

Background: 3-Deazauridine inhibits CTP synthetase as its 5′-triphosphate derivative and selectively depletes CTP pools.
Results: 3-Deazauridine fluorination shifts its antimetabolic target from CTP-S to orotidylate decarboxylase, resulting in depletion of intracellular UTP and CTP.
Conclusion: Such a target enzyme inhibition shift has a profound impact on the pyrimidine nucleotide pools.
Significance: A hydrogen to fluorine replacement in an antimetabolite drug results in a target enzyme shift.
Keywords: Cancer, Enzyme Inhibitors, Nucleoside, Nucleoside Nucleotide Analogues, Nucleoside Nucleotide Metabolism, Pyrimidine
Abstract
The antimetabolite prodrug 3-deazauridine (3DUrd) inhibits CTP synthetase upon intracellular conversion to its triphosphate, which selectively depletes the intracellular CTP pools. Introduction of a fluorine atom at C3 of 3DUrd shifts its antimetabolic action to inhibition of the orotidylate decarboxylase (ODC) activity of the UMP synthase enzyme complex that catalyzes an early event in pyrimidine nucleotide biosynthesis. This results in concomitant depletion of the intracellular UTP and CTP pools. The new prodrug (designated 3F-3DUrd) exerts its inhibitory activity because its monophosphate is not further converted intracellularly to its triphosphate derivative to a detectable extent. Combinations with hypoxanthine and adenine markedly potentiate the cytostatic activity of 3F-3DUrd. This is likely because of depletion of 5-phosphoribosyl-1-pyrophosphate (consumed in the hypoxanthine phosphoribosyl transferase/adenine phosphoribosyl transferase reaction) and subsequent slowing of the 5-phosphoribosyl-1-pyrophosphate-dependent orotate phosphoribosyl transferase reaction, which depletes orotidylate, the substrate for ODC. Further efficient anabolism by nucleotide kinases is compromised apparently because of the decrease in pKa brought about by the fluorine atom, which affects the ionization state of the new prodrug. The 3F-3DUrd monophosphate exhibits new inhibitory properties against a different enzyme of the pyrimidine nucleotide metabolism, namely the ODC activity of UMP synthase.
Introduction
De novo biosynthesis of pyrimidine nucleotides is executed by a number of enzymes eventually leading to the formation of UMP (Fig. 1). Subsequent conversion of UMP to UDP and UTP provides the substrate for CTP synthetase (CTP-S),2 which is the sole branching point for de novo synthesis of cytosine nucleotides. UTP is also the starting point for production of UDP-sugar derivatives such as UDP-glucose (Fig. 1). Selective inhibitors exemplified by N-(phosphonoacetyl)-l-aspartate (1), A771726 (2), and brequinar (3) have been reported for the earliest enzymatic conversion steps in the de novo synthesis pathway of orotic acid (OA) (Fig. 2 and supplemental Fig. S1). Orotate phosphoribosyl transferase (OPRT), which is inhibited by potassium oxonate (4), catalyzes the further conversion of OA to OMP, which is further converted to UMP by OMP decarboxylase (ODC). The latter step is inhibited by several antimetabolites including pyrazofurin (5) and 6-azauridine (6AUrd) (6) following conversion to their respective 5′-monophosphates. The two-step conversion of OA to UMP by OPRT and ODC activity is catalyzed by a multifunctional enzyme (UMP synthase, UMPS) that consists of a 24-kDa OPRT and a 28-kDa ODC domain connected through a short amino acid linker (7). For CTP-S, two well known selective inhibitors exist: 3-deazauridine (3DUrd) (8–10) and cyclopentenyl cytosine (11), both of which exert inhibitory activity as their 5′-triphosphate metabolites (Fig. 2). Several of the abovementioned antimetabolites have been shown to exhibit potent cytostatic and/or antiviral activity in cell culture (12).
FIGURE 1.

Pyrimidine nucleoside and/or nucleotide metabolism in mammalian cells.
FIGURE 2.

Interaction of antimetabolite drugs with pyrimidine nucleotide metabolism.
Recently, we synthesized the 3-fluoro derivative (3F-3DUrd; Fig. 3) of 3DUrd and evaluated its antiviral and cytostatic activities. This 3F-3DUrd was devoid of significant antiviral activity but showed pronounced cytostatic activity (13). However, to our surprise, further studies revealed that administration of 3F-3DUrd not only depleted the cellular CTP pools as expected for a CTP-S inhibitor such as 3DUrd, but it also significantly decreased the UTP pools. That finding suggested an additional or alternative mechanism of cytostatic action for this novel compound beyond that of 3DUrd and cyclopentenyl cytosine. We now report that substitution of a fluorine atom for hydrogen at the C3 of 3DUrd shifts the mechanism of cytostatic action from selective inhibition of CTP synthetase (for 3DUrd) to inhibition of UMP synthase (for 3F-3DUrd).
FIGURE 3.

Structural formula of 3F-3DUrd and interconversion (equilibrium) of the (R)- and (S)-3F-3DUrd diastereomers. The enol form is favored over the keto form.
MATERIALS AND METHODS
Test Compounds
Urd, orotidine (Otd), 2′-deoxythymidine, hypoxanthine (Hpx), Gua, Ado, Ade, Guo, and Ino were purchased from Sigma; cytidine (Cyd) was from ICN (Cleveland, OH). Orotic acid was obtained from Serva (Heidelberg, Germany). 3-Deazauridine was from Sigma. 3-Fluoro-3-dezauridine (3F-3DU) was synthesized as described (13). K-oxonate (Acros, Morris Plains, NJ), 6-azauridine (6AU) (Sigma), pyrazofurin (Eli Lilly Co, Indianapolis, IN), and A771726 (Enzo Life Sciences, San Diego, CA) were used in the antimetabolic studies. The following radiolabeled compounds were obtained from Moravek Biochemicals (Brea, CA): [2-14C]orotic acid (58 mCi/mmol), [6-3H]5-fluorouracil (13 Ci/mmol), [6-3H]5-fluorouridine (11.1 Ci/mmol), and [5-3H]uridine (17 Ci/mmol).
Cytostatic Activity Assays
The cytostatic activities of the test compounds were evaluated against murine leukemia L1210 and mammary carcinoma FM3A cells, human T-lymphocyte CEM, mammary carcinoma MCF-7, prostate cancer PC-3, osteosarcoma OST thymidine kinase-deficient, cervix carcinoma HeLa, and embryonic lung fibroblast HEL cells. The cells were seeded in 96-well microtiter plates at 2 × 104–5 × 104 cells/200 μl-well in RPMI 1640 (Invitrogen), supplemented with 10% fetal calf serum (Invitrogen), 2 mm l-glutamine (Invitrogen), and 0.075% sodium bicarbonate (Invitrogen). After two (L1210 and FM3A) or 3 (CEM, MCF-7, PC-3, OST thymidine kinase-deficient, HeLa, and HEL) days, the cell number was determined with a Coulter Z1 Particle counter (Analis, Ghent, Belgium). The monolayer cells were first trypsinized for 10 min before counting. The IC50 or 50% inhibitory concentration was determined as the compound concentration required to inhibit cell proliferation by 50%.
Preparation of Crude CEM Cell Extracts as an Enzyme Source or to Determine the Intracellular Nucleotide Pools
Five-ml CEM cell cultures were seeded at ∼400,000 cells/ml RPMI 1640 culture medium (supplemented with 10% fetal calf serum, 2 mm l-glutamine, and 0.075% NaHCO3) and incubated at 37 °C in a CO2-controlled (5%) humidified atmosphere. After several time periods, the cells were harvested by centrifugation (10 min; 1,200 rpm) and washed twice with cold RPMI 1640 medium (without serum). The cells were then suspended in suspension buffer (PBS) and sonicated (3 × 10 s) to lyse >95% of the cells. After one more centrifugation step (10 min; 13,000 rpm) to remove the cell debris, the supernatants were divided into aliquots and frozen at −80 °C until used as enzyme (UMP synthase) source. Alternatively, the cells were exposed to 66% cold methanol after the last washing step and kept on ice for 10 min. Then the precipitated cell pellet was centrifuged for 10 min at 13,000 rpm, and the supernatant was used for determining the intracellular nucleotide pools by HPLC analysis.
