

Background: Annexin-A1 is an important regulator of VEGF-mediated endothelial cell migration and angiogenesis.
Results: We found that miR-196a targets Annexin-A1 to inhibit VEGF-mediated cell migration and angiogenesis. Moreover, VEGF decreases miR-196a and increases Annexin-A1.
Conclusion: VEGF-induced decrease of miR-196a expression may participate to the angiogenic switch.
Significance: These results bring important new insights in understanding the mechanisms underlying angiogenesis-associated pathologies.
Keywords: Angiogenesis, Annexin, Cell Motility, p38 MAPK, Vascular Endothelial Growth Factor (VEGF)
Abstract
Endothelial cell migration induced in response to vascular endothelial growth factor (VEGF) is an essential step of angiogenesis. It depends in part on the activation of the p38/MAPKAP kinase-2/LIMK1/annexin-A1 (ANXA1) signaling axis. In the present study, we obtained evidence indicating that miR-196a specifically binds to the 3′-UTR region of ANXA1 mRNA to repress its expression. In accordance with the role of ANXA1 in cell migration and angiogenesis, the ectopic expression of miR-196a is associated with decreased cell migration in wound closure assays, and the inhibitory effect of miR-196a is rescued by overexpressing ANXA1. This finding highlights the fact that ANXA1 is a required mediator of VEGF-induced cell migration. miR-196a also reduces the formation of lamellipodia in response to VEGF suggesting that ANXA1 regulates cell migration by securing the formation of lamellipodia at the leading edge of the cell. Additionally, in line with the fact that cell migration is an essential step of angiogenesis, the ectopic expression of miR-196a impairs the formation of capillary-like structures in a tissue-engineered model of angiogenesis. Here again, the effect of miR-196a is rescued by overexpressing ANXA1. Moreover, the presence of miR-196a impairs the VEGF-induced in vivo neo-vascularization in the Matrigel Plug assay. Interestingly, VEGF reduces the expression of miR-196a, which is associated with an increased level of ANXA1. Similarly, the inhibition of miR-196a with an antagomir results in an increased level of ANXA1. We conclude that the VEGF-induced decrease of miR-196a expression may participate to the angiogenic switch by maintaining the expression of ANXA1 to levels required to enable p38-ANXA1-dependent endothelial cell migration and angiogenesis in response to VEGF.
Introduction
Angiogenesis, the sprouting of new capillaries from pre-formed capillaries, contributes to the expansion of the vascular network in several normal and pathological processes (1, 2). Physiological angiogenesis is highly regulated during development, wound repair, and menstrual cycle progression (3). Persistent dysregulation of angiogenesis is a common denominator associated with various pathological disorders, including age-related macular degeneration, rheumatoid arthritis, tumor progression, and metastasis (4, 5). Angiogenesis is a multistep and multicellular process that involves proteolytic degradation of the extracellular matrix, followed by migration and proliferation of capillary endothelial cells, pericyte recruitment, and assembly of the mature vessel (6). The angiogenic process is regulated by a tight balance between pro- and anti-angiogenic agents. Endostatin is a typical antiangiogenic agent and vascular endothelial growth factor (VEGF)4 is the major promoter of both physiological and pathological angiogenesis (6).
Members of the VEGF family contain six structurally related proteins: VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placenta-derived growth factor (PIGF) (7, 8). Human VEGF-A monomers exist as 5 different isoforms, of which VEGF165, hereafter referred to as VEGF, is the most abundant and biological active form (6). VEGF binds two tyrosine kinase receptors on blood vessel endothelial cells, VEGF receptor-1 (VEGFR-1/Flt-1) and VEGF receptor-2 (VEGFR-2/KDR/Flk-1). Both receptors are essential for developmental neovascularization and their knock-out is embryonic lethal (9, 10).
VEGFR-2 is the major signaling endothelial cell receptor for VEGF in adults. Ligand binding to VEGFR-2 triggers its oligomerization, which activates its tyrosine kinase activity leading to phosphorylation of specific tyrosine residues located within its cytoplasmic domain. In particular, VEGFR-2-mediated endothelial actin remodeling and cell migration are triggered by the autophosphorylation of the receptor at Tyr-1214, upstream of the activation of the p38 MAP kinase pathway (7, 11, 12). Cell migration downstream of VEGFR-2 has also been shown to be mediated through Gab1, which acts as an adaptor for Grb2, PI3-kinase and the tyrosine phosphatase SHP2 (13, 14).
