% reduced) relative to wild type (Table 1), with essentially no flavin semiquinone build up.
FIGURE 7.

Redox states of enzyme flavin and heme centers during steady-state NADPH oxidation as described under “Experimental Procedures.” Panels contain wavelength scans of CaM-bound wild-type nNOS and hinge length extension mutants that were recorded before (black solid line) and after (dashed line) initiating the reaction with NADPH at room temperature. Spectra of fully reduced enzymes are also shown (solid gray line). Insets show difference spectra that were calculated by subtracting resting enzyme spectra from either the steady-state spectra (dashed line minus black solid line from main panel) or from the fully reduced spectra (gray solid line minus black solid line from main panel). Scans are representative of three independent experiments done under identical conditions.
The level of reduced flavins maintained in nNOS during the steady-state reaction is determined by the rates of electron input and electron exit from the nNOS flavoprotein domain. We therefore examined if the steady-state levels of reduced flavins would correlate with the rate of electron input (i.e. the flavin reduction rates) that we measured for the various nNOS proteins. Table 1 indicates that an inverse correlation exists between the second phase rates of flavin reduction and the levels of reduced flavins maintained in the proteins during the steady state. A similar correlation did not hold for the first phase rates of flavin reduction (Table 1). This suggests that the slower second phase of flavin reduction in the hinge extension mutants was helping to increase their reduced flavins build up relative to wild type.
NO Synthesis Activity and Corresponding NADPH Oxidation
We measured the steady-state NO synthesis activities with either l-Arg or Nω-hydroxy-l-arginine as substrate for each CaM-bound enzyme. All mutants had measurable NO synthesis activity with either substrate (Fig. 8, left panel; supplemental Table S3), albeit lower than wild-type nNOS in all cases. We also measured corresponding rates of NADPH oxidation during NO synthesis from l-arginine. In general, the rate of NADPH oxidation followed the NO synthesis activity of each enzyme (Fig. 8, right panel). However, all the mutants oxidized more NADPH per NO formed compared with wild type (Fig. 8, right panel, and supplemental Table S4). Under the CaM-free condition, the hinge mutants also oxidized NADPH at faster rates than wild-type nNOS (supplemental Fig. S6), but their increases here were less compared with the uncoupled NADPH consumption observed for the CaM-bound proteins during NO synthesis. These data show that the changes in FMN-FAD/NADPH hinge length increase flavin auto-oxidation and diminish coupling between NADPH consumption and NO synthesis in nNOS.
FIGURE 8.

Steady-state NO synthesis rates and concurrent NADPH oxidation rates versus FMN-FAD/NADPH hinge length. Bars indicate the rates of NO synthesis from Arg or Nω-hydroxy-l-arginine (NOHA) (left panel) and the corresponding NADPH oxidation rates during NO synthesis from Arg (right panel) measured at 25 °C. The proportion of the total NADPH oxidation rate that was coupled and uncoupled from NO synthesis is indicated in each bar (right panel). Wild-type values are indicated at the 25-residue hinge length position. Assay conditions are described under “Experimental Procedures.” Values represent the mean and standard deviation of at least five independent measurements.
DISCUSSION
In NOS the FMN domain is connected to both an electron-donating (FAD/NADPH) and an electron-accepting (NOSoxy) partner domain. Our study focused on the hinge connecting the FMN domain to its electron-donating partner (the FMN-FAD/NADPH hinge). We found that changing its length has multiple effects on electron transfer and the associated catalytic properties of nNOS, as discussed below.
Electron Flux through the nNOS Flavoprotein Domain to Cytochrome c
Shortening the FMN-FAD/NADPH hinge slowed down electron flux through the flavoprotein domain by about 50% in both the CaM-free and CaM-bound nNOS, as judged from the cytochrome c reductase activities. This occurred despite no apparent change in the electron input rate (i.e. their flavin reduction kinetics). Conceivably, a shorter hinge could inhibit productive alignment of the FAD/NADPH and FMN domains as is required for FMN reduction, or alternatively, it could restrict the reduced FMN domain from achieving conformational states that best facilitate cytochrome c reduction. The normal flavin reduction kinetics of the hinge-shortened mutants suggest the latter mechanism is operative.