Nucleotide Pool Measurements in CEM Cell Extracts
Five-ml CEM cell cultures (400,000 cells/ml) were exposed to fixed concentrations of test compounds for 6, 24, or 48 h, after which cell extracts were prepared as described above. In these cell extracts, the nucleotide pools were separated and quantified by HPLC analysis (Alliance 2690; Waters, Milford, MA) using a Partisphere-SAX anion exchange column (4.6 × 125 mm; Wattman International Ltd., Maidstone, UK). The following gradient was used: 5 min with 100% buffer A (5 mm NH4H2PO4, pH 5); a 15-min linear gradient of 100% buffer A to 100% buffer B (300 mm NH4H2PO4, pH 5); 20 min with 100% buffer B; a 5-min linear gradient to 100% buffer A; and equilibration at 100% buffer A for 5 min. The UV absorbance of the peaks was recorded at 262 nm.
Identification of Nucleosides and Nucleobases in CEM Cell Cultures Exposed to Test Compounds
CEM cell extracts of antimetabolite-exposed CEM cell cultures were prepared as described above. Concentrations of the pyrimidine nucleoside metabolites and their degradation products that eluted from the Partisil-SAX HPLC column within 4 min were determined using HPLC tandem mass spectrometry, essentially as described before (14).
Incorporation of [14C]Orotic Acid into Uridine Nucleotide Pools of CEM Cell Cultures in the Presence or Absence of Antimetabolite Drugs
CEM cell cultures (5-ml; 400,000 cells/ml) were incubated for 24 h in the presence of 3DUrd (10 μm), 3F-3DUrd (10 μm), or no drug at 37 °C. After the incubation period, the cell extracts were prepared and subjected to HPLC analysis as described above. The fraction numbers containing radiolabeled orotic acid/Otd, UMP, UDP, UTP, and UDPG were 4, 7, 14, 22, and 10 min, respectively. The identities of the peaks were confirmed in similar HPLC gradients with the corresponding nonradiolabeled compound standards.
Conversion of [14C]Orotic Acid into [14C]OMP and [14C]UMP in CEM Cell Extracts in the Presence or Absence of Drug
Exponentially growing CEM cell cultures in 75-cm2 culture flasks (20 ml) were centrifuged, and 108 cells were suspended in 1 ml of PBS and sonicated for 3 × 10 s. The lysed cells were centrifuged at 13,000 rpm for 10 min at 4 °C, and the supernatant was frozen at −80 °C until used. To 30 μl of nucleoside kinase mixture (8.3 mm MgCl2, 16.6 mm DTT, 0.08 mg of BSA, 1 mm ATP, and 16.6 mm NaF in 83 mm Tris·HCl, pH 8.0) was added: 5 μl of Milli-Q H2O, 5 μl of drug (i.e., K-oxonate, 6AUrd, or 3F-3DUrd) and 10 μl of uridine/cytidine kinase-1 (UCK-1) enzyme (10 μg). The 50-μl reaction mixture was incubated overnight at 37 °C (∼20 h) to allow full conversion of 6AUrd and 3F-3DUrd to their respective 5′-monophosphates. To this reaction mixture was then added 50 μl of OPRT reaction mixture containing 10 μl of 5-phosphoribosyl-1-pyrophosphate (10 mm) (MP Biochemicals, Solon, OH), 10 μl of [14C]orotic acid (1 μCi), and 10 μl of unlabeled orotic acid (1 mm). Incubation was continued for 30, 60, and 120 min at 37 °C. After the different incubation time periods, 20 μl of the reaction mixture was withdrawn and added to 40 μl of ice-cold MeOH to precipitate the proteins and the nucleic acids. This mixture was then centrifuged for 10 min at 13,000 rpm and 4 °C, and the supernatants were injected into the HPLC column using the same gradient as described above for separation and quantification of orotic acid and the radiolabeled nucleotides (i.e., OMP, UMP, UDP, and UTP). [14C]OA, [14C]OMP, and [14C]UMP corresponded to the radiolabeled fractions 4–5, 7–8, and 13–14. In another set of experiments, 10 μl of the [14C]OA solution was replaced by 10 μl of the [3H]5-FU (1 μCi) solution to measure formation of [3H]5-FUMP in the CEM cell extracts in the presence or absence of 6AUrd, 3F-3DUrd, or K-oxonate.
Preparation of Purified UCK and Uridylate-Cytidylate Kinase (UMP-CMP Kinase)
The UCK-1 and UMP-CMP kinase expression plasmids were constructed as described earlier (15, 16). The expression plasmids were transformed into the Escherichia coli strain BL21(DE3) (Novagen), and single colonies were inoculated into LB medium supplemented with 100 μg/ml ampicillin. The bacteria were grown at 37 °C, and protein expression was induced at A600 ≈ 0.8 with 1 mm isopropyl-1-thio-β-d-galactopyranoside. The expressed UCK-1 protein was purified using a Talon metal affinity resin column (Clontech) as described (16), and the UMP-CMP kinase protein was purified using glutathione-Sepharose 4B (GE Healthcare) as described (15). The purities of the recombinant proteins were verified by SDS-PAGE, and the protein concentrations were determined with the Bradford Protein Assay (Bio-Rad) using bovine serum albumin as the concentration standard.
RESULTS
Cytostatic Activity of 3F-3DUrd
The cytostatic properties of the new 3-fluoro-substituted 3DUrd derivative (3F-3DUrd) (Fig. 3) were compared with those of 3-deazauridine (3DUrd, a CTP synthetase inhibitor), 6AUrd (an ODC inhibitor), pyrazofurin (an ODC inhibitor), K-oxonate (an OPRT inhibitor), and the leflunomide derivative A771726 (a DHO-DH inhibitor) (supplemental Fig. S1) against a variety of tumor cell lines. 3F-3DUrd showed a 2–10-fold higher antiproliferative activity than K-oxonate and was ∼ 2–7-fold less or 2-fold more potent than A771726, depending the nature of the tumor cell line. 3F-3DUrd was 2-fold (CEM) to 30-fold (MCF-7) less cytostatic than 3DUrd. Pyrazofurin and 6AUrd were 30–80-fold more cytostatic than 3F-3DUrd against the panel of tumor cell lines (supplemental Table S1).
Effect of Natural Pyrimidine and Purine Derivatives on the Cytostatic Action of 3F-3DUrd, 3DUrd, and Other Antimetabolites
To obtain more information on the nature of the antimetabolic action of 3F-3DUrd and 3DUrd, natural pyrimidine and purine derivatives were administered to drug-exposed CEM cell cultures. A number of well known antimetabolites acting on different enzymes of the de novo pyrimidine nucleotide synthesis pathways were included as controls (Table 1). Among the natural pyrimidine derivatives, Urd and Cyd annihilated the cytostatic activity of all of the antimetabolites. In contrast, the addition of deoxythymidine, OA, and Otd to the antimetabolite-containing tumor cell cultures at subtoxic concentrations did not markedly affect the antiproliferative activity. The purine derivatives Hpx, Ade, Ado, and Ino markedly increased the cytostatic potential of 3F-3DUrd by 20–70-fold, whereas they had modest or negligible effects on the cytostatic activity of 3DUrd. The pronounced increase of cytostatic activity of 3F-3DUrd but not 3DUrd by these purine derivatives was also observed for the ODC inhibitors pyrazofurin and 6AUrd, and the OPRT inhibitor K-oxonate, but not for the DHO-DH inhibitor A771726 (Table 1). These data provided definitive evidence that 3F-3DUrd behaved differently in its antimetabolic action than its parent 3DUrd analogue and that the behavior of 3F-3DUrd was similar to that of the UMPS (OPRT/ODC) inhibitors.