Annexin-1 (Lipocortin 1, Calpactin II herein abbreviated as ANXA1) is one of the 13 members of the annexin superfamily. The members of the annexin family are characterized by a highly conserved protein core domain that harbors Ca2+ and phospholipid-binding sites (15). ANXA1 is a steroid-regulated protein that transduces the glucocorticoid anti-inflammatory action by inhibiting phospholipase A2 (16). In line with this, ANXA1−/− mice show a partial or complete resistance to the anti-inflammatory effects of glucocorticoids (17). ANXA1 further regulates the inflammatory response by mediating the neutrophil responsiveness to fMetLeuPhe (18). On the other hand, many studies have shown that ANXA1 is phosphorylated on tyrosine residues following the activation of EGF and HGF receptor-tyrosine kinases or on serine residues by protein kinase c (19, 20). In both cases, these phosphorylation events target the PI3 kinase and ERK MAP kinase glucocorticoid signaling pathways and play important signaling functions in cell proliferation, differentiation and apoptosis (21, 22). A role of “signal amplifier” has also been attributed to ANXA1, because it promotes the release of second messengers that affect cell proliferation and migration (19). In this context, there is a growing body of evidence indicating that ANXA1 may interact with motile cytoskeletal proteins such as tubulin and actin (23–25). Along these lines, we recently reported that phosphorylation of ANXA1 by LIM kinase downstream of the p38 MAP kinase pathway is an important regulator of endothelial cell migration in response to VEGF (7). Moreover, the cell invasion potential of intestinal epithelial cell line SKCO-15 is increased by 2-fold in the presence of extracellularly added full-length rANXA1 (26). This effect seems to be mediated via autocrine/paracrine signaling involving formyl peptide receptors. This role of ANXA1 in tumor invasion is consistent with previous reports showing that ANXA1 is associated with metastasis in several invasive malignancies (26–28). Accordingly, ANXA1 null mice present defects in tumor growth, metastasis, angiogenesis, and wound healing suggesting the importance of ANXA1 in regulating tumor progression (29, 30).
miRNAs are single-stranded, evolutionarily conserved, small (21–23 nucleotides) non-coding RNAs. They are generated from sequential processing of primary miRNAs (pri-miR) into mature miRNAs from the nucleus to the cytoplasm (31). miRNAs are transcribed at first as pri-miRNAs from mainly intergenic or intronic regions by the RNA polymerase II. The pri-miRs are then processed by the ribonuclease III DROSHA in the nucleus to form the precursor miRNAs (pre-miRs). These pre-miRs are exported to the cytoplasm by exportin-5 where the ribonuclease III DICER processes them again to give the mature microRNAs that will be loaded onto the RNA-induced silencing complex (RISC) (32). It is as part of the RISC complex that these small RNAs regulate gene expression by binding to a target messenger RNA in its 3′-UTR region, thereby repressing translation and/or degrading mRNA (33, 34). Functional miRNAs should be associated with several proteins including members of the Argonaute (Agos) family that are part of an active RISC complex and are considered as major regulators of silencing (35). A large body of evidence suggests that the multigene regulatory capacity of miRNAs is dysregulated in cancer. In most cases, a global reduction of miRNA abundance appears as a general trait of human cancers (36–38). Yet, little is known on the underlying mechanisms or the phenotypic advantages afforded to cells by reduced miRNA expression, and on the clinical relevance of this phenomenon. Over the past 3 years, miR-196a was implicated in numerous types of cancers (39). In particular, miR-196a showed a significant inverse correlation with ANXA1 mRNA levels in 12 cancer cell lines of esophageal, breast and endometrial origin (40). These findings support the potential role of miR-196a in the regulation of ANXA1 expression. In the present study, we show that ANXA1 is post-transcriptionally regulated by miR-196a in response to VEGF, which contributes to regulate endothelial cell migration.
EXPERIMENTAL PROCEDURES
Chemicals
All treatments were done using recombinant human VEGF-A165 (herein named VEGF) produced by R&D Systems (Minneapolis, MN) and kindly provided by the NCI Biological Resources Branch (Rockville, MD).
Cells
Human umbilical vein endothelial cells (HUVECs) were isolated by collagenase digestion of umbilical veins from undamaged sections of fresh cords (41). The cords have been obtained after approbation of the CRCHUQ Ethic Committee. Subcultures were maintained in EGM2 media (LONZA, Allendale, NJ). Replicated cultures were obtained by trypsination and were used at passages <5. Treatments were done on HUVECs cultivated on gelatin and made quiescent by serum-starvation using M199 media containing 5% heat-inactivated fetal bovine serum (FBS), l-glutamine, and antibiotics. Normal Human Dermal Fibroblasts (NHDFs) were obtained from LONZA and maintained in DMEM supplemented with 10% FBS and antibiotics. Human embryonic kidney cells (HEK293T) were cultivated in DMEM containing 10% FBS and antibiotics.
