

Background: PKCθ plays a key role in T lymphocyte activation, but its regulatory mechanism is not understood.
Results: Phosphotyrosine binds the PKCθ C2 domain and activates PKCθ.
Conclusion: The PKCθ C2 domain-phosphotyrosine binding is important for PKCθ activation in T cells.
Significance: This study provides new mechanistic insight into the regulation of PKCθ in T cells.
Keywords: Lipid-binding Protein, Phosphotyrosine, Protein Kinase C (PKC), Signal Transduction, T Cell Biology, C2 Domain
Abstract
Protein kinase Cθ (PKCθ) is a novel PKC that plays a key role in T lymphocyte activation. To understand how PKCθ is regulated in T cells, we investigated the properties of its N-terminal C2 domain that functions as an autoinhibitory domain. Our measurements show that a Tyr(P)-containing peptide derived from CDCP1 binds the C2 domain of PKCθ with high affinity and activates the enzyme activity of the intact protein. The Tyr(P) peptide also binds the C2 domain of PKCδ tightly, but no enzyme activation was observed with PKCδ. Mutations of PKCθ-C2 residues involved in Tyr(P) binding abrogated the enzyme activation and association of PKCθ with Tyr-phosphorylated full-length CDCP1 and severely inhibited the T cell receptor/CD28-mediated activation of a PKCθ-dependent reporter gene in T cells. Collectively, these studies establish the C2 domain of PKCθ as a Tyr(P)-binding domain and suggest that the domain may play a major role in PKCθ activation via its Tyr(P) binding.
Introduction
Protein kinases C (PKCs) comprise a family of serine/threonine kinases that mediate a wide variety of cellular processes (1–3). All PKCs contain an N-terminal regulatory domain and a C-terminal catalytic domain. PKCs are typically subdivided into three classes: conventional PKC (α, βI, βII, and γ subtypes), novel PKC (δ, ϵ, η, and θ subtypes), and atypical PKC (ζ and λ/ι subtypes), based upon structural differences in the regulatory domain. Conventional and novel PKCs have two types of membrane-targeting domains, a tandem repeat of C1 domains (C1A and C1B) and a C2 domain, in the regulatory domain. The C1 domain (∼50 residues) is a cysteine-rich compact structure that was identified as the interaction site for sn-1,2-diacylglycerol (DAG)4 and phorbol ester (4, 5). C1 domains display different affinities for DAG and phorbol esters (6–9), the basis of which has not been fully elucidated, and also bind alcohols (10–14). The C2 domain (∼130 residues) is an eight-stranded β sandwich protein involved in Ca2+-dependent membrane binding for conventional isoforms (15–17). All novel PKCs contain a Ca2+-independent C2 domain, which has been reported to interact with other proteins (18, 19) or anionic phospholipids in the case of PKCϵ (20–22).
PKCθ is a novel PKC expressed exclusively in T lymphocytes and muscle cells (23, 24). T cell activation requires sustained interaction of the T cell receptor (TCR) with MHC-bound peptide antigen presented by antigen-presenting cells. The T-antigen-presenting cell contact region, where this interaction occurs, referred to as the immunological synapse, serves as a signaling platform where activated TCR molecules, accessory/costimulatory receptors, and intracellular adaptors and enzymes are clustered (25). Generation of DAG by phospholipase Cγ1 in the plasma membrane in response to TCR triggering is a key signal in the initiation of T cell activation, which culminates in cell proliferation and the execution of T cell effector functions (26). Although T cells express many PKC isoforms (α, βI, δ, ϵ, η, θ, and ζ) (23, 24), PKCθ plays a non-redundant and obligatory function in mature T cell activation (but not in T cell development), as indicated by findings that PKCθ-deficient T cells display a grossly defective proliferation and interleukin-2 production in response to TCR/CD28 costimulation, resulting from impaired activation of the transcription factors nuclear factor (NF)-κB, AP-1, and, to a lesser extent, NFAT (27, 28).
We recently performed rigorous analysis of the mechanism of DAG-dependent membrane targeting and activation of PKCθ (29). This study revealed that PKCθ followed a unique mode of DAG-dependent activation among PKC isoforms (6, 8, 9, 30), where the C1B domain is predominantly involved in the membrane binding and activation. Additionally, we found that the C2 domain of PKCθ (PKCθ-C2) plays a unique autoinhibitory role in the resting state of PKCθ, suppressing its membrane binding and activation until phosphorylation of Tyr90, which was reported to be induced by TCR stimulation (31), unlocks an inhibitory intramolecular tether. The C2 domain of PKCδ, the closest relative of PKCθ in the PKC family, was found to be a novel Tyr(P)-binding domain, which could specifically bind a Tyr(P)-containing peptide derived from CDCP1, a protein known to be phosphorylated on tyrosine by activated Src kinase (18). However, the physiological significance of this interaction has not been demonstrated. High structural similarity between PKCδ-C2 and PKCθ-C2 and the unique autoinhibitory role of PKCθ-C2 raise an interesting possibility that PKCθ-C2 may also bind Tyr(P) and that this interaction may play an important role in the activation of PKCθ in T cells. To test this hypothesis, we measured the binding of PKCθ-C2 to a Tyr(P) peptide and the effect of this interaction on PKC activation in vitro and in T cells. Our results show that PKCθ-C2 is a genuine Tyr(P)-binding module and that this interaction modulates the in vitro enzyme activity of PKCθ and its function in T cells.
EXPERIMENTAL PROCEDURES
Materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS), and sn-1,2-dioleoylglycerol (DiC18) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and used without further purification. Triton X-100 and ATP were from Sigma. Phospholipids concentrations were determined by a modified Bartlett analysis (32). [γ-32P]ATP (3 Ci/μmol) and L1 sensor chip were from GE Healthcare. Two peptides (GGALYSIYQPYVFAKKK and GGALYSIpYQPYVGAKKK) were synthesized using an Applied Biosystems (ABI) 433A peptide synthesizer.
Expression Vector Construction and Mutagenesis
For bacterial expression of the C2 domain, the expression vector was created by subcloning the cDNA sequence encoding the C2 domain (amino acids 1–123) of human PKCθ into the pET28a vector (Novagen, Madison, WI) between the restriction sites NcoI and XhoI by overlap extension PCR using Pfu polymerase (Stratagene, La Jolla, CA). This vector is designed to introduce an N-terminal His6 tag for affinity purification of the expressed proteins. Mutants of the C2 domain were generated using four primers via the overlap extension method (33). For mammalian expression, wild-type (WT) PKCθ and C2-mutated cDNAs were cloned into the pEGFP-N1 vector (Clontech) between the restriction sites XhoI and BamHI. Mutants of the C2 domain were generated using four primers via the overlap extension method (33, 35). For insect cell expression of WT or mutated PKCθ, baculovirus transfer vectors encoding the cDNA of PKCθ were generated by PCR using the pVL1392 PKCθ plasmid as a template (36). The mutation was verified by DNA sequencing.