TABLE 1.
Effect of natural pyrimidine and purine derivatives on the cytostatic activity of 3F-3DUrd and other antimetabolite drugs in CEM cell culture
| Addition of natural DNA/RNA precursors | IC50a |
|||||
|---|---|---|---|---|---|---|
| 3F-3DUrd | 3-DUrd | Pyrazofurin | 6-Aza-Urd | K-oxonate | A77 1726 | |
| μm | ||||||
| As such | 19 ± 8.6 | 7.4 ± 3.1 | 0.24 ± 0.09 | 0.63 ± 0.28 | 204 ± 43 | 47 ± 41 |
| + Urd 500 μm | >250 | >250 | >250 | >250 | >250 | >250 |
| + Urd 100 μm | >250 | >250 | >250 | >250 | >250 | >250 |
| + Cyd 500 μm | >250 | >250 | 123 ± 121 | >250 | >250 | >250 |
| + Cyd 100 μm | >250 | >250 | 99 ± 85 | >250 | >250 | >250 |
| + Orotic acid 500 μm | 13 ± 8.5 | ≥27 | 0.24 ± 0.15 | 0.62 ± 0.30 | >250 | >250 |
| + Orotic acid 100 μm | 10 ± 8.6 | 12 ± 5.8 | 0.26 ± 0.17 | 0.53 ± 0.25 | ≥250 | 153 |
| + Orotidine 100 μm | 6.8 ± 2.7 | 17 ± 10.3 | 0.079 ± 0.01 | 0.37 ± 0.17 | 149 ± 47 | 132 |
| + dThd 10 μm | 11 ± 1.4 | 9 ± 4.0 | 0.20 ± 0.01 | 0.69 ± 0.06 | 82 ± 9.9 | 32 ± 6.1 |
| + dThd 5 μm | 19 ± 8.6 | 9.6 ± 0.64 | 0.18 ± 0.11 | 0.75 ± 0.17 | 121 ± 82 | 61 ± 45 |
| + Hpx 100 μm | 0.79 ± 0.44 | 3.55 ± 0.29 | 0.013 ± 0.007 | 0.045 ± 0.002 | 13 ± 2.3 | 114 |
| + Hpx 20 μm | 2.2 ± 0.50 | 4.31 ± 0.88 | 0.12 ± 0.009 | 0.15 ± 0.01 | 105 ± 80 | 110 |
| + Gua 100 μm | 4.9 ± 2.8 | 0.89 ± 0.16 | 0.17 ± 0.07 | 0.15 ± 0.07 | 42 ± 6.0 | >50 |
| + Gua 20 μm | 9.3 ± 0.47 | 10 ± 2.4 | 0.25 ± 0.13 | 0.54 ± 0.13 | ≥250 | 106 |
| + Ado 500 μm | 0.31 ± 0.13 | 2.0 ± 1.5 | 0.026 ± 0.015 | 0.031 ± 0.008 | 12 ± 2.1 | 18 ± 13 |
| + Ado 100 μm | 0.87 ± 0.02 | 2.9 ± 0.69 | 0.082 ± 0.014 | 0.089 ± 0.012 | 76 ± 16 | 62 ± 48 |
| + Ade 500 μm | 0.25 ± 0.18 | 2.3 ± 1.9 | 0.011 ± 0 | 0.013 ± 0 | 6.2 ± 1.1 | 31 13 |
| + Ade 100 μm | 0.53 ± 0.33 | 4.0 ± 0.95 | 0.020 ± 0.005 | 0.031 ± 0.003 | 19 ± 13 | 31 ± 10 |
| + Ade 20 μm | 3.2 ± 2.9 | 4.2 ± 2.4 | 0.14 ± 0.07 | 0.22 ± 0.09 | 150 ± 141 | 22.4 |
| + Guo 20 μm | 16 ± 11 | 3.3 ± 3.1 | 0.29 ± 0.01 | 0.32 ± 0.22 | ≥208 | 117 |
| + Ino 100 μm | 1.3 ± 0.19 | 3.3 ± 0.0 | 0.031 ± 0.005 | 0.054 ± 0.021 | 29 ± 11 | 105 |
| + Ino 20 μm | 3.5 ± 0.47 | 3.3 ± 0.05 | 0.11 ± 0 | 0.22 ± 0.007 | 110 ± 9.9 | 126 ± 9.9 |
a 50% Inhibitory concentration or compound concentration required to inhibit CEM cell proliferation by 50%.
Impact of 3F-3DUrd on the Intracellular Nucleotide Pools of Drug-exposed Lymphocyte CEM Tumor Cells
The impact of exposure of CEM tumor cell cultures to 3DUrd and the new 3F-3DUrd drug on the intracellular nucleotide pools was investigated. The data are shown in a representative anion exchange HPLC chromatogram for 24 h drug-exposed cell cultures (supplemental Fig. S2, top panel, control; middle panel, 3DUrd-treated; and bottom panel, 3F-3DUrd-treated) and in Table 2 for 24 and 48 h 3F-3DUrd-exposed cell cultures (upper part for each time course experiment). It is apparent that 3DUrd caused markedly decreased CTP (peak 17) but not UTP (peak 16) and UDPG (peak 8) pools (supplemental Fig. S2, compare top and middle panels), whereas 3F-3DUrd caused significant decreases in all three CTP, UTP, and UDPG pools (supplemental Fig. S2, compare top and bottom panels). These effects were dose-dependent and were most pronounced after a 24-h incubation time. The nucleotide pools were (partially) restored after 48 h (Table 2). The effect on the NTP pools observed for 3F-3DUrd is not expected to occur for a CTP synthetase inhibitor but rather for an inhibitor of an early step in de novo UTP synthesis catalyzed by DHO-DH, OPRT, or ODC (Figs. 1 and 2).
TABLE 2.
Effect of 3F-3DUrd on the intracellular CEM nucleotide pools
| Drug concentration and incubation time | Percentage of control (without drug) nucleotide poolsa |
||||
|---|---|---|---|---|---|
| UTP | CTP | ATP | GTP | UDPG | |
| 24 h | |||||
| 10 μm 3F-3DUrd | 8.6 ± 3.5 | 12 ± 6.1 | 77 ± 6.4 | 71 ± 21 | 17 ± 0.7 |
| 2 μm 3F-3DUrd | 30 ± 3.5 | 29 ± 4.2 | 103 ± 4.9 | 81 ± 33 | 39 ± 3.5 |
| 0.5 μm 3F-3DUrd | 75 ± 2.8 | 76 ± 9.9 | 101 ± 7.8 | 54 ± 13 | 80 ± 0.7 |
| 48 h | |||||
| 10 μm 3F-3DUrd | 63 ± 48 | 50 ± 26 | 74 ± 9.9 | 81 ± 35 | 64 ± 22 |
| 2 μm 3F-3DUrd | 132 ± 81 | 98 ± 35 | 89 ± 1.4 | 94 ± 27 | 151 ± 36 |
| 0.5 μm 3F-3DUrd | 94 ± 13 | 102 ± 9.2 | 119 ± 1.4 | 108 ± 39 | 128 ± 11 |
a The data are the means ± S.D. of three independent experiments.