Lengthening the FMN-FAD/NADPH hinge increased electron flux through the flavoprotein domain in the CaM-free condition but either had no impact on the CaM response or (in one case) appeared to diminish it. These different outcomes may be reconciled by considering how CaM impacts the conformational and catalytic behaviors of nNOS. Electron flux through the flavoprotein domain is repressed in the CaM-free state and is relieved upon CaM binding (14–16, 18). The repression involves unique C-terminal tail and autoinhibitory insert protein elements (see Fig. 2A) that help stabilize an FMN-shielded conformation that is unreactive toward cytochrome c (46, 47). CaM eliminates the repression through a specific bridging interaction (40) that (i) shifts the conformational equilibrium to favor FMN-deshielded states that are reactive toward cytochrome c (see Fig. 1) (21) and that (ii) likely increases the speed of the conformational transitions (48). In the CaM-free enzymes, a longer hinge may help the FMN domain achieve the conformational states that facilitate cytochrome c reduction, basically the opposite effect of hinge shortening (as discussed above). The 6-residue hinge extension presents an extreme example in that its conformational equilibrium under CaM-free conditions is already shifted to a set point that is more like the CaM-bound enzyme. When the hinge extension mutants bind CaM, however, there can be a combined impact on their conformational equilibrium parameters, which in one case (6-residue hinge extension mutant) has a deleterious impact on electron flux to cytochrome c. Apparently, CaM further shifts the conformational equilibrium parameters of this protein beyond the optimal range for electron flux.
Our kinetic study of flavin reduction in the CaM-bound hinge extension mutants showed they have no problem accepting the first hydride equivalent from NADPH into their flavins, but then they progressively develop a kinetic barrier for further flavin reduction as hinge length increases, as manifested by their slower second phases of flavin reduction. This could also negatively impact electron flux. Presumably, when CaM binds to the 6-residue extension mutant, the kinetic barrier may reach a level at which FMN reduction becomes slow enough to be rate-limiting for electron flux through the enzyme to cytochrome c. We suspect that a longer FMN-FAD/NADPH hinge diminishes the ability of the FMN domain to interact with the FAD/NADPH domain and achieve the closed or FMN-shielded conformation that is required for interflavin electron transfer during catalysis. This phenomenon could also explain why the hinge extension mutants maintain a greater level of reduced flavins (50–60%) during the steady-state reaction compared with wild-type nNOS (33%); their FAD groups are nearly fully reduced and are waiting to transfer electrons to the FMN domain. A similar “conformational closing” problem is thought to explain the slow reductase activities reported for an nNOS mutant whose interdomain salt bridge interaction was eliminated (23) and for a CPR mutant whose FMN-FAD/NADPH hinge was partly deleted (27).
Model for Flavin Reduction and Impact of Hinge Length Changes
We assume that the initial rate of flavin reduction at 485 nm (k1 in our study) approximates the observed rate of hydride transfer from NADPH to FAD, whereas the second phase (k2) approximates the combined rates of electron distribution between the FADH2 and FMN and the rate of flavin reduction by a second molecule of NADPH. A similar interpretation of 485 nm kinetic data was suggested in previous reports on NOS or CPR enzymes (23, 27, 34, 46, 49, 50). Given this model, k1 cannot represent more than the first 50% of the total absorbance change at 485 nm, and k2 should therefore represent the second 50% of the total absorbance change. Our 485 nm stopped-flow traces show that the hinge extension mutants behave normally during reduction by the first hydride from NADPH but then increasingly develop a kinetic barrier toward accepting further electrons as hinge length is increased. How might this occur? We know that the two-electron reduced nNOS flavoprotein domain can formally exist in three states as follows: FADH2/FMN, the di-semiquinone FADH/FMNH, and FAD/FMNH2. The flavin midpoint couples in nNOS are poised to favor a blend of the 2nd and 3rd states (21, 51, 52), and populating the 3rd state enables a second hydride transfer from NADPH to FAD in the second phase of flavin reduction. Because the hinge extensions are unlikely to impact the flavin midpoint potentials, we assume that the flavin midpoint potentials in the hinge mutants remain similar to wild type. Under this circumstance, a slow second phase of flavin reduction could be due to a slow inter-flavin electron transfer (i.e. between FADH2 and FMN and/or between FADH and FMNH) or, alternatively, by a slow hydride transfer into the FAD/FMNH2 species after it forms. Although our data cannot definitively distinguish between these two mechanistic possibilities,4 we believe a slow inter-flavin electron transfer is most consistent with our data as explained below.
Regarding the hinge extensions possibly slowing the rate of the second hydride transfer from NADPH into FAD, it is difficult to imagine how the hinge extensions could allow the initial hydride transfer to proceed normally and then inhibit the second hydride transfer into FAD, without also invoking a concurrent problem in the inter-flavin electron transfer steps needed to form the requisite FAD/FMNH2 hydride acceptor species. For example, this would have the hinge extensions causing the hydride transfer into FAD to become sensitive to the reduction level of the FMN domain (i.e. to differ when it contains FMN versus FMNH2). There is no clear precedent for this type of influence on FAD reduction that does not also invoke an effect on the inter-flavin electron transfer. In addition, a problem with FAD reduction would be expected to decrease the level of reduced flavins that build up during the steady-state electron flux, which is the opposite of what was observed, i.e. the hinge extension mutants all had a greater build up of reduced flavins relative to WT nNOS (see Fig. 7).