Protein Expression and Purification
Escherichia coli strain BL21(DE3) (Novagen) was used as a host for C2 domain expression. Two liters of Luria broth was inoculated with a 50-ml overnight culture of cells. Cells were grown at 37 °C with shaking (250 rpm) to an optical density of 0.8 at 600 nm, at which time the protein production was induced by adding 1 mm isopropyl β-d-1-thiogalactopyranoside. Cells were then incubated with shaking (250 rpm) at 25 °C for 4 h. For harvesting, cells were centrifuged at 5000 × g for 10 min, and the pellet was washed once with sodium phosphate buffer (pH 8.0) and resuspended in 25 ml of extraction buffer containing 50 mm sodium phosphate, pH 8.0, 300 mm NaCl, 10 mm imidazole, 1% Triton X-100, and 0.2 mm phenylmethylsulfonyl fluoride. The suspension was homogenized in a hand-held homogenizer chilled on ice. The extract was centrifuged at 50,000 × g for 45 min at 4 °C. One ml of nickel-nitrilotriacetic acid-agarose (Qiagen, Valencia, CA) was added to the supernatant, and the mixture was incubated on ice while shaking at 80 rpm for 1 h. The mixture was poured onto a 10-ml column, and the column was washed first with 20 ml of 50 mm sodium phosphate, pH 8.0, containing 300 mm NaCl and 10 mm imidazole and subsequently with 15 ml of 50 mm sodium phosphate, pH 8.0, containing 300 mm NaCl and 15 mm imidazole. The column was washed with an additional 10 ml of the same buffer containing 20 mm imidazole. The protein was then eluted in seven fractions of 0.5 ml in 50 mm sodium phosphate buffer containing 300 mm imidazole. Eluted fractions were then analyzed on a 14% SDS-polyacrylamide gel. Fractions containing the PKCθ C2 domain were concentrated and desalted in an Ultrafree-15 centrifugal filter device (Millipore, Bedford, MA).
Full-length PKCθ and mutants were expressed in baculovirus-infected Sf9 cells. The pVL1392-PKCθ DNA was prepared for transfection by using an EndoFree Plasmid Midi kit (Qiagen) to avoid endotoxin contamination and transfected into Sf9 cells with the BaculogoldTM transfection kit (BD Pharmingen). Cells were incubated for 4 days at 27 °C, and the supernatant was collected and used for more cells for amplification of virus. After three cycles of amplification, high titer virus stock was obtained. Sf9 cells were maintained as monolayer cultures in TMN-FH medium (Invitrogen) containing 10% fetal bovine serum (Invitrogen). For expression of the proteins, cells were grown to 2 × 106 cells/ml in 300-ml suspension cultures and infected with a multiplicity of infection of 10. The cells were then incubated for 60 h at 27 °C. For protein purification, cells were harvested at 1000 × g for 10 min, the pellet was then washed once with sodium phosphate buffer, pH 8.0, and resuspended in 22 ml of extraction buffer containing 50 mm sodium phosphate, pH 8.0, 300 mm NaCl, 10 mm imidazole, 1% Triton X-100, 0.2 mm phenylmethylsulfonyl fluoride, and a protease inhibitor mixture tablet EDTA-free (Roche Applied Science). The suspension was homogenized in a handheld homogenizer chilled on ice. The extract was centrifuged at 50,000 × g for 45 min at 4 °C. One ml of nickel-nitrilotriacetic acid-agarose (Qiagen) was added and incubated on ice while shaking at 80 rpm for 1 h. The mixture was poured into a 10-ml column and washed first with 10 ml of 50 mm sodium phosphate, pH 8.0, containing 300 mm NaCl and 10 mm imidazole and subsequently with 10 ml of 50 mm sodium phosphate, pH 8.0, containing 300 mm NaCl and 15 mm imidazole. The column was then washed with 6 ml of the same buffer containing 20 mm imidazole and next with 3 ml of the same buffer containing 25 mm imidazole. The protein was then eluted from the column with six 0.75-ml fractions of 50 mm sodium phosphate, pH 8.0, containing 300 mm NaCl and 300 mm imidazole. The eluted fractions were then analyzed on an 8% SDS-polyacrylamide gel. Fractions containing PKCθ were concentrated and desalted in an Ultrafree-15 centrifugal filter device (Millipore). Protein concentration was determined by the bicinchoninic acid method (Pierce).
Determination of PKC Activity
Activity of PKCδ and PKCθ was assayed by measuring the initial rate of [32P]phosphate incorporation from [γ-32P]ATP (50 mm, 0.6 mCi/tube) into myelin basic protein (200 mg/ml) (9). The reaction mixture contained large unilamellar vesicles (0.2 mm total lipid concentration), 0.16 m KCl, and 5 mm MgCl2 in 20 mm HEPES, pH 7.4. The reaction was started by adding 50 mm MgCl2 to the mixture and left for 10 min at room temperature. The reaction was then quenched with 5% phosphoric acid. Next, 75 μl of the quenched reaction mixture was transferred to P-81 ion exchange paper and washed three times with 5% phosphoric acid followed by one wash with 95% ethanol. The papers were dried in an oven at 60 °C for 10 min. The papers were then transferred into scintillation vials containing 4 ml of scintillation fluid (Fisher), and radioactivity was determined by liquid scintillation counting. To rule out the possibility that the control or the Tyr(P) peptide is phosphorylated during our assay, the phosphorylation of these peptides was assessed under the same conditions without myelin basic protein (i.e. with and without POPC/POPS/DiC18 (59:40:1) vesicles). Under these conditions, phosphorylation of the peptides was not detectable, demonstrating that phosphorylation of the control or the Tyr(P) peptide does not contribute to the [32P]phosphate signal in our PKC activity assays.
Surface Plasmon Resonance (SPR) Analysis
PKC-peptide binding was measured by the SPR analysis using a BIAcore X biosensor system and the L1 chip as described previously (37–39). The Tyr(P) peptide was amine-coupled to a CM5 chip (BIAcore) to a density of 200 resonance units according the manufacturer's protocol. Likewise, the control peptide was coupled to a second sensor surface and served as a control. The equilibrium dissociation constant (Kd) was determined from steady-state binding measurements as described previously (40, 41). Briefly, varying concentrations of PKCδ, PKCθ, and their respective C2 domains (typically within a 10-fold range above or below the Kd) were injected at 5 μl/min at 23 °C, and data were analyzed using BIAevaluation 3.0 software (Biacore) as described previously (37, 38). In separate experiments, both PKCθ and PKCδ were amine-coupled to the sensor surface, and Tyr(P) peptide or non-Tyr(P) peptide was injected at varying concentrations. Kd values obtained under these conditions were the same as those determined with the peptide coupled to the sensor surface.
Molecular Modeling
The homology model of human PKCθ-C2 was built with the Nest (42) and Modeler (43) programs as described previously (44). The sequence alignment was performed with BLAST, and the crystal structure of the PKCδ-C2 (Protein Data Bank entry 1YRK) was used as the template for homology model building. The quality of the model was tested using Verify3D (45). The electrostatic property of PKCδ-C2 was calculated with a modified version of the program DelPhi and visualized in the program GRASP (46).
Co-immunoprecipitation
Jurkat-TAg T cells were cotransfected with a FLAG-tagged CDCP1 plasmid (18) plus green fluorescence protein (GFP)-tagged wild-type or mutated PKCθ vectors and cultured for 72 h. The efficiency of transfection was determined by flow cytometry analysis of GFP-positive cells. Transfected cells were stimulated with 1 mm of sodium orthovanadate (Na3VO4), a phosphatase inhibitor, for 10 min at 37 °C in order to induce global cellular tyrosine phosphorylation. Cells were then lysed in a buffer containing 50 mm Tris, 50 mm NaCl, 5 mm EDTA, 1% Nonidet P-40, and protease inhibitors. Immunoprecipitation was performed using an anti-GFP antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and precipitated proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were immunoblotted with an anti-FLAG antibody (Sigma-Aldrich).
Reporter Gene Activation
Jurkat-TAg T cells (REF) were cotransfected with GFP-tagged wild-type or mutated PKCθ vectors and RE/AP-Luc, a luciferase reporter plasmid, based on the CD28 response element (RE/AP) in the il2 gene promoter (47), and a β-galactosidase (β-gal) reporter plasmid. Cells were left unstimulated or stimulated with anti-CD3 (OKT3) plus anti-CD28 (CD28.2) monoclonal antibodies for 6 h. Luciferase activity normalized to β-Gal activity was determined in triplicates. Transfection efficiency was determined by GFP fluorescence using flow cytometry.