In these experiments, 3F-3DUrd was converted into its 5′-monophosphate but not into di- or triphosphates in detectable amounts (peak 11) (supplemental Fig. S2, bottom panel), whereas 3DUrd was converted into the mono-, di-, and 5′-triphosphates that were consistently detected in HPLC chromatograms (supplemental Fig. S2, middle panel, peaks 6, 14, and 20). These observations strongly suggest that 3F-3DUrd 5′-monophosphate is the active antimetabolite, whereas 3DUrd 5′-triphosphate is the active CTP synthetase inhibitor. A marked accumulation of several other cellular metabolites was observed in the first 4-min elution profiles of extracts with 3F-3DUrd-treated but not in the 3DUrd-treated CEM tumor cell cultures (see HPLC chromatograms in supplemental Fig. S2 (bottom panel, peaks 2 and 3) compared with supplemental Fig. 2 (top and middle panels)).
Because the purine bases Ade and Hpx and the purine nucleosides Ado and Ino were found to markedly potentiate the cytostatic activity of 3F-3DUrd (Table 1), the effect of Hpx (100 μm) on the NTP pools of 3F-3DUrd-exposed CEM tumor cell cultures was investigated (Table 3). The addition of Hpx further decreased both the UTP and CTP pools when measured after 24 h, and this effect was still pronounced after 48 h of incubation.
TABLE 3.
Effect of hypoxanthine on the nucleotide pools of 3F-3DUrd-exposed CEM tumor cell cultures
| Drug concentration and incubation time | Percentage of control (without drug) nucleotide poolsa |
||||
|---|---|---|---|---|---|
| UTP | CTP | ATP | GTP | UDPG | |
| 24 h | |||||
| 10 μm 3F-3DUrd + Hpx 100 μm | 8.7 ± 2.0 | 12 ± 6.3 | 105 ± 7.8 | 136 ± 39 | 15 ± 5.8 |
| 2 μm 3F-3DUrd + Hpx 100 μm | 17 ± 4.4 | 14 ± 7.3 | 101 ± 15 | 83 ± 13 | 27 ± 2.1 |
| 0.5 μm 3F-3DUrd + Hpx 100 μm | 37 ± 3.6 | 32 ± 5.9 | 103 ± 6.7 | 79 ± 16 | 48 ± 13 |
| 48 h | |||||
| 10 μm 3F-3DUrd + Hpx 100 μm | 6.3 ± 0.6 | 9.4 ± 1.6 | 86 ± 18 | 106 ± 7.6 | 14 ± 5.9 |
| 2 μm 3F-3DUrd + Hpx 100 μm | 18 ± 0.6 | 14 ± 1.5 | 86 ± 13 | 82 ± 17 | 18 ± 4.0 |
| 0.5 μm 3F-3DUrd + Hpx 100 μm | 82 ± 27 | 78 ± 31 | 111 ± 10 | 84 ± 28 | 100 ± 46 |
a The data are the means ± S.D. of three independent experiments.
Identification of Pyrimidine Precursor Metabolites in Extracts of CEM Tumor Cell Cultures Exposed to Antimetabolites
HPLC/mass spectrometry analyses revealed pronounced intracellular accumulations of N-carbamoyl aspartate, DHO, OA, and Otd in CEM cell cultures treated with 3F-3DUrd (Table 4). Such enhanced pyrimidine precursor accumulations were also observed for pyrazofurin, 6-aza-Urd, and K-oxonate but not for the CTP synthetase inhibitor 3DUrd nor for the DHODH inhibitor A771726 (Table 3). These findings strongly suggest a change from inhibition of CTP synthetase (with 3DUrd) to inhibition of an early event in the de novo pyrimidine nucleotide synthesis pathway (with 3F-3dDUrd) that occurs after the DHODH step but before UMP production. Uridylate synthase (OPRT/ODC) appears to qualify as the most likely intracellular target enzyme for 3F-3DUrd (presumably as its 5′-monophosphate).
TABLE 4.
Quantification of intracellular pyrimidine precursors in CEM tumor cell cultures exposed to a variety of antimetabolic drugs
| Metabolite | Intracellular pyrimidine metabolite concentrationsa |
||||||
|---|---|---|---|---|---|---|---|
| As such | 3F-3DUrd (20 μm) | 3DUrd (10 μm) | A771726 (50 μm) | K-oxonate (250 μm) | Pyrazofurin (0.5 μm) | 6AU (0.5 μm) | |
| nmol/106 CEM cells | |||||||
| NC-aspartateb | <0.09 | 47 | <0.08 | 62 | 33 | 35 | 32 |
| Dihydroorotic acid | <0.09 | 1.4 | <0.08 | 5.5 | 1.4 | 1.1 | 0.92 |
| Orotic acid | <0.09 | 4.4 | <0.08 | <0.05 | 4.5 | 4.0 | 3.1 |
| Orotidine | <0.18 | 7.6 | 0.27 | <0.09 | 5.6 | 7.5 | 5.8 |
| Uridine | <0.18 | <0.12 | <0.15 | <0.09 | <0.01 | 0.10 | <0.08 |
| Uracil | <0.36 | <0.23 | <0.31 | <0.18 | <0.02 | 0.20 | <0.17 |
a Intracellular metabolite concentrations expressed in nmol/106 CEM cells upon exposure of CEM tumor cell cultures to several antimetabolite drugs for 24 h.
b N-1-Phosphocarbonyl-l-aspartate.
Labeling of the Intracellular Uracil Nucleotide Pools in CEM Tumor Cell Cultures by [14C]Orotic Acid in the Presence of 3DUrd and 3F-3DUrd
CEM tumor cell cultures were exposed to 10-μm 3DUrd or 3F-3DUrd for 24 h in the presence of [14C]orotate ([14C]OA). After the incubation period, the cell extracts were prepared, and the radiolabeled UMP, UDP, UTP, and UDPG pools were quantified by HPLC analysis (Fig. 4). The addition of 3DUrd resulted in significantly increased labeling of the UTP and UDPG pools, which is indicative of CTP synthetase inhibition. In contrast, the addition of 3F-3DUrd resulted in virtually complete prevention of UMP, UDP, UTP, and UDPG pool labeling by [14C]OA with markedly increased formation of 14C-labeled OA and OA metabolites that had an early retention time in HPLC chromatograms (i.e., UV-visible peak (5) at 3–4 min) (Fig. 4). These findings again suggest inhibition of the conversion of [14C]OA to [14C]UMP and are in agreement with the HPLC/mass spectrometry results that show intracellular accumulations of DHO, OA, and Otd in 3F-3DUrd-exposed CEM tumor cell cultures.
FIGURE 4.

Incorporation of [14C]OA in uridine nucleotide pools of antimetabolite-exposed CEM tumor cell cultures. The 14C labeling of uridine nucleotide pools by [14C]orotic acid in CEM tumor cell cultures was performed for 24 h in the presence of 10-μm of 3DUrd or 10-μm of 3F-DUrd or in the absence of drug.
3F-3DUrd Inhibits the ODC Activity of UMPS (OPRT/ODC) in Crude CEM Cell Extracts
To investigate whether the presumed 5′-monophosphate of 3F-3DUrd inhibits UMPS (OPRT/ODC), 3F-3DUrd was incubated overnight with UCK to completely convert it to its 5′-monophosphate derivative. CEM cell extracts were then exposed to the 3F-3DUrd 5′-monophosphate, and [14C]OA was added to start the sequence of synthetic reactions. Upon addition of [14C]OA to the cell extracts, time-dependent conversion of [14C]OA to [14C]OMP and [14C]UMP occurred. After 30, 60, and 120 min of incubation, UMP was the predominant metabolite derived from [14C]OA (∼5-fold more UMP than OMP was formed at each time point). In the presence of K-oxonate, OMP was detected only in trace amounts, and UMP was virtually undetected (Fig. 5). This is compatible with the known inhibition of OPRT by K-oxonate. In the presence of the ODC inhibitor 6-AUrd, OMP was formed in a time-dependent manner, whereas only trace amounts of UMP were detected. Thus, the efficient OPRT-catalyzed conversion of OA to OMP was no longer followed by the ODC-catalyzed decarboxylation of OMP to UMP (Fig. 5). In the presence of 3F-3DU (monophosphate), a similar product profile was observed as obtained with 6-AUrd; both OMP and UMP were formed from [14C]OA, but the ratio of OMP/UMP was much higher in the 3F-3DU-exposed cell extracts than in the controls. This indicates a markedly increased inhibition of orotidine decarboxylase activity relative to an essentially unaffected level of orotidylate phosphoribosyl transferase activity (Fig. 5).