Antibodies
Anti-ANXA1 monoclonal mouse antibody was purchased from BD Transduction Laboratories (Mississauga, ON, Canada). Anti-α-tubulin monoclonal antibody was obtained from Sigma-Aldrich. The anti-mouse-IgG-horseradish peroxidase (HRP) was from The Jackson Laboratory (Bar Harbor, ME). Anti-IgG antibody coupled to IRDye800 or IRDye680 were obtained from Rockland Immunochemicals (Gilbertsville, PA) or from Invitrogen (Carlsbad, CA). A mouse γ-globulin was purchased from The Jackson Laboratory.
Plasmids, siRNA, miRNA Mimics, and Antagomirs
The pmiR-196a expressing plasmid was obtained from System Biosciences (Mountain View, CA). GFP expressing vector (pmiR-empty) and plasmids necessary for lentiviral particles production were a kind gift of Dr. Manuel Caruso (Laval University, QC, Canada), and CSII-EF-MCS-IRES2-Venus (CSII-Venus) vector was a kind gift of Dr. Hiroyuki Miyoshi (RIKEN Tsukuba Institute, Japan). ANXA1 cDNA without 3′-UTR region was subcloned in CSII-Venus vector using NotI and BamHI restriction sites (CSII-Venus-ANXA1). psiCHECK-2 vector was obtained from Promega (Madison, WI). ANXA1 coding sequence and 3′-UTR region was amplified from HUVECs total RNA following cDNA production using the following primers: Forward primer 5′-CTTTGCAAGAAGGTAGAGATAAAGACAC-3′; Reverse primer 5′-CTTGTGACGTCATTTTATTTTCAGCTACATAG-3′ and KOD Hot Start DNA polymerase according to the manufacturer protocol (EMD, Philadelphia, PA). Then, ANXA1 3′-UTR was cloned downstream of the Renilla luciferase stop codon on psiCHECK-2 vector using XhoI/NotI restriction sites (Forward primer: 5′-CACTCGAGACATTCCCTTGATGGTCTC-3′; Reverse primer: 5′-CTGCGGCCGCTCATTTTATTTTCAGCTACATAG-3′). After sequencing, the resultant vector (psiCHECK-2-ANXA1–3′UTR-wt) containing Renilla luciferase under the control of ANXA1 3′-UTR and firefly luciferase as a control were used to produce psiCHECK-2-ANXA1–3′UTR-mutated by directed mutagenesis using the following primers: Forward primer: 5′-CAGTGTAGGTTCGTACATGCTGAAAAATAT-3′; Reverse primer: 5′-CATGTACGAACCTACACTGTAATCCTG-3′). Validated ANXA1 siRNA 7 was purchased from Qiagen (Mississauga, ON, Canada). siRNA 7 was designed to target the mRNA of human ANXA1 (GenBankTM accession no. NM_000700). The target sequence of ANXA1 siRNA 7 is as follows: sense, 5′-ATGCCTCACAGCTATCGTGAA-3′ and antisense, 5′-TTCACGATAGCTGTGAGGCAT-3′. Mature miR-196a mimic or antagomir and control were obtained from Dharmacon (Lafayette, CO).
Lentiviral Particle Preparation
HEK293T were plated at 3 × 106 per 100 mm Petri dish in 10 ml DMEM supplemented with 10% FBS. Co-transfection was perfomed by Ca2+ phosphate-mediated transfection using 2.5 μg of pRSV-Rev, 6.5 μg of pMDLg-pRRE, 3.5 μg of pMD2.G together with 10 μg of transgene expressing vectors (pmiR-196, pmiR-empty, CSII-Venus or CSII-Venus-ANXA1 vectors). After overnight incubation, media were replaced with 5 ml of lentiviral particle collection media (M199 medium containing 20% heat-inactivated FBS, ECGS (60 μg/ml) (Sigma-Aldrich), l-glutamine, heparin, and antibiotics). After 24 h, the media were collected and centrifuged at 4 °C, 1200 rpm, for 5 min. The supernatant was stored in aliquots at −80 °C. The Multiplicity of Infection (MOI) known as the ratio of infectious virus particles per cell, was determined by a 48 h transduction of HUVECs in a cascade dilution experiment. The percentage of GFP-positive cells was determined by FACS using EPICS-XL-MCL flow cytometer (Beckman-Coulter, Ramsey, MN).