RESULTS
PKCθ-C2 Binding to Tyr(P)
The recent x-ray structure of PKCδ-C2 in complex with a Tyr(P) peptide elucidated how this domain achieved the stereospecific recognition of a CDCP1-derived Tyr(P)-containing peptide (18). Alignment of the C2 domain sequences from PKCδ and PKCθ demonstrates a high degree of similarity (i.e. 52% sequence identity by T-coffee (48); see Fig. 1A). Most importantly, PKCθ-C2 harbors the same residues in PKCδ-C2 that interact with the Tyr(P) peptide (see Fig. 1, C and E). These residues are not found in other novel PKCs, PKCϵ and PKCη. To determine if PKCθ-C2 could also bind a Tyr(P)-containing peptide, we monitored the binding of PKCθ-C2 to the CDCP1-derived Tyr(P) peptide by SPR analysis (see Fig. 2 and Table 1). As a positive control, we first monitored the binding of PKCδ-C2, and it bound the peptide with Kd = 240 nm (see Fig. 2, A and B, and Table 1), which agrees with the reported value determined using isothermal titration calorimetry (18). PKCθ-C2 also exhibited robust binding to the peptide and bound with 2-fold higher affinity (Kd = 120 nm) than PKCδ-C2 (see Fig. 2, C and D, and Table 1). Neither C2 domain showed significant binding to the control peptide containing Tyr in place of Tyr(P) (data not shown). We also monitored binding of full-length PKCδ and PKCθ to the peptide. In both instances, the affinity of the full-length proteins for the peptide was within the experimental error ranges of that of their respective C2 domains (Table 1). This suggests that the Tyr(P)-binding site is not cryptic in either protein, at least in PKCs expressed in Sf9 cells.
FIGURE 1.

Sequence alignment and structures of the PKCθ and PKCδ C2 domains. A, Sequence alignment of the C2 domains of rat PKCδ and human PKCθ C2 domains performed in T-Coffee (48) shows 52% sequence identity. Identical residues are shown in red, highly similar residues in blue, similar residues in green, and nonconserved residues in black. Residues deemed important for Tyr(P) peptide binding are marked with blue arrows, and Tyr90 that is phosphorylated in PKCθ is marked with an asterisk. B, crystal structure of the PKCδ-C2-CDCP1 peptide complex (PDB entry 1YRK) (18). The protein is shown in a surface representation using PyMOL, with blue and red qualitatively indicating positive and negative surface electrostatic potential. The bound Tyr(P) peptide is shown in a stick representation. Notice that the cationic groove interacts with the Tyr(P) moiety shown in red. C, a ribbon diagram of the PKCδ-C2 model in the same molecular orientation as Fig. 1B. The side chains of residues that are involved in Tyr(P) binding of PKCδ-C2 are highlighted in a space-filling representation and labeled. D, homology model of the PKCθ C2 domain built using the PKCδ C2 domain as a template. The surface representation shows the presence of a cationic groove that is comparable with the Tyr(P)-binding pocket of PKCδ-C2. E, ribbon diagram of the PKCθ-C2 model in the same molecular orientation as Fig. 1D. The side chains of residues that are involved in Tyr(P) binding of PKCθ-C2 are highlighted in a space-filling representation and labeled.
FIGURE 2.

Determination of PKCθ-C2-peptide binding by equilibrium SPR analysis. A, SPR binding sensorgrams are shown for PKCδ-C2 at 35, 75, 150, 400, and 1000 nm. To test for nonspecific binding the control surface utilized the non-phosphorylated peptide, which was subtracted from the Tyr(P) peptide binding response to correct for refractive index changes and yield the displayed sensorgrams. B, saturation response (Req) from PKCδ-C2 binding at each respective protein concentration (P0) in A was plotted versus [PKCδ-C2] to fit with a nonlinear least squares analysis of the binding isotherm (Req = Rmax/(1 + Kd/P0) to determine the Kd (see Table 1). C, SPR binding sensorgrams are shown for PKCθ-C2 at 25, 45, 110, 275, and 650 nm to a Tyr(P)-coated surface with the nonphosphorylated peptide control surface subtracted out for refractive index correction. D, as in B, the saturation response (Req) from PKCθ-C2 binding at each respective protein concentration (P0) in C was plotted to determine the Kd. For both PKCδ-C2 and PKCθ-C2, binding signals from the non-phosphorylated peptide were typically less than 5% of those from the Tyr(P) peptide at a given PKC concentration.
TABLE 1.
Phosphotyrosine peptide (GGALYSIpYQPYVFAKKK) binding of PKCθ, PKCθ-C2, and mutants
All binding measurements were performed in 10 mm HEPES, pH 7.4, containing 0.16 m KCl.
| Protein | Kd | Increase in Kda |
|---|---|---|
| nm | -fold | |
| PKCθ-C2 WT | 120 ± 30 | 1.0 |
| PKCδ-C2 WT | 240 ± 50 | 0.5 |
| PKCθ-C2 K49A | NDb | |
| PKCθ-C2 M52A | 780 ± 100 | 6.5 |
| PKCθ-C2 T59A | 1200 ± 100 | 10 |
| PKCθ-C2 H63A | ND | |
| PKCθ-C2 R68A | ND | |
| PKCθ-C2 Y90E | 91 ± 4 | 0.76 |
| PKCθ full-length | 73 ± 20 | 0.63 |
| PKCθ full-length Y90E | 75 ± 4 | 0.63 |
| PKCθ full-length Y90F | 82 ± 3 | 0.68 |
| PKCδ full-length | 150 ± 30 | 6.7 |
a -Fold increase in Kd relative to the binding of PKCθ-C2 to Tyr(P) peptide.
b ND, not detectable.
Origin of Tyr(P) Specificity of PKCθ-C2
To account for the Tyr(P)-binding properties of PKCθ-C2, we built a model structure (Fig. 1, D and E) based upon its homology to PKCδ-C2 (Fig. 1B). The model structure suggests that the Tyr(P)-binding pocket of PKCδ-C2 is well conserved in PKCθ-C2. On the basis of this structure, we prepared a panel of mutants of PKCθ-C2 and measured their binding to the Tyr(P) peptide to identify Tyr(P)-binding residues. Specifically, we mutated Arg68, Lys49, Met52, Thr59, and His63, whose counterparts in PKCδ-C2 are involved in Tyr(P) binding. Results summarized in Table 1 show that Lys49, His63, and Arg68 are critical for Tyr(P) binding because the mutations of these residues abolished the peptide binding. Also, Met52 and Thr59 seem to contribute to the Tyr(P) binding. Interestingly, Y90E and Y90F mutations had no effect on Tyr(P) binding of both full-length PKCθ and PKCθ-C2. This indicates that although the phosphorylation of Tyr90 is important for the DAG-dependent translocation and activation of PKCθ, it is not a prerequisite for the Tyr(P) binding.
In order to assess the importance of the critical Tyr(P)-binding residues in PKCθ-C2 in intact T cells, we cotransfected Jurkat T cells with FLAG-tagged CDCP1 (18) and GFP-tagged WT or C2-mutated PKCθ vectors (H63A and R68A as Tyr(P) binding-deficient mutants and M52A as a mutant with reduced Tyr(P) binding). We then stimulated the cells with Na3VO4, a Tyr(P) phosphatase inhibitor that causes a global increase in cellular tyrosine phosphorylation, in order to induce phosphorylation of CDCP1 on tyrosine. Proteins were immunoprecipitated with anti-GFP and then analyzed for the presence of CDCP1. No PKCθ association of CDCP1 was observed in unstimulated T cells (data not shown). However, Na3VO4 stimulation caused CDCP1 to associate with WT PKCθ. Under the same conditions, all three mutants showed dramatically reduced interaction with CDCP1 (Fig. 3). These results indicate that the same C2 residues that are involved in in vitro association between PKCθ and the CDCP1 Tyr(P) peptide are also required for the interaction between PKCθ and intact CDCP1 in stimulated T cells.