FIGURE 5.
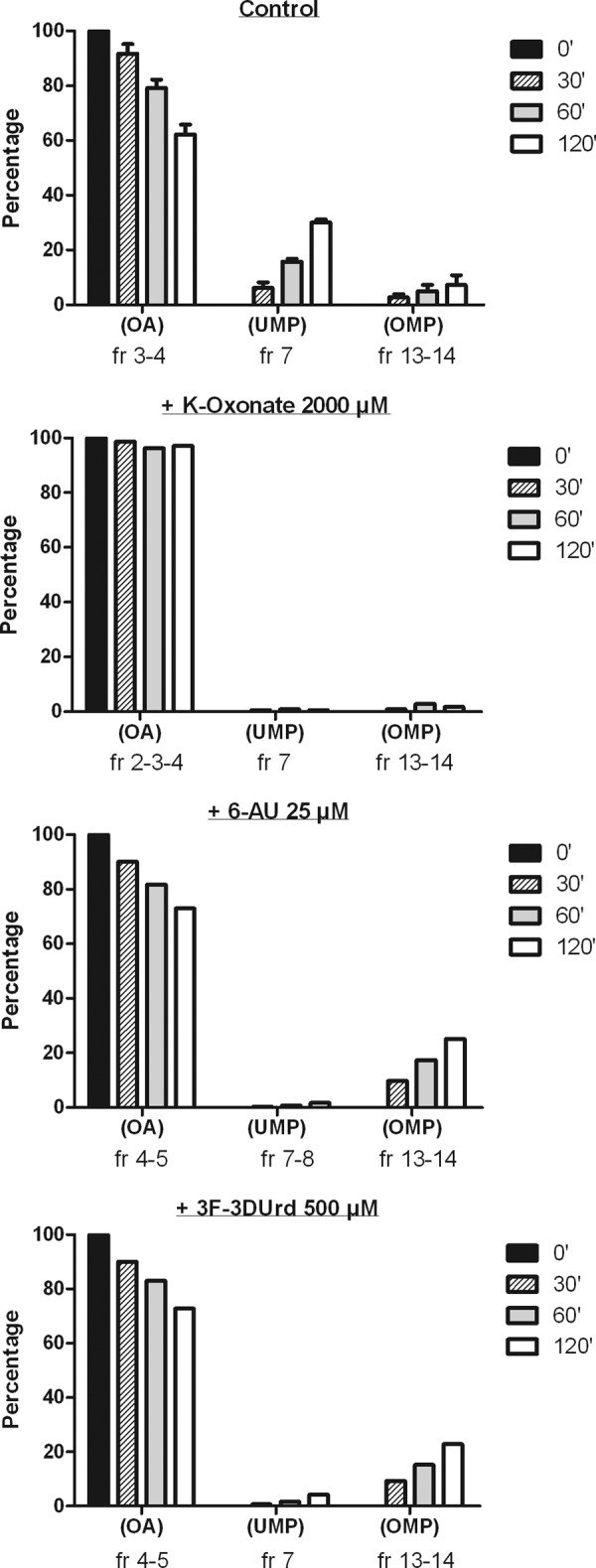
Conversion of [14C]OA to [14C]OMP by OPRT and [14C]UMP by ODC in CEM cell extracts exposed to K-oxonate, 6AUrd, or 3F-3DUrd and in the presence of exogenously added UCK.
Because 5-FU is converted to 5-FUMP by the OPRT activity of UMPS (17) but 5-FUMP is not a substrate for the ODC activity of UMPS, it was logical to perform similar experiments with [3H]5-FU in place of [14C]OA. It is noteworthy that CEM cell extracts converted 5-FU to 5-FUMP equally well in the presence or absence of 100-μm 6AUMP or 500-μm 3F-3DUMP, whereas 2-mm K-oxonate efficiently blocked 5-FUMP formation (Fig. 6). This again suggests that 3F-3DUrdMP is inhibitory for the orotidylate decarboxylase activity, but not the orotate phosphoribosyl transferase activity of the uridylate synthase enzyme complex.
FIGURE 6.
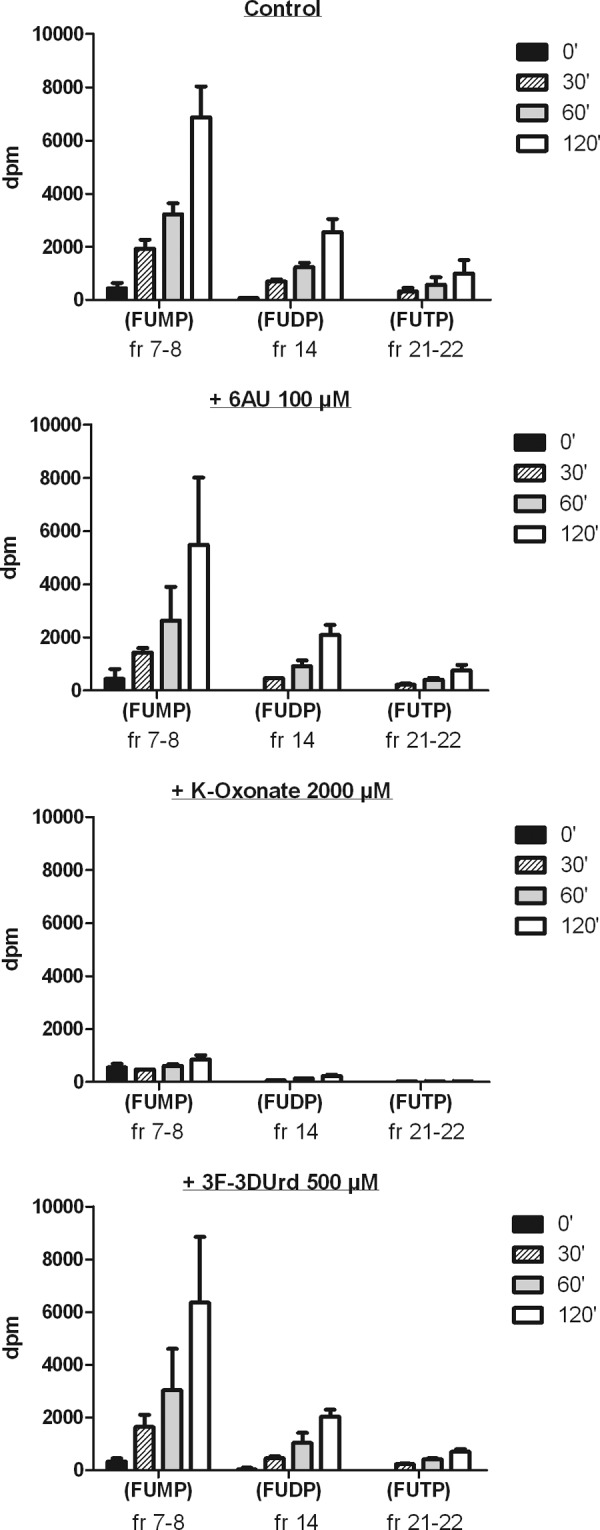
Conversion of [3H]5-FU to [3H]5-FUMP by OPRT in CEM cell extracts in the absence or presence of 6AUMP, 3F-3DUMP, or K-oxonate.