Transfection and Transduction
HUVECs were transfected with miRNA mimics, antagomirs, siRNA, or plasmid vectors using X-tremeGene HP Transfection Reagent obtained from Roche (Laval, QC, Canada) according to the manufacturer's protocol or by electroporation. HUVECs were transduced with miR-196a precursor expressing vector (pmiR-196a) or with empty vector (pmiR-empty) together or not with CSII-Venus or CSII-Venus-ANXA1 using lentivirus-mediated infection in the presence of 8 μg/ml hexadimethrine bromide (Sigma-Aldrich).
RNA Extraction and RT-qPCR
RNA Extraction
Total RNAs were extracted using Mirvana Isolation kit (Ambion Applied Biosystems Streetville On Canada). Total RNA concentration was determined using Nanodrop 1000 spectophometer (Thermo Fisher Scientific) and quality was assessed using a 1% agarose gel before its utilization for cDNA production.
TaqMan RT-qPCR
For miRNA expression quantification, reverse transcription was performed using TaqMan MicroRNA Reverse Transcription Kit following the manufacturer's procedure and using specific microRNAs primers for reverse transcription from microRNA assays (hsa-miR-196a :241070 and the housekeeping short non-coding RNA U6 snRNA : 001973) (Invitrogen). microRNA level of expression was determined using Universal PCR Master Mix, No AmpErase UNG according to the manufacturer protocol (Invitrogen, AB 7900). The 2ΔΔCt method was used to determine the Fold Changes (FC).
Immunoprecipitation
After treatments, cells were washed with phosphate-buffered saline and were lysed in B buffer containing 150 mm NaCl, 50 mm Tris-HCl, pH 7.5, 0.5% Triton X-100, 0.1% sodium deoxycholate, 2 mm EDTA, 2 mm EGTA, 1 mm NA3VO4, 1 mm benzamide, 1 μm leupeptin, 50 mm NaF, and 1 mm phenylmethylsulfonyl fluoride. Cells were centrifuged at 13,000 rpm for 10 min and proteins were precleared with 15 μl 50% (v/v) protein G-Sepharose for 45 min. Supernatants were incubated on ice for 16 h with appropriate antibody. Then, 15 μl of 50% (v/v) protein G-Sepharose were added, and the incubation was extended for 45 min on ice with shaking. Antibody-antigen complexes were washed three times with B buffer and then SDS-PAGE loading buffer was added.
Western Blotting
After treatments, cells were lysed using SDS-PAGE loading buffer. Equal amounts per well (30 μg) of total proteins were separated by SDS-PAGE, and the gels were transferred onto nitrocellulose membranes for Western blotting. After incubating nitrocellulose membranes with the appropriate primary antibodies, antigen-antibody complexes were detected with an anti-IgG antibody coupled to horseradish peroxidase and then revealed using an enhanced chemiluminescence kit. Alternatively, antigen-antibody complexes were detected with an anti-IgG antibody coupled to IRDye800 or IRDye680 and revealed using an infrared imaging system (Li-Cor, Lincoln, NE). Quantification of the immunoreactive bands was done by densitometric scanning using the Image J software.
Luciferase Reporter Assay
HUVECs (2.5 × 105 cells) were electropored with psiCHECK-2-ANXA1–3′UTR-wt or psiCHECK-ANXA1–3′-UTR-mutated (4 μg) together with miR-196a mimic or a control mimic (200 nm). The following day, cells were lysed and luciferase activity was evaluated using Dual-Luciferase Reporter Assay System following manufacturer's protocol (Promega). Renilla luciferase activity which is under the control 3′-UTR insert was evaluated and firefly luciferase was measured as a loading control in each condition using a Glomax luminometer 20/20 (Promega).
Wound Closure Assay
HUVECs were plated in a 12-well-plates at 100,000 cells/wells. The day after, cells were transfected using X-tremeGENE HP DNA Transfection Reagent. Cells were transfected with a mature miR-196a mimic or a control mimic. Forty-eight hours later, cells were serum-starved for 4 h. Then, a scratch was applied manually to the confluent cell monolayer. After three washes to remove the detached cells, the remaining cells were placed in serum-starved medium with or without VEGF treatment (10 ng/ml). The plate was then incubated in the living chamber of a Nikon-TE2000 inverted microscope equipped with 10× lens (numerical aperture of 0.30), with a Metamorph (7.7.5 version) imaging system and with a Photometrics CoolSNAP HQ2 camera. The pictures were captured using both transmitted light together or not with fluorescence. The total number of cells that filled the scratch in each condition was manually counted in three to four different fields at 8, 12, and 18 h. For each time point, the number of cells in VEGF-treated fields was normalized against the mean of corresponding untreated fields to represent the relative VEGF-induced migration.