FIGURE 3.
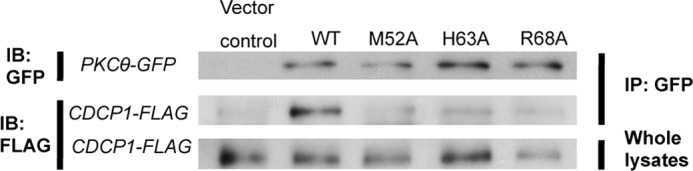
Association of PKCθ with CDCP1. Jurkat-TAg T cells were cotransfected with a FLAG-tagged CDCP1 expression vector plus vectors encoding GFP-tagged wild-type or C2-mutated PKCθ vectors (or empty vector), as indicated. The cells were stimulated with Na3VO4 (1 mm) for 10 min and lysed. Anti-GFP immunoprecipitates (IP) or whole lysates were resolved by SDS-PAGE, transferred to nitrocellulose, and immunoblotted (IB) with the indicated antibodies.
Effect of Tyr(P) Binding on PKCθ Enzyme Activity
Although PKCδ-C2 was shown to specifically bind the CDCP1-derived Tyr(P) peptide, biochemical and physiological effects of this interaction were not reported (18). To establish the physiological significance of the PKCθ-C2-Tyr(P) binding, we first monitored the activity of both PKCθ and PKCδ in the presence of the Tyr(P) peptide using myelin basic protein as a substrate. As shown in Fig. 4, the Tyr(P) peptide greatly increased PKCθ activity even in the absence of lipid activators while showing a statistically insignificant effect on PKCδ activity. Because PKCδ has higher activity than PKCθ in the presence of 200 μm POPC/POPS/DiC18 (59:40:1) vesicles under our assay conditions (see Fig. 5, A and B), this difference is not due to low intrinsic enzyme activity of PKCδ.
FIGURE 4.
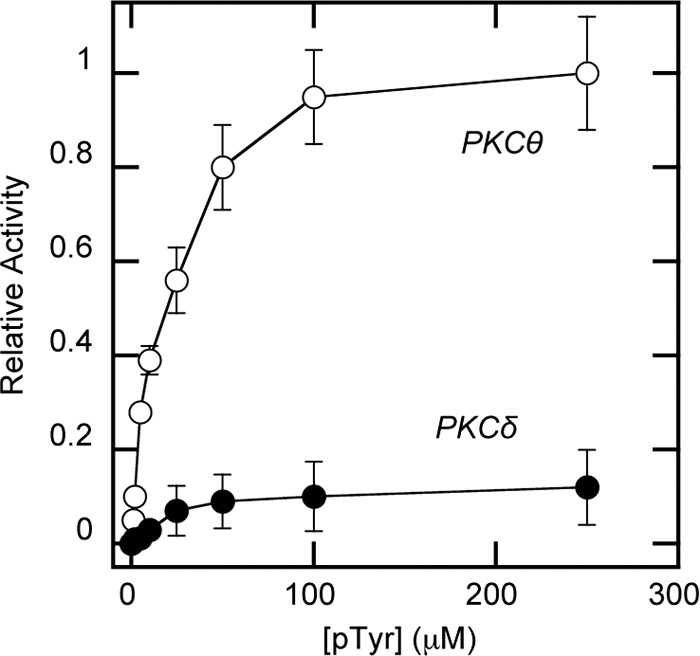
Enzymatic activity of PKCθ and PKCδ in the presence of Tyr(P) peptide. The kinase activity of 30 nm PKC was measured in the presence of varying concentrations of Tyr(P) peptide in 20 mm HEPES, pH 7.4, containing 0.16 m KCl, 5 mm MgCl2, and myelin basic protein (200 μg/ml) but in the absence of lipid cofactors. Each data point represents an average of triplicate measurements. Relative activity was calculated in comparison with the maximal activity of WT PKCθ achieved with 250 μm peptide (0.02 μmol/min/mg). Notice that PKCδ shows a much lower degree of activation by the peptide than PKCθ. Because PKCδ has higher activity than PKCθ in the presence of 200 μm POPC/POPS/DiC18 (59:40:1) vesicles under our assay conditions (see Fig. 5, A and B), this difference is not due to low intrinsic enzyme activity of PKCδ. Also, p < 0.0001 for PKCθ and p < 0.33 for PKCδ between 0 and 100 μm Tyr(P) (pTyr), indicating that the degree of activation of PKCδ by 100 μm Tyr(P) is not statistically meaningful. Error bars, S.D.
FIGURE 5.

PS- and Tyr(P)-dependent activity of PKCδ, PKCθ, and PKCθ mutants. The kinase activity of 30 nm PKCδ (A) or PKCθ (B) was measured in 20 mm HEPES buffer, pH 7.4, containing 0.16 m KCl, 5 mm MgCl2, myelin basic protein (200 μg/ml), and 200 μm POPC/POPS/DiC18 ((89 − x):x:1) vesicles, in the presence or absence of 5 μm peptide. For PKCθ mutants, the enzyme assay was performed under the same conditions except that a fixed concentration of PS (i.e. POPC/POPS/DiC18 = 89:10:1 (C) or POPC/POPS/DiC18 = 59:40:1 (D)) was used in the vesicles. Each bar represents an average of triplicate measurements. Relative activity was calculated in comparison with the maximal activity of WT PKCθ (0.08 μmol/min/mg) achieved with POPC/POPS/DiC18 (59:40:1) vesicles and 250 μm peptide. White bars, kinase activity measured in the absence of the Tyr(P); shaded bars, kinase activity measured with 5 μm Tyr(P) peptide. When PKCθ was activated by 250 μm peptide alone (0.07 μmol/min/mg), the addition of 200 μm POPC/POPS/DiC18 (59:40:1) vesicles modestly increased the activity (0.08 μmol/min/mg). Note that PKCδ had higher intrinsic activity (0.067 μmol/min/mg) than PKCθ (0.039 μmol/min/mg) in the presence of POPC/POPS/DiC18 (59:40:1) but without the Tyr(P) peptide. Specific activity (in terms of μmol/min/mg) of PKCθ mutants in the presence of POPC/POPS/DiC18 (59:40:1) vesicles alone was 0.038 for M52A, 0.037 for H63A, and 0.037 for R68A, showing that lipid-dependent activity was not altered for these mutations. Error bars, S.D.
We also measured the effect of the Tyr(P) peptide on PKC activity in the presence of different concentration of lipid activators. Specifically, we measured the enzyme activity with and without the Tyr(P) peptide while varying phosphatidylserine (PS) composition in POPC/POPS/DiC18 ((89 − x):x:1) vesicles. PS has been shown to activate PKCs by specifically releasing their interdomain tethering as well as enhancing their membrane affinity (6, 8, 9, 30, 37, 49). As shown in Fig. 5A, PKCδ activity was increased with the increase in PS; however, 5 μm Tyr(P) peptide had little effect on PKCδ activity regardless the PS concentration. In contrast, 5 μm Tyr(P) peptide greatly enhanced the PKCθ activity at different PS concentrations (i.e. 4-fold at 10–20 mol % and 2.5-fold at 40 mol % PS). The fact that the degree of PKCθ activation by Tyr(P) decreases as the PS concentration rises suggests that PKCθ activation by PS and by Tyr(P) complement each other. This notion was supported by the finding that when PKCθ was activated by 250 μm Tyr(P) peptide instead of 5 μm, the addition of 40 mol % PS (i.e. 200 μm of POPC/POPS/DiC18 (59:40:1) vesicles) only modestly increased the activity (i.e. from 0.07 to 0.08 μmol/min/mg). Last, a non-phosphorylated peptide activated neither PKC under any condition (data not shown), demonstrating the specific nature of PKCθ activation by the Tyr(P) peptide.