Poor Conversion of 3F-3DUrd to Its 5′-Triphosphate Derivative in the Presence of Uridine/Cytidine Kinase and UMP/CMP Kinase in a Cell-free Enzyme Assay
The state of base ionization in 3F-3DUrd (and 5′-phosphate derivatives) relative to that of the 3-DUrd analogues in the cultures/extracts might significantly affect binding at enzyme active sites and/or allosteric sites. 3-DUrd has a pKa of ∼6.2–6.5 (18, 19). The lower pKa of 3F-3DUrd could have a marked effect on both the extent of ionization to the anionic enolate and also on the electronic distribution in the keto-enol tautomeric neutral forms. Calculations3 indicate that a 3DUrd solution at pH 7.0 has ∼96.5% in the 4-enol form and ∼3.5% in the ionized form, whereas only ∼32.1% of 3F-3DUrd is in its 4-enol form and ∼67.9% in the ionized form at pH 7.0. Transport of the more extensively ionized 3F-3DUrd across cell membranes, and binding to UMP kinase- and NDP kinase-type enzymes might be much weaker than with 3DUrd (and its 5′-phosphate). If 3F-3DUrd does not (adequately) bind to UMP kinase/NDP kinase-type enzymes, this could be a major factor in failing to reach significant intracellular 3F-3DUDP and 3F-3DUTP levels, and subsequent lack of CTP synthetase inhibition. An experiment was performed in which 500-μm 3F-3DUrd was exposed to a mixture of uridine/cytidine kinase and UMP/CMP kinase (in the presence of 2.5 mm ATP) for 24 h at 37 °C, and the resulting products were quantified by HPLC. All of the 3F-3DUrd was converted into its 5′-monophosphate (93%), 5′-diphosphate (5%), and 5′-triphosphate (2%) derivatives. The highly predominant presence of the 5′-monophosphate and lack of effective conversion to the 5′-di- and 5′-triphosphates are in agreement with the undetected levels of 3F-3DUrd di- and 5′-triphosphate in the HPLC chromatograms of the metabolites (supplemental Fig. S2) and point to a likely restriction of significant conversion of the drug into its 5′-triphosphate derivative in intact tumor cells. Consequently, significant CTP synthetase inhibition, if any, would not be expected to occur in 3F-3DUrd-exposed tumor cells.
Structural Considerations
High level quantum mechanical calculations (BHandHLYP/aug-cc-pVTZ) on N-methyl-3-deazauracil favor the 4-hydroxy-2-pyridone enol tautomer over the keto form (data not shown), and the same is true for N-methyl-3-deaza-3F-uracil. In both cases, the ring structure is planar. These results for N-methyl-3-deazauracil are consistent with published data for 3DUrd in solution and in the solid state (20). 3DUrd was regarded as a cytidine analogue because of the similar arrangement of single and double bonds in the heterocyclic rings (20). A proton acceptor is lacking at position 3, although its absence might be compensated in some binding sites by the greater proton donor strength of the significantly acidic phenolic 4-hydroxyl group (Fig. 3). Similar considerations apply with 3F-3DUrd; but in this case the fluorine atom can function as a weak acceptor of hydrogen bonds from NH or OH groups in proteins. It also could participate in favorable dipolar interactions with carbonyl carbons (so-called fluorophilic sites) (21). The fluorine atom at C3 would also significantly enhance the acidity of the neighboring phenolic hydroxyl group at C4.
ODC activity in human cells is harbored in the C-terminal domain of the bifunctional UMPS enzyme that is also endowed with OPRT activity in its N-terminal domain (7). Human UMPS is an obligate dimer, and capping of one monomer by the other is strictly required to constitute a functional ODC catalytic center (22). The mechanism of decarboxylation involves tight binding and conformational restraint of the entire OMP molecule (phosphate, ribose, and base) in the active site to ensure substrate destabilization along the reaction coordinate by forcing the carboxylate group out of the plane of the pyrimidine ring. To achieve this, the carboxamide nitrogen of Gln-430 in the human ODC donates hydrogen bonds to O2 of OMP (UMP in Fig. 7, left panel) and one of the phosphate oxygens. In turn, the carboxamide oxygen accepts a hydrogen bond from the side chain hydroxyl of Ser-372, and that hydroxyl oxygen accepts a hydrogen bond from N3 of OMP (UMP in Fig. 7, left panel). Finally, the NH of Ser-372 donates a hydrogen bond to O4 of OMP (UMP in Fig. 7, left panel). This obligate pattern is consistent with the fact that ODC activity is not inhibited by 3DUrd, which lacks a NH hydrogen bonding functionality at position 3. However, our finding that 3F-3DUMP is a good inhibitor of ODC suggests that the fluorine-containing analogue somehow circumvents that limitation, perhaps by the creation of a fluorophilic site (21) (Fig. 7, right panel). A crystallographic complex of ODC with 3F-3DUMP will enable clarification of this issue.
FIGURE 7.

Molecular interaction of UMP and 3F-3DUMP with human orotidine 5′-monophosphate decarboxylase (ODC). Left panel, interaction of the reaction product UMP in the active site of ODC, as seen in the x-ray crystal structure deposited with Protein Data Bank code 2QCD (22). Hydrogen bonds are shown as broken lines. Crosses represent water molecules. Right panel, structure of 3F-3DUMP modeled into the same active site. The fluorine atom is colored cyan. Hydrogens are omitted for clarity.
DISCUSSION
It was surprising to discover that introduction of a fluorine atom at C3 of the 3-deazauracil ring resulted in a shift of the enzyme target of 3DUrd (as its 5′-triphosphate) from inhibition of CTP-S to inhibition of the ODC activity of UMPS by 3F-3DUrd (as its 5′-monophosphate). Introduction of a fluorine atom at C3 of 3DUrd allows formation of two (R and S) diastereomeric keto tautomers of the fluorinated molecule (in equilibrium with the enol form) (Fig. 3). The enol form of 3DUrd predominates in neutral aqueous solution, but the pKa-lowering effect of the fluoro substituent on 3F-3DUrd could alter the neutral tautomeric as well as the ionization equilibria significantly. In fact, Nesnow et al. (23) found that the introduction of a fluorine atom at the 5-position of 3-deazauridine (pKa = ∼6.5) resulted in the reduction of the pKa by two units for 5-fluoro-3-deazauridine (pKa = ∼4.5). A similar reduction in pKa would be expected for 3-fluoro-3-deazauridine and is in full agreement with our quantum mechanical calculation. The pKa-lowering effect of the fluoro substituent on 3F-3DUrd might result in less efficient recognition/binding of 3F-3DUrd 5′-monophosphate for further phosphorylation by UMP/CMP kinase. Our cell-free enzyme (Urd/Cyd kinase + UMP/CMP kinase) and metabolic experiments are in harmony with this possibility, which predicts that significant levels of 3F-3DUrd 5′-triphosphate were not present in the intact tumor cells. Therefore, it remains possible that if 3F-3DUrd 5′-triphosphate were to have been formed intracellularly at sufficiently high levels, it might also have inhibited CTP synthetase. Kinetic analysis of purified CTP synthetase in the presence of synthetically generated 3F-3DUrd 5′-triphosphate should clarify this issue.
A large part of the substrate and inhibitor binding energies in their complexes with ODC is thought to arise from interactions of the enzyme with the 5′ phosphate and ribose hydroxyl groups (Fig. 7). These strong attractive forces, together with hydrogen bonds that hold the pyrimidine base firmly in position and precisely oriented, make it possible for the side chain carboxylate of Asp-312 to exert electrostatic repulsion on the leaving carboxylate group of OMP and to catalyze the reaction very efficiently (“Circe effect”) (24). The resulting loss of entropy causes a decrease in affinity, but it provides a sufficient free energy advantage for the huge rate acceleration of OMP decarboxylation exerted by this highly proficient enzyme (25). The loss of entropy also affects nucleotide analogues that inhibit ODC, which likely explains why inhibition constants are all in the high micromolar range (except for those inhibitors that bind covalently to the enzyme, e.g. 6-iodo-UMP) (26).