Regarding the hinge extensions possibly slowing down the inter-flavin electron transfer, our previous work (21, 53) and this study (flavin fluorescence measures) imply that the CaM-bound wild-type and the hinge mutant enzymes are predominantly (80–90%) in an open or FMN-deshielded conformation at the point of mixing with NADPH, such that their FAD and FMN cofactors are held apart and not in electrical contact. Despite the open conformational state of CaM-bound wild-type nNOS, its rate of conformational change must be rapid enough to quickly form the closed or FMN-shielded conformation that is needed for electron transfer from FADH2 (or FADH) to the FMN domain (see Fig. 1), because its k1 and k2 rates of NADPH-dependent flavin reduction are fairly similar. However, we suspect that the hinge extensions must slow the rates of conformational transitions and/or increase the freedom of motion of the FMN domain such that it takes longer for them to form the closed or FMN-shielded conformation, and thus they display slower k2 rates of flavin reduction. This interpretation is consistent with their having a greater level of reduced flavins during steady-state electron flux (because the FAD is reduced and waiting to transfer electrons to FMN domain) and is consistent with the conformational model that was proposed to explain slower FMN reduction in a CPR hinge deletion mutant (27, 54).
There were some apparent inconsistencies between the flavin reduction rates we measured and the cytochrome c reductase activities for some of the hinge extension mutants. For example, the 2- and 4-residue hinge extension mutants showed decreased flavin reduction k2 values but had normal reductase activities. On the surface, this implies that the flavin reduction rate is not limiting and apparently can be slowed somewhat relative to wild type without negatively impacting the steady-state electron flux to cytochrome c. Only in the 6-residue hinge extension mutant is the second phase of flavin reduction slowed down enough to finally limit the reductase activity. In addition, the apparent discrepancies may reflect that the reductase assay contains excess cytochrome c that reacts quickly with the FMNH2 species and thus drives flavin reduction, whereas in the stopped-flow reaction there is no external electron acceptor present to drive flavin reduction, and thus it approaches an equilibrium where significant FMNH2 build up is achieved.
Electron Transfer from FMN Domain to the NOS Heme
NOS heme reduction requires movement of the FMN domain followed by precise interactions with the NOSoxy domain (KB in Fig. 1), and it is therefore more constrained compared with cytochrome c reduction. This likely explains why the heme reduction rate is 2 orders of magnitude slower than cytochrome c reduction in nNOS (supplemental Table S2), despite the FMN and NOSoxy domains being physically linked. Laser flash photolysis and pulsed EPR studies suggest that the FMN and heme cofactors in NOS enzymes can come within 19 Å of one another for electron transfer (55, 56). The crystal structure of the reductase domain of nNOS suggests that the FMN-FAD/NADPH hinge is an unstructured loop that could be capable of full extension (30). With this in mind, it is surprising that shortening the FMN-FAD/NADPH hinge by as much as 6 of its 25 total residues caused no more than a 30% drop in the heme reduction rate in our study. We estimate that deleting six residues would shorten the 25-residue polypeptide from about 32 to 24 Å when it is fully extended. Thus, the FMN-FAD/NADPH hinge in nNOS is actually longer than it needs to be to support a normal rate of heme reduction. This is consistent with the FMN-FAD/NADPH hinge being typically two residues longer in nNOS than in other NOS isoforms, and it establishes that hinge length in wild-type nNOS does not limit its rate of heme reduction.
Lengthening the FMN-FAD/NADPH hinge by 4 or 6 residues either decreased or increased the rate of heme reduction depending on how the enzyme was poised before initiating the reaction. Of the two methods we used (NADPH-triggered versus CaM-triggered heme reduction), the results from the CaM-triggered experiments may be more straightforward because in that case the CaM-free enzymes were given NADPH to reduce their flavins prior to triggering heme reduction with CaM. This means that their FMN domains already contained FMNhq at the point of CaM triggering, and they just needed to make a productive contact with the oxygenase domain for heme reduction to occur. Hinge lengthening could increase the probability in two ways. It might do so purely by physical means (i.e. lengthening the FMN-FAD/NADPH hinge allows a better approach for a reduced FMN domain). Hinge lengthening also may shift the conformational equilibrium of the reduced reductase domain of nNOS toward the FMN-deshielded state, thereby increasing the population of a reduced FMN domain that is in a conformation available to contact the oxygenase domain once CaM binds. As noted above, any effect on the conformational equilibrium would add to the changes that naturally occur when CaM binds, which in the reduced wild-type nNOS results in a shift from a 50:50 mix to an 80:20 mix of open versus closed conformations (21). The relative contribution of these or other potential mechanisms of action await further study. In any case, we conclude that a longer FMN-FAD/NADPH hinge is beneficial for heme reduction only after the nNOS flavins become reduced.