We then measured the effects of mutating Tyr(P)-binding residues in the C2 domain of PKCθ on enzyme activation. As described above, PKCθ WT was activated 4-fold by 5 μm Tyr(P) peptide with 10 mol % PS in the vesicles. Under the same conditions, H63A and R68A showed no activation by 5 μm Tyr(P) peptide, whereas M52A exhibited modest activation (Fig. 5C). This shows that binding of Tyr(P) to the C2 domain is directly responsible for Tyr(P)-dependent PKCθ activation. Interestingly, all three mutants and WT showed the same higher activity at 40 mol % PS as at 10 mol % PS in the absence of Tyr(P), demonstrating that the three mutated residues are not involved in lipid (i.e. PS and DAG)-dependent PKCθ activation (Fig. 5D). Unlike WT, however, H63A and R68A were not activated by the addition of 5 μm Tyr(P) peptide under these conditions.
Collectively, these results clearly show that binding of Tyr(P) to PKCθ-C2 directly and greatly enhances the enzymatic activity of PKCθ, which may complement lipid-dependent PKCθ under physiological conditions. The disparate effect of Tyr(P) binding on the enzyme activity of PKCθ and PKCδ is also consistent with our recent studies showing that their C2 domains have distinct roles in enzyme activation; i.e. PKCθ-C2 plays an autoinhibitory role (29), whereas PKCδ-C2 is non-inhibitory (9).
Requirement of Tyr(P) Binding for PKCθ Function in Intact T Cells
In order to establish the functional relevance of Tyr(P) binding by the PKCθ C2 domain in the context of T cell activation, we analyzed the ability of the C2-mutated PKCθ (i.e. M52A, H63A, and R68A) to activate known PKCθ downstream targets. Because the transcription factors NF-κB and AP-1 are known targets of activated PKCθ (27, 28, 50–52), we used a reporter gene based on the CD28 response element (RE/AP) in the il2 gene promoter, which contains a combined binding site for NF-κB and AP-1 (43). Hence, wild-type or C2-mutated PKCθ vectors were tested for their ability to stimulate this reporter gene in cotransfected Jurkat T cells costimulated with anti-CD3 and -CD28 antibodies.
By comparison with empty vector-transfected cells, WT PKCθ greatly stimulated the reporter gene activity in anti-CD3/CD28-stimulated T cells; in contrast, the His63 and Arg68 mutants and, to a somewhat lesser extent, the Met52 mutant were incapable of activating the reporter gene (Fig. 6A). The results shown in Fig. 6B demonstrate that all of the transfected PKCθ proteins were expressed at similar levels, as determined by the expression of GFP fluorescence. Thus, the Tyr(P)-binding residues in PKCθ-C2 are required for the downstream functions of PKCθ, suggesting that binding of a putative Tyr(P)-containing protein, whose phosphorylation is induced by TCR/CD28 costimulation, to the PKCθ C2 domain positively regulates its activation.
FIGURE 6.

Effect of Tyr(P) binding site mutations of PKCθ on its T cell regulatory activity. A, Jurkat-TAg T cells were cotransfected with vectors encoding GFP-tagged wild-type or C2-mutated PKCθ vectors (or empty vector) together with an RE/AP-Luc and β-gal reporter genes. Cells were left unstimulated or stimulated with anti-CD3 plus anti-CD28 monoclonal antibodies for 6 h. Luciferase activity normalized to β-gal activity was determined in triplicates. RLU, relative luciferase units. B, expression of the transfected GFP-PKCθ proteins was analyzed by flow cytometry, demonstrating similar expression levels of all proteins. Error bars, S.D.
DISCUSSION
It has long been known that the subcellular targeting pattern of PKCs varies in an isoform-, cell type-, and activator-specific manner (3). Extensive studies have been performed to understand the mechanisms by which PKC isoforms achieve unique modes of cellular localization and activation because deciphering these mechanisms will lead to better means of controlling specific PKC isoforms and cellular processes associated with their activity. The unique role of PKCθ in T cell activation (23, 24) makes it an attractive model for mechanistic studies. Currently, structural information on intact PKCθ is unavailable, and the mechanism underlying its unique localization and action in T cells remains poorly understood. The present study reveals that the C2 domain of PKCθ interacts with a CDCP1-derived Tyr(P) peptide with high affinity and specificity and that this binding directly and specifically enhances the in vitro enzyme activity of PKCθ. Mutations of the Tyr(P)-binding residues in the PKCθ C2 domain abrogate Tyr(P)-dependent PKCθ activation. The same peptide also tightly binds the highly homologous PKCδ C2 domain but shows little effect on the activity of PKCδ. The biological relevance of the interaction between the C2 domain of PKCθ and Tyr(P)-containing peptide ligands is evidenced by our finding that PKCθ associates with Tyr(P)-CDCP1 in stimulated intact T cells. Also, mutations of the Tyr(P)-binding residues essentially abolished the ability of PKCθ to activate a RE/AP reporter gene in anti-CD3/CD28-stimulated T cells, this reporter being a well characterized target of TCR/CD28-induced, PKCθ-dependent activation (46).
The regulation of cellular localization and activation of PKCθ is complex (24) and may involve multiple mechanisms in addition to canonical C1 domain-mediated binding to DAG-containing membranes. This notion is supported by at least two findings. First, inhibition of DAG-generating PLCγ1 only partially inhibited CD3/CD28-stimulated PKCθ membrane translocation but fully blocked PKCα from translocating to the membrane (53). Second, despite having highly similar catalytic domains, PKCθ shows only about half of the activity of PKCδ in the presence of POPC/POPS/DiC18 (59:40:1), but the former is 40% more active than the latter in the presence of POPC/POPS/DiC18 (59:40:1) and 5 μm CDCP1-derived Tyr(P) peptide (see Fig. 5, A and B). This finding, coupled with the observation that localization of PKCθ to the immunological synapse is positively correlated with kinase activity (54), raises the tempting possibility that, in addition to a high local DAG concentration documented at the immunological synapse (55), some Tyr(P)-containing protein(s) at the immunological synapse recruits PKCθ via its C2 domain and promotes its activation even under conditions of low DAG production. Our preliminary results show that a glutathione S-transferase-PKCθ-C2 fusion protein can pull down several Tyr(P)-containing proteins from stimulated T cell lysates.5 Undoubtedly, further studies are needed to identify all interaction partners for the PKCθ C2 domain, which is beyond the scope of this investigation. However, it is evident from our study using PKCθ and its Tyr(P) binding-deficient mutants that binding of a Tyr(P)-containing protein to the PKCθ C2 domain is important for the signaling activity of PKCθ in T cells under physiological conditions.