In contrast, some competitive inhibitors of ODC such as CMP and xanthosine 5′-monophosphate have been shown to bind in the active site of the highly homologous Methanobacterium thermoautotrophicum ODC in an unusual fashion (27). Solid state x-ray crystal structures indicate that the phosphate-binding loop is disordered, and the nucleobases form hydrogen bonds only with a conserved Ser residue (directly for xanthosine 5′-monophosphate or through a water molecule for CMP) (27). CMP, unlike OMP and UMP, does not have a hydrogen bond donor at position 3 but has a nitrogen atom that can accept a hydrogen bond. It also has a hydrogen bond donor (amino group) at C4 analogous to the phenolic hydroxyl group at C4 of 3F-3DUrd. It is possible that 3F-3DUrd binds to human ODC by occupying its active site in a different manner than other potent inhibitors such as 5′-phosphoribofuranosyl barbituric acid and 6AUrd that are anchored through an extended hydrogen-bonded network involving all of the phosphate oxygens, the ribose hydroxyl groups, and the nucleobase (26).
It is remarkable that substitution of fluorine for hydrogen at C3 of 3DUrd (a prodrug that undergoes phosphorylation to 3DUTP, which inhibits CTP-S) produces a new prodrug 3F-3DUrd that is monophosphorylated to 3F-3DUMP (which is an inhibitor of ODC). Variables that might alter binding and/or phosphorylation of these two closely related prodrugs (especially at the nucleotide relative to nucleoside kinase levels) include: 1) enhancement of the acidity of the phenolic hydroxyl group at C4 of 3F-3DUrd relative to that of 3DUrd with correspondingly increased ratios of the ionized enolate form, 2) polarization effects with the electronegative fluorine substituent, 3) altered ratios of the neutral keto/enol tautomers, and 4) preferential binding of one diastereomeric keto tautomer of 3F-3DUrd. Answers to two questions would provide important information: 1) does 3DUMP also inhibit ODC? and 2) does 3F-3DUTP also inhibit CTP-S? Studies in progress and evaluation of additional prodrug analogues will pursue answers to these questions.
Introduction of fluorine into nucleosides has generated analogues with potent antimetabolite activities. Examples are 5-fluoro-2′-deoxyuridine (fludoxuridine), 2′,2′-difluorocytidine (gemcitabine) and 2′-fluoro-araA (fludarabine) that inhibit thymidylate synthase, ribonucleotide reductase, DNA polymerase, and/or incorporation/excision of nucleoside (28). We have demonstrated that the introduction of fluorine into the antimetabolite drug 3-deazauridine mechanistically shifts its antimetabolic (inhibitory) action from CTP synthetase to OMP decarboxylase. By doing so, it both decreases CTP pools (as does its parent drug 3DUrd) and drops UTP pools. Although anticancer selectivity is potentially compromised by the existence of the molecular targets in both tumor cells and host tissues, compounds such as 3F-3DUrd may demonstrate an adequate degree of selectivity resulting from differences between tumor and normal cells in metabolic and proliferative states (28). Primary colon tumors and xenographs contain higher specific activities of enzymes of salvage and de novo pathways of UMP synthesis in comparison with normal colon cells (specifically, UCK (the activating enzyme for 3F-3DUrd) and ODC (the target enzyme for 3F-3DUrd)) (29). Furthermore, most normal cells in a patient are quiescent and therefore less sensitive to cytostatic agents (28).
3F-3DUrd, 3DUrd, pyrazofurin, and 6-AUrd must be converted/activated to at least their 5′-monophosphates by UCK before they can inhibit the target enzyme OPRT/ODC (for 3F-3DUrd, pyrazofurin, and 6-AUrd as 5′-monophosphates) or CTP-S (for 3DUrd 5′-triphosphate). The addition of Urd or Cyd would directly compete with these antimetabolites for phosphorylation and thus would delay their conversion to the active 5′-phosphate derivatives. Because the overall antiproliferative effects of the antimetabolites should result from depletion of UTP and/or CTP pools, addition of Urd or Cyd to the drug-exposed cell cultures would restore the UTP/CTP pools and annihilate the cytostatic drug activity. The latter effect may also explain the decreased antiproliferative activity of the OPRT inhibitor K-oxonate and the DHO-DH inhibitor A771726, neither of which require activation by UCK to exert their cytostatic activities.
Our findings that the natural purine bases Ade and Hpx and their ribonucleosides Ado and Ino markedly increased the cytostatic potential of 3F-3DUrd in a dose-dependent manner, but not that of 3DUrd (nor A771726) in the drug-exposed CEM tumor cell cultures, are also in agreement with ODC inhibition by 3F-DUrd monophosphate. These observations can indeed be rationalized by assuming that the addition of an excess of Hpx, Ade, Ino (upon purine nucleoside phosphorylase-driven intracellular hydrolysis to Hpx), and Ado (upon deamination to Ino by adenosine deaminase and subsequent hydrolysis to Hpx by purine nucleoside phosphorylase) substantially depletes the intracellular 5-phosphoribosyl-1-pyrophosphate pools by formation of IMP and AMP through the action of hypoxanthine phosphoribosyl transferase and adenine phosphoribosyl transferase, respectively. As a result, concentrations of the depleted 5-phosphoribosyl-1-pyrophosphate pools might become rate-limiting for the OPRT/ODC-catalyzed synthesis of OMP/UMP from orotate (supplemental Fig. S3). The reduced levels of OMP, which is a competing substrate for binding to ODC, might also result in increased levels of inhibition of ODC by monophosphorylated 3F-3DUrd, pyrazofurin, and 6AUrd. A similar phenomenon was noted earlier with pyrazofurin and 6AUrd in CHO cells (30). The lack of such pronounced potentiating activity on the cytostatic action of 3DUrd with the noted purine derivatives is in harmony with its different mechanism of cytostatic action (i.e., CTP-S for 3DUrd versus OPRT/ODC for 3F-3DUrd).
In conclusion, introduction of a fluorine atom at the C3 position of the CTP synthetase-targeting 3DUrd resulted in a novel compound (3F-3DUrd) that gained new inhibitory properties as its 5′-monophosphate against the ODC activity of UMP synthase. The marked lowering of the pKa of 3F-3DUrd and resulting effects on the tautomerism and ionization state of the 4-hydroxyl group most likely affect its interaction with this enzyme that catalyzes an early event in pyrimidine nucleotide biosynthesis. This inhibition shift in the antimetabolic target enzyme was confirmed by concomitant depletion of intracellular UTP and CTP pools in 3F-3DUrd-exposed tumor cells as well as by the markedly increased cytostatic potential of 3F-3DUrd upon 5-phosphoribosyl-1-pyrophosphate pool depletion in the presence of the natural purine bases and their ribonucleosides.
Supplementary Material
Acknowledgments
We thank Ria Van Berwaer, Kristien Minner, and Lizette van Berckelaer for excellent technical assistance and Christiane Callebaut for dedicated editorial help.
The work was supported by Katholieke Universiteit Leuven Grant GOA 10/14.

This article contains supplemental Table S1 and Figs. S1–S3.
C. McGuigan and J. Balzarini, unpublished data.
- CTP-S
- CTP synthetase
- 6AUrd
- 6-azauridine
- DHO-DH
- dihydroorotate-dehydrogenase
- 3-DUrd
- 3-deazauridine
- 3F-3DUrd
- 3-fluoro-3-DUrd
- 5-FU
- 5-fluorouracil
- 5-FUMP
- 5-fluoro-uridylate
- Hpx
- hypoxanthine
- OA
- orotic acid
- ODC
- OMP decarboxylase
- OMP
- orotidylate
- OPRT
- orotate phosphoribosyl transferase
- Otd
- oritidine
- UCK
- uridine/cytidine kinase
- UDPG
- UDP-glucose
- UMPS
- UMP synthase.