Our results from the NADPH-triggered reactions showed that lengthening the FMN-FAD/NADPH hinge caused a slower heme reduction. This implies that the formation/availability of FMNhq is perturbed in the hinge extension mutants and becomes rate-limiting. Under this particular circumstance, heme reduction requires that NADPH transfer a hydride to FAD followed by electron transfer between FAD and FMN to form FMNhq, which is the species capable of heme reduction. In wild-type nNOS, all these initial flavin reduction steps are fast relative to the observed rate of heme reduction and do not limit the process. However, in certain mutants like D1393V nNOS, flavin reduction is rate-limiting for heme reduction due to a slow hydride transfer between NADPH and FAD (34, 57). Thus, if increasing the length of the FMN-FAD/NADPH hinge somehow slows the flavin reduction step(s) leading to FMNhq formation, it could become rate-limiting for the NADPH-triggered heme reduction reaction but would not affect the rate seen in a CaM-triggered heme reduction reaction, as we observed. As discussed above, our data support this model and specifically point to a kinetic barrier arising for FMN reduction in the hinge extension mutants. Thus, in the NADPH-triggered heme reduction reactions of the hinge extended mutants, FAD reduction is relatively normal and fast (1st phase of flavin reduction), but most of the FMN domain is not in contact with the FAD/NADPH domain, and the conformational closing rate is slowed down enough to limit the speed at which FMNhq can build up (2nd phase of flavin reduction). The hinge extension mutants also have a faster flavin auto-oxidation rate, which would further diminish their FMNhq build up. To summarize, our current results suggest that the hinge-shortened mutants have a mildly slower heme reduction due to it being physically more difficult for their reduced FMN domains to reach the oxygenase domain of nNOS. In contrast, the hinge extension mutants can reach the NOSoxy domain better than in wild type, but they ultimately have slower heme reduction because they have trouble forming FMNhq in a timely manner. It is interesting that the negative effect of hinge lengthening on the heme reduction rate correlates very well with the decreases in electron flux through the heme to O2 as measured in the steady-state reactions (see Fig. 6). This good correlation implies that the factors limiting FMNhq formation/availability in the single turnover reactions (conformational closing) continue to limit electron flux through the nNOS heme to O2 in the steady-state reaction.
NO Synthesis and Uncoupled O2 Reduction
Changes in the FMN-FAD/NADPH hinge length led to lower NO synthesis activity in all cases. Lower activity was associated with slower heme reduction in most but not all of the mutants. However, all of the hinge mutants had increased uncoupled NADPH oxidation during their NO synthesis. In the worst cases, the amount of uncoupled NADPH oxidation was 3 to 4 times greater than the coupled NADPH oxidation used for NO synthesis. Exactly why this occurs is unclear, as it seemed to only partly arise from the reduced flavins having an increased propensity to air-oxidize. At this point, our results suggest the native FMN-FAD/NADPH hinge length in nNOS helps to maximize the coupling between NADPH oxidation, heme reduction, and NO synthesis, and thus minimize production of reduced oxygen species during catalysis.
Relation to Other Proteins
The FMN-FAD/NADPH hinge length varies between 19 and 25 residues among NOS enzymes, and nNOS-like isoforms typically have longer hinges than the iNOS- or eNOS-like isoforms (supplemental Fig. S7). In comparison, the FMN-FAD/NADPH hinge length in related dual-flavin reductases is ∼14 residues in CPR (27), 27 residues in NR1 (58), and 94 residues in methionine synthase reductase (28). Thus, hinge lengths in NOS are intermediate compared with these related enzymes and do not vary much relative to what is seen among the larger family. This may reflect that NOS enzymes have the added constraint of an attached electron acceptor domain (i.e. NOSoxy) that restricts the allowable range of FMN domain motion during catalysis.