This study also shows that although PKCs are generally considered to have a high degree of homology, intact PKCs and their individual lipid binding domains, including the C2 domain, have distinctly different mechanisms of membrane and protein interactions. PKCθ is similar to PKCδ (18) in one aspect; i.e. they both bind a Tyr(P)-containing peptide from CDCP1 with nanomolar affinity. However, the functional consequences of this binding are completely different. Although Tyr(P) binding by PKCθ leads to increased enzymatic activity in the presence or absence of DAG-containing vesicles, Tyr(P) binding by PKCδ has a statistically insignificant effect under the same conditions. This difference may derive from the different mechanisms by which their C2 domains mediate membrane binding and activation. For example, the C2 domain of PKCδ is not involved in membrane binding and activation of PKCδ (9), whereas the C2 domain of PKCθ is involved in both processes (29). Also, the PKCθ C2 domain is involved in autoinhibition to suppress the membrane binding of the rest of the molecule (29), whereas the PKCδ C2 domain is not (9). Indeed, the PKCθ C2 domain shows the most pronounced autoinhibitory effect among PKC C2 domains implicated in autoinhibition (56–58). Thus, Tyr(P) binding may simply function in recruiting PKCδ to a binding partner protein, whereas it may play a more direct role in enzyme activation for PKCθ.
In this case, how does Tyr(P) binding induce PKCθ activation? Because displacement of the pseudosubstrate from the active site is a prerequisite of PKC activation, it is likely that Tyr(P) binding to the C2 domain of PKCθ also displaces the pseudosubstrate. The pseudosubstrate of novel PKCs, including PKCθ, is located between the C2 domain and the C1A domain (Fig. 7A), and thus any major conformational change of the flanking C2 or C1A domain would induce the movement of the neighboring pseudosubstrate, triggering its displacement from the active site of the enzyme. Binding of a Tyr(P)-containing protein to the C2 domain would probably cause a conformational change of the C2 domain that could lead to the displacement of the pseudosubstrate, either directly or through conformational changes of other domains. PKCθ is unique among PKCs because its C1B domain has much higher DAG affinity than its C1A domain (29). In all other PKC isoforms studied, the C1A domain has intrinsic DAG affinity that is higher than (or at least comparable with) that of the C1B domain (6, 8, 9, 30). Thus, the C1A domain usually plays a role more dominant than (or at least comparable with) that of the C1B domain in PKC activation (6, 8, 9, 30), and the conformational change of the C1A domain associated with its DAG-dependent membrane binding induces the displacement of the neighboring pseudosubstrate. For PKCθ, membrane binding of the remote C1B domain should have a less direct conformational effect on the pseudosubstrate, as evidenced by a smaller degree of lipid-dependent activation for PKCθ than for PKCδ (see Fig. 5, A and B). For this reason, the Tyr(P)-C2 interaction may serve as a key complementary regulatory mechanism that can even override the lipid-dependent activation of PKCθ under certain conditions. This notion is supported by the findings that the Tyr(P) peptide can induce PKCθ activity with and without lipid activators, PS and DAG, and that mutations of residues involved in Tyr(P) binding (H63A and R68A) abrogate Tyr(P)-dependent activation but not lipid-dependent activation.
FIGURE 7.

Schematic domain structure of PKCθ (A) and a proposed mechanism of membrane binding and activation of PKCθ (B). A, domain organization of PKCθ is shown schematically with the location of the Tyr(P) (pTyr) binding pocket in the C2 domain indicated by a red arrow. B, in the resting state of PKCθ, Tyr90 is unphosphorylated, and neither the C2 nor C1B domain is fully exposed for optimal membrane interactions. In the canonical activation mechanism, phosphorylation of Tyr90 induces conformational changes that promote the membrane recruitment of PKCθ by lipid binding. Binding of the C2 domain to PS (yellow) and subsequent membrane penetration and DAG (red) binding of the C1B domain pull the pseudosubstrate region from the active site, resulting in enzyme activation. Regardless of Tyr90 phosphorylation, the C2 domain can bind Tyr(P) of a membrane-bound protein, which causes the activation of PKCθ through conformational changes that also lead to the removal of pseudosubstrate from the active site. PS and DAG binding may further activate the protein or prolong its membrane residence and/or activation.
Although our model of PKCθ activation by Tyr(P) binding is consistent with our data, it is possible that a more complex interplay of C1A, C1B, and C2 domains is involved in membrane binding and activation of PKCθ. For example, recent studies have implicated the C1B domain in activation of PKCs (58–60). In particular, a recent crystal structure of PKCβII indicates that its C1B domain keeps the protein in an inactive state by interacting with a C-terminal region of the protein and that this intramolecular tethering is relieved upon DAG binding to the C1B domain (60). Although it remains to be seen how the intramolecular tethering by the C1B domain would be relieved for most PKCs whose C1B domain has low DAG affinity, at least for PKCθ whose C1B domain has high DAG affinity, DAG-dependent untethering of the C1B domain would certainly cause the global conformational changes, presumably including the C2 domain. It is also possible that the Tyr(P)-C2 domain interaction can take place intramolecularly or between two PKC molecules as an autoinhibitory mechanism, which is then relieved by competitive binding of a Tyr(P)-containing protein to the C2 domain. PKCθ has been shown to be phosphorylated only on Tyr90 in the C2 domain (31), which precludes the possibility of intramolecular Tyr(P)-C2 domain interaction; however, the Tyr(P)-C2 domain interaction between two PKCθ molecules is still possible. In the case of PKCδ that has been shown to be phosphorylated on Tyr52, Tyr155, Tyr187, Tyr311, Tyr332, and Tyr565 (34), autoinhibitory Tyr(P)-C2 domain interaction can take place both intramolecularly and between two PKCδ molecules.
On the basis of our previous (29) and current results, we propose a new hypothetical model of PKCθ activation that involves two different modes of enzyme activation (see Fig. 7). A canonical mode of PKCθ activation is triggered by Tyr90 phosphorylation (31) that causes conformational changes, which in turn promotes the membrane recruitment of PKC via its lipid binding domains. PS binding by the C2 domain and subsequent membrane penetration and DAG binding of the C1B domain relieve the intramolecular tethering and pull the pseudosubstrate region from the active site, resulting in enzyme activation. This lipid-mediated pathway may not lead to full enzyme activation, especially when the local DAG concentration is not high. Whether Tyr90 is phosphorylated or not, the C2 domain can bind Tyr(P) of a membrane-bound protein, which causes the activation of PKCθ through as yet undefined conformational changes that also lead to the removal of pseudosubstrate from the active site. We speculate that Tyr(P) binding of PKCθ could increase the membrane binding as well because Tyr(P)-containing proteins, such as CDCP1, are membrane-embedded proteins. In other words, Tyr(P) binding increases the effective concentration of PKCθ at the membrane, hence enhancing membrane affinity. In this non-canonical activation mode, PS and DAG binding by C2 and C1B domain, respectively, may further activate PKCθ and/or prolong its activation. Our mutational study indicates that the Tyr(P) binding site and membrane binding site of the PKCθ C2 domain are topologically separate and functionally independent of each other. However, the study also shows that the activation by Tyr(P) complements lipid-dependent activation (and vice versa) of PKCθ under certain in vitro conditions. It is unclear at present whether or not lipid binding is still required for full activation of PKCθ that is recruited to the membrane via Tyr(P) binding under physiological conditions. This hypothetical model should serve as a framework with which to investigate the complex mechanism of cellular membrane recruitment and activation of this important signaling enzyme.
Acknowledgments
We thank Sandra Chimon for assistance with peptide synthesis. R. V. S. also thanks Dr. Yi Xue for excellent technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants CA035299 (to A. A.), CA035299 and GM76581 (to W. C.), and AI081077 (to R. S.). This work was also supported by the World Class University Program Grant R31-2008-000-10105-0 (to W. C.) through the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology.
K. Hayashi and A. Altman, unpublished observation.
- DAG
- sn-1,2-diacylglycerol
- DiC18
- 1–2-dioleoyl-sn-glycerol
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine
- PS
- phosphatidylserine
- PKCθ-C2 and PKCδ-C2
- C2 domain of protein kinase Cθ and Cδ, respectively
- SPR
- surface plasmon resonance
- TCR
- T cell receptor.