REFERENCES
- 1. Tsuboi K. K., Kwong L. K. (1978) Antiproliferative agents and differential survival between normal and cancer cells. Cancer Res. 38, 3745–3750 [PubMed] [Google Scholar]
- 2. Greene S., Watanabe K., Braatz-Trulson J., Lou L. (1995) Inhibition of dihydroorotate dehydrogenase by the immunosuppressive agent leflunomide. Biochem. Pharmacol. 50, 861–867 [DOI] [PubMed] [Google Scholar]
- 3. Vyas V. K., Ghate M. (2011) Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) inhibitors. Mini Rev. Med. Chem. 11, 1039–1055 [DOI] [PubMed] [Google Scholar]
- 4. Shirasaka T., Shimamoto Y., Fukushima M. (1993) Inhibition by oxonic acid of gastrointestinal toxicity of 5-fluorouracil without loss of its antitumor activity in rats. Cancer Res. 53, 4004–4009 [PubMed] [Google Scholar]
- 5. Dix D. E., Lehman C. P., Jakubowski A., Moyer J. D., Handschumacher R. E. (1979) Pyrazofurin metabolism, enzyme inhibition, and resistance in L5178Y cells. Cancer Res. 39, 4485–4490 [PubMed] [Google Scholar]
- 6. Handschumacher R. E. (1960) Orotidylic acid decarboxylase. Inhibition studies with azauridine 5′-phosphate. J. Biol. Chem. 235, 2917–2919 [PubMed] [Google Scholar]
- 7. Evans D. R., Guy H. I. (2004) Mammalian pyrimidine biosynthesis. Fresh insights into an ancient pathway. J. Biol. Chem. 279, 33035–33038 [DOI] [PubMed] [Google Scholar]
- 8. Robins M. J., Lee A. S. (1975) Nucleic acid related compounds. 17. 3-Deazuridine. Stannous chloride catalysis of cis-diol vs. phenolic base methylation with diazomethane. J. Med. Chem. 18, 1070–1074 [DOI] [PubMed] [Google Scholar]
- 9. Robins M. J., Currie B. L. (1968) The synthesis of 3-deazauridine. Chem. Commun. 1547–1548 [Google Scholar]
- 10. McPartland R. P., Wang M. C., Bloch A., Weinfeld H. (1974) Cytidine 5′-triphosphate synthetase as a target for inhibition by the antitumor agent 3-deazauridine. Cancer Res. 34, 3107–3111 [PubMed] [Google Scholar]
- 11. Glazer R. I., Knode M. C., Lim M. I., Marquez V. E. (1985) Cyclopentenyl cytidine analogue. An inhibitor of cytidine triphosphate synthesis in human colon carcinoma cells. Biochem. Pharmacol. 34, 2535–2539 [DOI] [PubMed] [Google Scholar]
- 12. De Clercq E. (1987) Targets for the antiviral activity of pyrimidine and purine nucleoside analogues. Nucleosides Nucleotides 6, 197–207 [Google Scholar]
- 13. Robins M. J., Yang H., Miranda K., Peterson M. A., De Clercq E., Balzarini J. (2009) Synthesis and biological evaluation of 3,3-difluoropyridine-2,4(1H,3H)-dione and 3-deaza-3-fluorouracil base and nucleoside derivatives. J. Med. Chem. 52, 3018–3027 [DOI] [PubMed] [Google Scholar]
- 14. van Kuilenburg A. B., van Lenthe H., Löffler M., van Gennip A. H. (2004) Analysis of pyrimidine synthesis “de novo” intermediates in urine and dried urine filter-paper strips with HPLC-electrospray tandem mass spectrometry. Clin. Chem. 50, 2117–2124 [DOI] [PubMed] [Google Scholar]
- 15. Van Rompay A. R., Norda A., Lindén K., Johansson M., Karlsson A. (2001) Phosphorylation of uridine and cytidine nucleoside analogs by two human uridine-cytidine kinases. Mol. Pharmacol. 59, 1181–1186 [DOI] [PubMed] [Google Scholar]
- 16. Van Rompay A. R., Johansson M., Karlsson A. (1999) Phosphorylation of deoxycytidine analog monophosphates by UMP-CMP kinase. Molecular characterisation of the human enzyme. Mol. Pharmacol. 56, 562–569 [DOI] [PubMed] [Google Scholar]
- 17. Sakamoto E, Nagase H, Kobunai T, Oie S, Oka T, Fukushima M, Oka T. (2007) Orotate phosphoribosyltransferase expression level in tumors is a potential determinant of the efficacy of 5-fluorouracil. Biochem. Biophys. Res. Commun. 363, 216–222 [DOI] [PubMed] [Google Scholar]
- 18. Dahlig-Harley E., Paterson A. R., Robins M. J., Cass C. E. (1984) Transport of uridine and 3-deazauridine in cultured human lymphoblastoid cells. Cancer Res. 44, 161–165 [PubMed] [Google Scholar]
- 19. Gassen H. G., Schetters H., Matthaei H. (1972) Codon-anticodon interaction studied with oligonucleotides containing 3-deazauridine, 4-deoxyuridine or 3-deaza-4-deoxyuridine. II. Ribosome binding of oligonucleotides and phenylalanyl-tRNA. Biochim. Biophys. Acta 272, 560–567 [DOI] [PubMed] [Google Scholar]
- 20. Schwalbe C. H., Gassen H. G., Saenger W. (1972) 3-Deazauridine. Crystal structure and conformation. Nat. New Biol. 238, 171–173 [DOI] [PubMed] [Google Scholar]
- 21. Böhm H. J., Banner D., Bendels S., Kansy M., Kuhn B., Müller K., Obst-Sander U., Stahl M. (2004) Fluorine in medicinal chemistry. Chembiochem. 5, 637–643 [DOI] [PubMed] [Google Scholar]
- 22. Wittmann J. G., Heinrich D., Gasow K., Frey A., Diederichsen U., Rudolph M. G. (2008) Structures of the human orotidine-5′-monophosphate decarboxylase support a covalent mechanism and provide a framework for drug design. Structure 16, 82–92 [DOI] [PubMed] [Google Scholar]
- 23. Nesnow S., Miyazaki T., Khwaja T., Meyer R. B., Jr., Heidelberger C. (1973) Pyridine nucleosides related to 5-fluorouracil and thymine. J. Med. Chem. 16, 524–528 [DOI] [PubMed] [Google Scholar]
- 24. Jencks W. P. (1975) Binding energy, specificity, and enzymic catalysis. The Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 43, 219–410 [DOI] [PubMed] [Google Scholar]
- 25. Radzicka A., Wolfenden R. (1995) A proficient enzyme. Science 267, 90–93 [DOI] [PubMed] [Google Scholar]
- 26. Heinrich D., Diederichsen U., Rudolph M. G. (2009) Lys314 is a nucleophile in non-classical reactions of orotidine-5′-monophosphate decarboxylase. Chemistry 15, 6619–6625 [DOI] [PubMed] [Google Scholar]
- 27. Wu N., Pai E. F. (2002) Crystal structures of inhibitor complexes reveal an alternate binding mode in orotidine-5′-monophosphate decarboxylase. J. Biol. Chem. 277, 28080–28087 [DOI] [PubMed] [Google Scholar]
- 28. Parker W. B. (2009) Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 109, 2880–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahmed N. K. (1984) Enzymes of the de novo and salvage pathways for pyrimidine biosynthesis in normal colon, colon carcinoma, and xenografts. Cancer 54, 1370–1373 [DOI] [PubMed] [Google Scholar]
- 30. Gupta R. S., Moffat M. R. (1982) Synergistic effect of purine derivatives on the toxicity of pyrazofurin and 6-azauridine towards cultured mammalian cells. J. Cell. Physiol. 111, 291–294 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.