So far, the impact of altering FMN-FAD/NADPH hinge length or composition in dual-flavin reductases has only been studied in CPR (27, 54), eNOS (19), and nNOS (this study). Our current results show that the 2-residue difference in hinge length between nNOS and eNOS does not explain why eNOS supports a much slower electron flux through its flavoprotein domain (19, 37). Thus, the difference may instead relate to the different compositions of their FMN-FAD/NADPH hinges. This is consistent with the eNOS hinge containing a deleterious proline residue, and with eNOS showing greater electron flux when its FMN-FAD/NADPH hinge is replaced with the hinge from nNOS (19). In CPR, extending the FMN-FAD/NADPH hinge length by 2, 4, 5, or 10 residues increased electron flux to cytochrome c by 35% (27, 54), and in certain cases it slightly increased its rate of cytochrome P450 heme reduction (27). This is similar to how our hinge extensions improved cytochrome c reduction by CaM-free nNOS. In contrast, deleting 2–7 consecutive residues of the CPR hinge diminished its electron flux by 84–99% (27, 54). We also found a slower electron flux in our hinge-shortened nNOS mutants, although the magnitude of the effect was considerably less in our case (electron flux was reduced by only 15–75% for nNOS). The difference may reflect that CPR has a shorter FMN-FAD/NADPH hinge and thus had a greater proportion of its hinge removed in the studies (up to 7 of 14 total residues) compared with nNOS (up to 6 of 25 residues). In CPR, the FMN-FAD/NADPH hinge deletions were also found to stabilize open conformations of the enzyme (FMN-deshielded) to the point where it is difficult for the enzyme to cycle between its closed and open conformations during steady-state electron transfer to cytochrome c (27, 54). In our case, it is hinge extensions that appear to have a similar negative impact on nNOS conformational cycling. In sum, several features of nNOS catalysis are sensitive to the FMN-FAD/NADPH hinge length. Changing hinge length can improve some of these features but diminishes others at the same time. The native hinge length appears to strike a good balance between the conformational interactions that are required for flavin and heme reduction during catalysis, so that nNOS can achieve maximal NO synthesis with minimal flavin auto-oxidation and uncoupled NADPH consumption.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants GM51491, CA53914, and HL58883 (to D. J. S.).

This article contains supplemental Figs. S1–S7 and Tables S1–S4.
The electron flux value we obtained with l-agmatine yields an estimate of 8.7 s−1 for heme reduction rate at 25 °C, in good agreement with our 5 s−1 rate measure for pre-steady-state heme reduction at 10 °C (Fig. 6), when the temperature difference is taken into account.
We could not extract meaningful absorbance changes at 600 nm from the diode array spectra of our nNOS proteins, and so could not follow the build up or decay of flavin semiquinone species during the reduction by NADPH.
- NOS
- nitric-oxide synthase
- eNOS
- endothelial nitric-oxide synthase
- iNOS
- inducible nitric-oxide synthase
- nNOS
- neuronal nitric-oxide synthase
- CPR
- cytochrome P450 reductase
- CaM
- calmodulin
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid
- H4B
- (6R)-5, 6, 7, 8-tetrahydro-l-biopterin
- FMNhq
- FMN hydroquinone
- NOSoxy
- N-terminal oxygenase domain
- DTT
- dithiothreitol.
REFERENCES
- 1. Griffith O. W., Stuehr D. J. (1995) Nitric-oxide synthases. Properties and catalytic mechanism. Annu. Rev. Physiol 57, 707–736 [DOI] [PubMed] [Google Scholar]
- 2. Knowles R. G., Moncada S. (1994) Nitric-oxide synthases in mammals. Biochem. J. 298, 249–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moncada S., Bolaños J. P. (2006) Nitric oxide, cell bioenergetics, and neurodegeneration. J. Neurochem. 97, 1676–1689 [DOI] [PubMed] [Google Scholar]
- 4. Schmidt H. H., Walter U. (1994) NO at Work. Cell 78, 919–925 [DOI] [PubMed] [Google Scholar]
- 5. Knowles R. G. (1997) Nitric oxide biochemistry. Biochem. Soc. Trans. 25, 895–901 [DOI] [PubMed] [Google Scholar]
- 6. Masters B. S., McMillan K., Sheta E. A., Nishimura J. S., Roman L. J., Martasek P. (1996) Neuronal nitric-oxide synthase, a modular enzyme formed by convergent evolution. Structure studies of a cysteine thiolate-liganded heme protein that hydroxylates l-arginine to produce NO• as a cellular signal. FASEB J. 10, 552–558 [DOI] [PubMed] [Google Scholar]