REFERENCES
- 1. Newton A. C. (2001) Protein kinase C. Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 101, 2353–2364 [DOI] [PubMed] [Google Scholar]
- 2. Parekh D. B., Ziegler W., Parker P. J. (2000) Multiple pathways control protein kinase C phosphorylation. EMBO J. 19, 496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shirai Y., Saito N. (2002) Activation mechanisms of protein kinase C. Maturation, catalytic activation, and targeting. J. Biochem. 132, 663–668 [DOI] [PubMed] [Google Scholar]
- 4. Brose N., Rosenmund C. (2002) Move over protein kinase C, you've got company. Alternative cellular effectors of diacylglycerol and phorbol esters. J. Cell Sci. 115, 4399–4411 [DOI] [PubMed] [Google Scholar]
- 5. Yang C., Kazanietz M. G. (2003) Divergence and complexities in DAG signaling. Looking beyond PKC. Trends Pharmacol. Sci. 24, 602–608 [DOI] [PubMed] [Google Scholar]
- 6. Ananthanarayanan B., Stahelin R. V., Digman M. A., Cho W. (2003) Activation mechanisms of conventional protein kinase C isoforms are determined by the ligand affinity and conformational flexibility of their C1 domains. J. Biol. Chem. 278, 46886–46894 [DOI] [PubMed] [Google Scholar]
- 7. Slater S. J., Ho C., Kelly M. B., Larkin J. D., Taddeo F. J., Yeager M. D., Stubbs C. D. (1996) Protein kinase Cα contains two activator binding sites that bind phorbol esters and diacylglycerols with opposite affinities. J. Biol. Chem. 271, 4627–4631 [DOI] [PubMed] [Google Scholar]
- 8. Stahelin R. V., Digman M. A., Medkova M., Ananthanarayanan B., Melowic H. R., Rafter J. D., Cho W. (2005) Diacylglycerol-induced membrane targeting and activation of protein kinase Cϵ. Mechanistic differences between protein kinases Cδ and Cϵ. J. Biol. Chem. 280, 19784–19793 [DOI] [PubMed] [Google Scholar]
- 9. Stahelin R. V., Digman M. A., Medkova M., Ananthanarayanan B., Rafter J. D., Melowic H. R., Cho W. (2004) Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cδ. J. Biol. Chem. 279, 29501–29512 [DOI] [PubMed] [Google Scholar]
- 10. Das J., Addona G. H., Sandberg W. S., Husain S. S., Stehle T., Miller K. W. (2004) Identification of a general anesthetic binding site in the diacylglycerol-binding domain of protein kinase Cδ. J. Biol. Chem. 279, 37964–37972 [DOI] [PubMed] [Google Scholar]
- 11. Das J., Zhou X., Miller K. W. (2006) Protein Sci. 15, 2107–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Slater S. J., Cook A. C., Seiz J. L., Malinowski S. A., Stagliano B. A., Stubbs C. D. (2003) Effects of ethanol on protein kinase C α activity induced by association with Rho GTPases. Biochemistry 42, 12105–12114 [DOI] [PubMed] [Google Scholar]
- 13. Slater S. J., Kelly M. B., Larkin J. D., Ho C., Mazurek A., Taddeo F. J., Yeager M. D., Stubbs C. D. (1997) Interaction of alcohols and anesthetics with protein kinase Cα. J. Biol. Chem. 272, 6167–6173 [DOI] [PubMed] [Google Scholar]
- 14. Slater S. J., Stagliano B. A., Seiz J. L., Curry J. P., Milano S. K., Gergich K. J., Stubbs C. D. (2001) Effects of ethanol on protein kinase C activity induced by filamentous actin. Biochim. Biophys. Acta 1544, 207–216 [DOI] [PubMed] [Google Scholar]
- 15. Cho W. (2001) Membrane targeting by C1 and C2 domains. J. Biol. Chem. 276, 32407–32410 [DOI] [PubMed] [Google Scholar]
- 16. Cho W., Stahelin R. V. (2006) Membrane binding and subcellular targeting of C2 domains. Biochim. Biophys. Acta 1761, 838–849 [DOI] [PubMed] [Google Scholar]
- 17. Manna D., Bhardwaj N., Vora M. S., Stahelin R. V., Lu H., Cho W. (2008) Differential roles of phosphatidylserine, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 in plasma membrane targeting of C2 domains. Molecular dynamics simulation, membrane binding, and cell translocation studies of the PKCα C2 domain. J. Biol. Chem. 283, 26047–26058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benes C. H., Wu N., Elia A. E., Dharia T., Cantley L. C., Soltoff S. P. (2005) The C2 domain of PKCδ is a phosphotyrosine binding domain. Cell 121, 271–280 [DOI] [PubMed] [Google Scholar]
- 19. Sundberg C., Thodeti C. K., Kveiborg M., Larsson C., Parker P., Albrechtsen R., Wewer U. M. (2004) Regulation of ADAM12 cell surface expression by protein kinase C ϵ. J. Biol. Chem. 279, 51601–51611 [DOI] [PubMed] [Google Scholar]
- 20. Corbalán-Garcia S., Sánchez-Carrillo S., García-Garcia J., Gómez-Fernández J. C. (2003) Characterization of the membrane binding mode of the C2 domain of PKC ϵ. Biochemistry 42, 11661–11668 [DOI] [PubMed] [Google Scholar]
- 21. Jose Lopez-Andreo M., Gomez-Fernandez J. C., Corbalan-Garcia S. (2003) The simultaneous production of phosphatidic acid and diacylglycerol is essential for the translocation of protein kinase Cϵ to the plasma membrane in RBL-2H3 cells. Mol. Biol. Cell 14, 4885–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ochoa W. F., Garcia-Garcia J., Fita I., Corbalan-Garcia S., Verdaguer N., Gomez-Fernandez J. C. (2001) Structure of the C2 domain from novel protein kinase Cϵ. A membrane binding model for Ca2+-independent C2 domains. J. Mol. Biol. 311, 837–849 [DOI] [PubMed] [Google Scholar]
- 23. Hayashi K., Altman A. (2007) Protein kinase C θ (PKCθ). A key player in T cell life and death. Pharmacol. Res. 55, 537–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Isakov N., Altman A. (2002) Protein kinase Cθ in T cell activation. Annu. Rev. Immunol. 20, 761–794 [DOI] [PubMed] [Google Scholar]
- 25. Grakoui A., Bromley S. K., Sumen C., Davis M. M., Shaw A. S., Allen P. M., Dustin M. L. (1999) The immunological synapse. A molecular machine controlling T cell activation. Science 285, 221–227 [DOI] [PubMed] [Google Scholar]
- 26. Downward J., Graves J. D., Warne P. H., Rayter S., Cantrell D. A. (1990) Stimulation of p21ras upon T-cell activation. Nature 346, 719–723 [DOI] [PubMed] [Google Scholar]
- 27. Sun Z., Arendt C. W., Ellmeier W., Schaeffer E. M., Sunshine M. J., Gandhi L., Annes J., Petrzilka D., Kupfer A., Schwartzberg P. L., Littman D. R. (2000) PKC-θ is required for TCR-induced NF-κB activation in mature but not immature T lymphocytes. Nature 404, 402–407 [DOI] [PubMed] [Google Scholar]
- 28. Pfeifhofer C., Kofler K., Gruber T., Tabrizi N. G., Lutz C., Maly K., Leitges M., Baier G. (2003) Protein kinase C θ affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J. Exp. Med. 197, 1525–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Melowic H. R., Stahelin R. V., Blatner N. R., Tian W., Hayashi K., Altman A., Cho W. (2007) Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cθ. J. Biol. Chem. 282, 21467–21476 [DOI] [PubMed] [Google Scholar]