Boyden Chamber Assay
Endothelial cell migration in Boyden chambers was assayed as previously reported (7). Briefly, 48 h-transfected or 96-h-transduced cells were serum-starved overnight using M199 media supplemented with 5% heat-inactivated fetal bovine serum, l-glutamine and antibiotics. Then, cells were harvested with trypsin, counted, centrifuged, and resuspended at 1.5 × 106 cells/ml in migration buffer (199 medium, 10 mm HEPES pH 7.4, 1 mm MgCl2, and 0.5% bovine serum albumin). Cells (1.5 × 105) were added on the upper part of 8.0-μm pore size gelatin-coated migration chambers (Corning, NY) separating the upper and lower chambers of a 6.5-mm transwell. Cells were left to adhere for 1 h. Then, 10 ng/ml VEGF were added in the lower chamber for 4 h. In transfection experiments pEGFP was co-transfected to visualize and count transfected cells that crossed the membrane of the Boyden chamber. The cells in the upper part of the chamber were removed with a cotton swab. Then, cells that crossed the membrane were counted manually in five fields using a 20× lens (numerical aperture of 0.45) on a Nikon-TE300 inverted microscope. In transduction experiments, 4 h after VEGF treatment, the sum of fluorescent transduced cells that crossed the membrane of the Boyden chamber was manually counted from five fields per chamber using the same microscopic facilities as described above. All experiments were performed three times at least in duplicates.
Angiogenesis Assays
HUVECs were transduced with pmiR-196 containing miR-196 precursor (pmiR-196a) or with an empty vector (pmiR-empty) (MOI of 65) together or not with CSII-Venus or CSII-Venus-ANXA1 (MOI of 33) using lentivirus-mediated infection. After 96 h, 50,000 transduced cells were plated on a 12-days monolayer of NHDFs. NHDFs were grown and maintained in DMEM supplemented with 10% fetal bovine serum and 50 μg/ml sodium l-ascorbate to facilitate the formation of a sheet (60,000 cells per well of a 12-well plate). Co-cultures were maintained for 6 days in DMEM supplemented with 10% fetal bovine serum, M199 supplemented with 5% fetal bovine serum and l-glutamine (v:v) and 50 μg/ml sodium l-ascorbate. Media were replaced every 48 h with or without VEGF treatment (10 ng/ml). This was performed for a total of 3 treatments (D0, D2, D4) (42). Capillary-like structures were observed at day 3 and day 5 using a Nikon-TE2000 inverted microscope equipted 4× lens (numerical aperture of 0.13) with a Metamorph (7.7.5 version) imaging system and with a Photometrics CoolSNAP HQ2 camera. Fifteen pictures per well from different fields were captured. Raw images were processed with ImageJ software using the following pipeline: images were converted to binary using a threshold value of 25 on 8 bit images, despeckle, convert to mask and skeletonize. We considered the number of remaining white pixels as the length of the capillary structures per field. Also the plugin AnalyseSkeleton in ImageJ was used to obtain the number of branches and the number of junctions. The total length of capillary like structures, the number of branches and the number of junctions were expressed as the mean of sums of 15 fields per replicate. Movies of these experiments were built with the pictures captured using a Metamorph imaging software (7.7.5 version).
Matrigel Plug Assay
Matrigel (BD Biosciences) was pre-mixed with 15 units of heparin (Sandoz, Boucherville, Qc, Canada) and 200 ng of VEGF or an equivalent volume of PBS. Then, miR-196a mimic (2 μm) or control mimic (2 μm) was added and the mixture was injected subcutaneously near the ventral midline of 6-week-old FVB/n male mice (Charles River). Plugs were harvested 12 days later and photographed. For the measure of hemoglobin (Hb) content, harvested plugs were weighted and incubated overnight in de-ionized water at 37 °C. Then, plugs were crushed with the rubber end of a syringe barrel and centrifuged. Supernatant Hb content was measured using the QuantiChrom Hemoglobin Assay Kit (BioAssay Systems, Hayward, CA) and normalized to plug weight. Neovascularization was determined after H&E staining of a 5 μm slice of paraffin-embedded plugs. Capillary structures were counted by region of interest (ROI) (five ROI per plug; at least five plugs per condition from two separate experiments).