- 7. Förstermann U., Boissel J. P., Kleinert H. (1998) Expressional control of the “constitutive” isoforms of nitric-oxide synthase (NOS I and NOS III). FASEB J. 12, 773–790 [PubMed] [Google Scholar]
- 8. Fulton D., Gratton J. P., Sessa W. C. (2001) Post-translational control of endothelial nitric-oxide synthase. Why isn't calcium/calmodulin enough? J. Pharmacol. Exp. Ther. 299, 818–824 [PubMed] [Google Scholar]
- 9. Kleinert H., Pautz A., Linker K., Schwarz P. M. (2004) Regulation of the expression of inducible nitric-oxide synthase. Eur. J. Pharmacol. 500, 255–266 [DOI] [PubMed] [Google Scholar]
- 10. Michel T., Feron O. (1997) Nitric-oxide synthases. Which, where, how, and why? J. Clin. Invest. 100, 2146–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stuehr D. J., Santolini J., Wang Z. Q., Wei C. C., Adak S. (2004) Update on mechanism and catalytic regulation in the NO synthases. J. Biol. Chem. 279, 36167–36170 [DOI] [PubMed] [Google Scholar]
- 12. Andrew P. J., Mayer B. (1999) Enzymatic function of nitric-oxide synthases. Cardiovasc. Res. 43, 521–531 [DOI] [PubMed] [Google Scholar]
- 13. Wei C. C., Crane B. R., Stuehr D. J. (2003) Tetrahydrobiopterin radical enzymology. Chem. Rev. 103, 2365–2383 [DOI] [PubMed] [Google Scholar]
- 14. Kobayashi K., Tagawa S., Daff S., Sagami I., Shimizu T. (2001) Rapid calmodulin-dependent interdomain electron transfer in neuronal nitric-oxide synthase measured by pulse radiolysis. J. Biol. Chem. 276, 39864–39871 [DOI] [PubMed] [Google Scholar]
- 15. Panda K., Ghosh S., Stuehr D. J. (2001) Calmodulin activates intersubunit electron transfer in the neuronal nitric-oxide synthase dimer. J. Biol. Chem. 276, 23349–23356 [DOI] [PubMed] [Google Scholar]
- 16. Sagami I., Daff S., Shimizu T. (2001) Intra-subunit and inter-subunit electron transfer in neuronal nitric-oxide synthase. Effect of calmodulin on heterodimer catalysis. J. Biol. Chem. 276, 30036–30042 [DOI] [PubMed] [Google Scholar]
- 17. Siddhanta U., Presta A., Fan B., Wolan D., Rousseau D. L., Stuehr D. J. (1998) Domain swapping in inducible nitric-oxide synthase. Electron transfer occurs between flavin and heme groups located on adjacent subunits in the dimer. J. Biol. Chem. 273, 18950–18958 [DOI] [PubMed] [Google Scholar]
- 18. Abu-Soud H. M., Yoho L. L., Stuehr D. J. (1994) Calmodulin controls neuronal nitric-oxide synthase by a dual mechanism. Activation of intra- and interdomain electron transfer. J. Biol. Chem. 269, 32047–32050 [PubMed] [Google Scholar]
- 19. Haque M. M., Panda K., Tejero J., Aulak K. S., Fadlalla M. A., Mustovich A. T., Stuehr D. J. (2007) A connecting hinge represses the activity of endothelial nitric-oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 104, 9254–9259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Craig D. H., Chapman S. K., Daff S. (2002) Calmodulin activates electron transfer through neuronal nitric-oxide synthase reductase domain by releasing an NADPH-dependent conformational lock. J. Biol. Chem. 277, 33987–33994 [DOI] [PubMed] [Google Scholar]
- 21. Ilagan R. P., Tiso M., Konas D. W., Hemann C., Durra D., Hille R., Stuehr D. J. (2008) Differences in a conformational equilibrium distinguish catalysis by the endothelial and neuronal nitric-oxide synthase flavoproteins. J. Biol. Chem. 283, 19603–19615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Konas D. W., Zhu K., Sharma M., Aulak K. S., Brudvig G. W., Stuehr D. J. (2004) The FAD-shielding residue Phe-1395 regulates neuronal nitric-oxide synthase catalysis by controlling NADP+ affinity and a conformational equilibrium within the flavoprotein domain. J. Biol. Chem. 279, 35412–35425 [DOI] [PubMed] [Google Scholar]
- 23. Welland A., Garnaud P. E., Kitamura M., Miles C. S., Daff S. (2008) Importance of the domain-domain interface to the catalytic action of the NO synthase reductase domain. Biochemistry 47, 9771–9780 [DOI] [PubMed] [Google Scholar]
- 24. Stuehr D. J., Tejero J., Haque M. M. (2009) Structural and mechanistic aspects of flavoproteins. Electron transfer through the nitric-oxide synthase flavoprotein domain FEBS J. 276, 3959–3974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Finn R. D., Basran J., Roitel O., Wolf C. R., Munro A. W., Paine M. J., Scrutton N. S. (2003) Determination of the redox potentials and electron transfer properties of the FAD- and FMN-binding domains of the human oxidoreductase NR1. Eur. J. Biochem. 270, 1164–1175 [DOI] [PubMed] [Google Scholar]