- 30. Medkova M., Cho W. (1999) Interplay of C1 and C2 domains of protein kinase C-α in its membrane binding and activation. J. Biol. Chem. 274, 19852–19861 [DOI] [PubMed] [Google Scholar]
- 31. Liu Y., Witte S., Liu Y. C., Doyle M., Elly C., Altman A. (2000) Regulation of protein kinase Cθ function during T cell activation by Lck-mediated tyrosine phosphorylation. J. Biol. Chem. 275, 3603–3609 [DOI] [PubMed] [Google Scholar]
- 32. Kates M. (1986) Techniques of Lipidology, 2nd Ed., pp. 114–115, Elsevier Science Publishers B.V., Amsterdam [Google Scholar]
- 33. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 34. Kikkawa U., Matsuzaki H., Yamamoto T. (2002) Protein kinase C δ (PKCδ). Activation mechanisms and functions. J. Biochem. 132, 831–839 [DOI] [PubMed] [Google Scholar]
- 35. Stahelin R. V., Subramanian P., Vora M., Cho W., Chalfant C. E. (2007) Ceramide-1-phosphate binds group IVA cytosolic phospholipase a2 via a novel site in the C2 domain. J. Biol. Chem. 282, 20467–20474 [DOI] [PubMed] [Google Scholar]
- 36. Lichtenbergova L., Yoon E. T., Cho W. (1998) Membrane penetration of cytosolic phospholipase A2 is necessary for its interfacial catalysis and arachidonate specificity. Biochemistry 37, 14128–14136 [DOI] [PubMed] [Google Scholar]
- 37. Bittova L., Stahelin R. V., Cho W. (2001) Roles of ionic residues of the C1 domain in protein kinase C-α activation and the origin of phosphatidylserine specificity. J. Biol. Chem. 276, 4218–4226 [DOI] [PubMed] [Google Scholar]
- 38. Stahelin R. V., Cho W. (2001) Differential roles of ionic, aliphatic, and aromatic residues in membrane-protein interactions. A surface plasmon resonance study on phospholipases A2. Biochemistry 40, 4672–4678 [DOI] [PubMed] [Google Scholar]
- 39. Stahelin R. V., Cho W. (2001) Roles of calcium ions in the membrane binding of C2 domains. Biochem. J. 359, 679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stahelin R. V., Burian A., Bruzik K. S., Murray D., Cho W. (2003) Membrane binding mechanisms of the PX domains of NADPH oxidase p40phox and p47phox. J. Biol. Chem. 278, 14469–14479 [DOI] [PubMed] [Google Scholar]
- 41. Stahelin R. V., Long F., Diraviyam K., Bruzik K. S., Murray D., Cho W. (2002) Phosphatidylinositol 3-phosphate induces the membrane penetration of the FYVE domains of Vps27p and Hrs. J. Biol. Chem. 277, 26379–26388 [DOI] [PubMed] [Google Scholar]
- 42. Petrey D., Xiang Z., Tang C. L., Xie L., Gimpelev M., Mitros T., Soto C. S., Goldsmith-Fischman S., Kernytsky A., Schlessinger A., Koh I. Y., Alexov E., Honig B. (2003) Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins 53, Suppl. 6, 430–435 [DOI] [PubMed] [Google Scholar]
- 43. Eswar N., John B., Mirkovic N., Fiser A., Ilyin V. A., Pieper U., Stuart A. C., Marti-Renom M. A., Madhusudhan M. S., Yerkovich B., Sali A. (2003) Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31, 3375–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stahelin R. V., Ananthanarayanan B., Blatner N. R., Singh S., Bruzik K. S., Murray D., Cho W. (2004) Mechanism of membrane binding of the phospholipase D1 PX domain. J. Biol. Chem. 279, 54918–54926 [DOI] [PubMed] [Google Scholar]
- 45. Lüthy R., Bowie J. U., Eisenberg D. (1992) Assessment of protein models with three-dimensional profiles. Nature 356, 83–85 [DOI] [PubMed] [Google Scholar]
- 46. Nicholls A., Sharp K. A., Honig B. (1991) Protein folding and association. Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11, 281–296 [DOI] [PubMed] [Google Scholar]
- 47. Shapiro V. S., Truitt K. E., Imboden J. B., Weiss A. (1997) CD28 mediates transcriptional upregulation of the interleukin-2 (IL-2) promoter through a composite element containing the CD28RE and NF-IL-2B AP-1 sites. Mol. Cell Biol. 17, 4051–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Notredame C., Higgins D. G., Heringa J. (2000) T-Coffee. A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 49. Ananthanarayanan B., Das S., Rhee S. G., Murray D., Cho W. (2002) Membrane targeting of C2 domains of phospholipase C-δ isoforms. J. Biol. Chem. 277, 3568–3575 [DOI] [PubMed] [Google Scholar]
- 50. Baier-Bitterlich G., Uberall F., Bauer B., Fresser F., Wachter H., Grunicke H., Utermann G., Altman A., Baier G. (1996) Protein kinase C-θ isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol. Cell Biol. 16, 1842–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Coudronniere N., Villalba M., Englund N., Altman A. (2000) NF-κB activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-θ. Proc. Natl. Acad. Sci. U.S.A. 97, 3394–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin X., O'Mahony A., Mu Y., Geleziunas R., Greene W. C. (2000) Protein kinase C-θ participates in NF-κB activation induced by CD3-CD28 costimulation through selective activation of IκB kinase β. Mol. Cell Biol. 20, 2933–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Villalba M., Bi K., Hu J., Altman Y., Bushway P., Reits E., Neefjes J., Baier G., Abraham R. T., Altman A. (2002) Translocation of PKCθ in T cells is mediated by a nonconventional, PI3K- and Vav-dependent pathway but does not absolutely require phospholipase C. J. Cell Biol. 157, 253–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Monks C. R., Freiberg B. A., Kupfer H., Sciaky N., Kupfer A. (1998) Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 395, 82–86 [DOI] [PubMed] [Google Scholar]
- 55. Spitaler M., Emslie E., Wood C. D., Cantrell D. (2006) Diacylglycerol and protein kinase D localization during T lymphocyte activation. Immunity 24, 535–546 [DOI] [PubMed] [Google Scholar]
- 56. Pepio A. M., Fan X., Sossin W. S. (1998) The role of C2 domains in Ca2+-activated and Ca2+-independent protein kinase Cs in aplysia. J. Biol. Chem. 273, 19040–19048 [DOI] [PubMed] [Google Scholar]
- 57. Slater S. J., Seiz J. L., Cook A. C., Buzas C. J., Malinowski S. A., Kershner J. L., Stagliano B. A., Stubbs C. D. (2002) Regulation of PKC α activity by C1-C2 domain interactions. J. Biol. Chem. 277, 15277–15285 [DOI] [PubMed] [Google Scholar]
- 58. Farah C. A., Nagakura I., Weatherill D., Fan X., Sossin W. S. (2008) Physiological role for phosphatidic acid in the translocation of the novel protein kinase C Apl II in Aplysia neurons. Mol. Cell Biol. 28, 4719–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dries D. R., Gallegos L. L., Newton A. C. (2007) A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J. Biol. Chem. 282, 826–830 [DOI] [PubMed] [Google Scholar]
- 60. Leonard T. A., Różycki B., Saidi L. F., Hummer G., Hurley J. H. (2011) Crystal structure and allosteric activation of protein kinase C βII. Cell 144, 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]