- 26. Laursen T., Jensen K., Møller B. L. (2011) Conformational changes of the NADPH-dependent cytochrome P450 reductase in the course of electron transfer to cytochromes P450. Biochim. Biophys. Acta 1814, 132–138 [DOI] [PubMed] [Google Scholar]
- 27. Hamdane D., Xia C., Im S. C., Zhang H., Kim J. J., Waskell L. (2009) Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem. 284, 11374–11384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wolthers K. R., Scrutton N. S. (2007) Protein interactions in the human methionine synthase-methionine synthase reductase complex and implications for the mechanism of enzyme reactivation. Biochemistry 46, 6696–6709 [DOI] [PubMed] [Google Scholar]
- 29. Crane B. R., Arvai A. S., Ghosh D. K., Wu C., Getzoff E. D., Stuehr D. J., Tainer J. A. (1998) Structure of nitric-oxide synthase oxygenase dimer with pterin and substrate. Science 279, 2121–2126 [DOI] [PubMed] [Google Scholar]
- 30. Garcin E. D., Bruns C. M., Lloyd S. J., Hosfield D. J., Tiso M., Gachhui R., Stuehr D. J., Tainer J. A., Getzoff E. D. (2004) Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J. Biol. Chem. 279, 37918–37927 [DOI] [PubMed] [Google Scholar]
- 31. Ghosh D. K., Holliday M. A., Thomas C., Weinberg J. B., Smith S. M., Salerno J. C. (2006) Nitric-oxide synthase output state. Design and properties of nitric-oxide synthase oxygenase/FMN domain constructs. J. Biol. Chem. 281, 14173–14183 [DOI] [PubMed] [Google Scholar]
- 32. Roman L. J., Masters B. S. (2006) Electron transfer by neuronal nitric-oxide synthase is regulated by concerted interaction of calmodulin and two intrinsic regulatory elements. J. Biol. Chem. 281, 23111–23118 [DOI] [PubMed] [Google Scholar]
- 33. Haque M. M., Fadlalla M., Wang Z. Q., Ray S. S., Panda K., Stuehr D. J. (2009) Neutralizing a surface charge on the FMN subdomain increases the activity of neuronal nitric-oxide synthase by enhancing the oxygen reactivity of the enzyme heme-nitric oxide complex. J. Biol. Chem. 284, 19237–19247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Panda K., Adak S., Konas D., Sharma M., Stuehr D. J. (2004) A conserved aspartate (Asp-1393) regulates NADPH reduction of neuronal nitric-oxide synthase. Implications for catalysis. J. Biol. Chem. 279, 18323–18333 [DOI] [PubMed] [Google Scholar]
- 35. Panda K., Haque M. M., Garcin-Hosfield E. D., Durra D., Getzoff E. D., Stuehr D. J. (2006) Surface charge interactions of the FMN module govern catalysis by nitric-oxide synthase. J. Biol. Chem. 281, 36819–36827 [DOI] [PubMed] [Google Scholar]
- 36. Ghosh D. K., Wu C., Pitters E., Moloney M., Werner E. R., Mayer B., Stuehr D. J. (1997) Characterization of the inducible nitric-oxide synthase oxygenase domain identifies a 49-amino acid segment required for subunit dimerization and tetrahydrobiopterin interaction. Biochemistry 36, 10609–10619 [DOI] [PubMed] [Google Scholar]
- 37. Adak S., Aulak K. S., Stuehr D. J. (2001) Chimeras of nitric-oxide synthase types I and III establish fundamental correlates between heme reduction, heme-NO complex formation, and catalytic activity. J. Biol. Chem. 276, 23246–23252 [DOI] [PubMed] [Google Scholar]
- 38. Tejero J., Hannibal L., Mustovich A., Stuehr D. J. (2010) Surface charges and regulation of FMN to heme electron transfer in nitric-oxide synthase. J. Biol. Chem. 285, 27232–27240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adak S., Ghosh S., Abu-Soud H. M., Stuehr D. J. (1999) Role of reductase domain cluster 1 acidic residues in neuronal nitric-oxide synthase. Characterization of the FMN-FREE enzyme. J. Biol. Chem. 274, 22313–22320 [DOI] [PubMed] [Google Scholar]
- 40. Tejero J., Haque M. M., Durra D., Stuehr D. J. (2010) A bridging interaction allows calmodulin to activate NO synthase through a bi-modal mechanism. J. Biol. Chem. 285, 25941–25949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guan Z. W., Iyanagi T. (2003) Electron transfer is activated by calmodulin in the flavin domain of human neuronal nitric-oxide synthase. Arch. Biochem. Biophys. 412, 65–76 [DOI] [PubMed] [Google Scholar]
- 42. Klatt P., Heinzel B., John M., Kastner M., Böhme E., Mayer B. (1992) Ca2+/calmodulin-dependent cytochrome c reductase activity of brain nitric-oxide synthase. J. Biol. Chem. 267, 11374–11378 [PubMed] [Google Scholar]
- 43. Adak S., Santolini J., Tikunova S., Wang Q., Johnson J. D., Stuehr D. J. (2001) Neuronal nitric-oxide synthase mutant (Ser-1412 → Asp) demonstrates surprising connections between heme reduction, NO complex formation, and catalysis. J. Biol. Chem. 276, 1244–1252 [DOI] [PubMed] [Google Scholar]
- 44. Adak S., Crooks C., Wang Q., Crane B. R., Tainer J. A., Getzoff E. D., Stuehr D. J. (1999) Tryptophan 409 controls the activity of neuronal nitric-oxide synthase by regulating nitric oxide feedback inhibition. J. Biol. Chem. 274, 26907–26911 [DOI] [PubMed] [Google Scholar]
- 45. Tejero J., Biswas A., Haque M. M., Wang Z. Q., Hemann C., Varnado C. L., Novince Z., Hille R., Goodwin D. C., Stuehr D. J. (2011) Mesohaem substitution reveals how haem electronic properties can influence the kinetic and catalytic parameters of neuronal NO synthase. Biochem. J. 433, 163–174 [DOI] [PubMed] [Google Scholar]
- 46. Guan Z. W., Haque M. M., Wei C. C., Garcin E. D., Getzoff E. D., Stuehr D. J. (2010) Lys-842 in neuronal nitric-oxide synthase enables the autoinhibitory insert to antagonize calmodulin binding, increase FMN shielding, and suppress interflavin electron transfer. J. Biol. Chem. 285, 3064–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tiso M., Tejero J., Panda K., Aulak K. S., Stuehr D. J. (2007) Versatile regulation of neuronal nitric-oxide synthase by specific regions of its C-terminal tail. Biochemistry 46, 14418–14428 [DOI] [PubMed] [Google Scholar]
- 48. Haque M. M., Kenney C., Tejero J., Stuehr D. J. (2011) A kinetic model linking protein conformational motions, interflavin electron transfer, and electron flux through a dual-flavin enzyme simulating the reductase activity of the endothelial and neuronal nitric-oxide synthase flavoprotein domains. FEBS J. 278, 4055–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Welland A., Daff S. (2010) Conformation-dependent hydride transfer in neuronal nitric-oxide synthase reductase domain. FEBS J. 277, 3833–3843 [DOI] [PubMed] [Google Scholar]
- 50. Xia C., Hamdane D., Shen A. L., Choi V., Kasper C. B., Pearl N. M., Zhang H., Im S. C., Waskell L., Kim J. J. (2011) Conformational changes of NADPH-cytochrome P450 oxidoreductase are essential for catalysis and cofactor binding. J. Biol. Chem. 286, 16246–16260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Daff S., Noble M. A., Craig D. H., Rivers S. L., Chapman S. K., Munro A. W., Fujiwara S., Rozhkova E., Sagami I., Shimizu T. (2001) Control of electron transfer in neuronal NO synthase. Biochem. Soc. Trans. 29, 147–152 [DOI] [PubMed] [Google Scholar]
- 52. Noble M. A., Munro A. W., Rivers S. L., Robledo L., Daff S. N., Yellowlees L. J., Shimizu T., Sagami I., Guillemette J. G., Chapman S. K. (1999) Potentiometric analysis of the flavin cofactors of neuronal nitric-oxide synthase. Biochemistry 38, 16413–16418 [DOI] [PubMed] [Google Scholar]
- 53. Ilagan R. P., Tejero J., Aulak K. S., Ray S. S., Hemann C., Wang Z. Q., Gangoda M., Zweier J. L., Stuehr D. J. (2009) Regulation of FMN subdomain interactions and function in neuronal nitric-oxide synthase. Biochemistry 48, 3864–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grunau A., Geraki K., Grossmann J. G., Gutierrez A. (2007) Conformational dynamics and the energetics of protein-ligand interactions. Role of interdomain loop in human cytochrome P450 reductase. Biochemistry 46, 8244–8255 [DOI] [PubMed] [Google Scholar]
- 55. Astashkin A. V., Elmore B. O., Fan W., Guillemette J. G., Feng C. (2010) Pulsed EPR determination of the distance between heme iron and FMN centers in a human inducible nitric-oxide synthase. J. Am. Chem. Soc. 132, 12059–12067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feng C., Dupont A. L., Nahm N. J., Spratt D. E., Hazzard J. T., Weinberg J. B., Guillemette J. G., Tollin G., Ghosh D. K. (2009) Intraprotein electron transfer in inducible nitric-oxide synthase holoenzyme. J. Biol. Inorg. Chem. 14, 133–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Konas D. W., Takaya N., Sharma M., Stuehr D. J. (2006) Role of Asp1393 in catalysis, flavin reduction, NADP(H) binding, FAD thermodynamics, and regulation of the nNOS flavoprotein. Biochemistry 45, 12596–12609 [DOI] [PubMed] [Google Scholar]
- 58. Paine M. J., Garner A. P., Powell D., Sibbald J., Sales M., Pratt N., Smith T., Tew D. G., Wolf C. R. (2000) Cloning and characterization of a novel human dual flavin reductase. J. Biol. Chem. 275, 1471–1478 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.