Fluorescence Microscopy
After treatment, cells were fixed with 3.7% formaldehyde and permeabilized with 0.1% saponin in phosphate buffer, pH 7.5. F-actin was detected using Rhodamin-phalloidin (Invitrogen, Carlsbad, CA). ANXA1 was detected using specific mouse monoclonal antibody (Cell Signaling, Danvers, MA). The cells were examined using Nikon-Eclipse 800 microscope equipped with a 40× objective lens (numerical aperture of 0.90).
Statistics
Values are expressed as fold changes or mean ± S.D. Unpaired Student's t-tests were used for comparison between two means. A p value <0.05 was considered statistically significant.
RESULTS
miR-196a Represses ANXA1 Expression Level
We recently reported that ANXA1 is an essential component of the p38-mediated endothelial cell migration in response to VEGF (7). In addition, it was shown that the expression of ANXA1 is inversely correlated with miR-196a level in several cancer cell lines (40). These findings prompted us to investigate the role of miR-196a in regulating ANXA1-dependent endothelial cell migration induced by VEGF. We first verified whether miR-196a regulates the expression of ANXA1 in endothelial cells. To this end, we overexpressed miR-196a by transfecting HUVECs with a specific miR-196a mimic and found that this was associated with a 40% decrease in the level of ANXA1 (Fig. 1, A–C). Reciprocally, the inhibition of miR-196a with a specific antagomir increased the level of ANXA1 by 1.5∼fold (Fig. 1D). Consistent with these findings, we observed that luciferase reporter carrying ANXA1 3′-UTR region was sensitive to miR-196a ectopic expression. In contrast, mutations of miR-196a complementary site within ANXA1 3′-UTR relieves miR-196a-dependent repression (Fig. 1E). This suggests that miR-196a represses the expression of ANXA1 by its specific binding to the 3′-UTR region of ANXA1 mRNA.
FIGURE 1.

miR-196a targets specifically ANXA1 in endothelial cells. A–C, HUVECs were transfected with miR-196a or control mimics. A, after 48 h, total RNA was extracted and miR-196a expression level was evaluated by TaqMan RT-qPCR. B and C, after 48 h, proteins were extracted, were separated by SDS-PAGE and transferred onto nitrocellulose membrane. ANXA1 and tubulin α were revealed by Western blot using specific antibodies. C, relative ANXA1 protein level was quantified (ANXA1/Tubulin) from two separated experiments by densitometry analysis using ImageJ software. D, HUVECs were transfected with antagomir-196a or control antagomir. After 48 h, proteins were extracted, were separated by SDS-PAGE and transferred onto nitrocellulose membrane. ANXA1 and Tubulin α were revealed by Western blot as in B. E, ANXA1 3′-UTR wild type (wt) or mutated (in the specific predicted miR-196a binding site) were subcloned in luciferase expressing vector (psiCHECK-2) to evaluated luciferase activity under the control of ANXA1 3′-UTR. These wt or mutated constructs were co-transfected in HUVECS together with miR-196a mimic or a control mimic. Renilla luciferase activity under the control of ANXA1 3′-UTR was evaluated and then normalized against firefly luciferase. The mean percentage of relative luciferase activity is represented (n = 3 at least in duplicate).
VEGF Decreases the Expression of miR-196a
Given that ANXA1 is importantly involved in VEGF-induced endothelial cell migration and that the level of ANXA1 is regulated by miR-196a, we next verified whether the expression of miR-196a and ANXA1 was modulated in the presence of VEGF. Overnight serum-starved HUVECs were treated or not with 10 ng/ml VEGF. Thereafter, total RNA was extracted and the relative level of miR-196a was determined. The results presented in Fig. 2A showed that VEGF induced a 2-fold decrease in the level of miR-196a, which was accompanied with an increase in ANXA1 already evident at 16 h and was still persistent at 36 h (Fig. 2B). Moreover, the inhibition of miR-196a with an antago-miR-196a raises the level of ANXA1 and further enhances the VEGF-increased expression of ANXA1 (Fig. 1D). These finding are consistent with the fact that the regulation of miR-196a by regulating the level of ANXA1 may be importantly involved in maintaining endothelial cells in a quiescent state in non-activated conditions.
FIGURE 2.
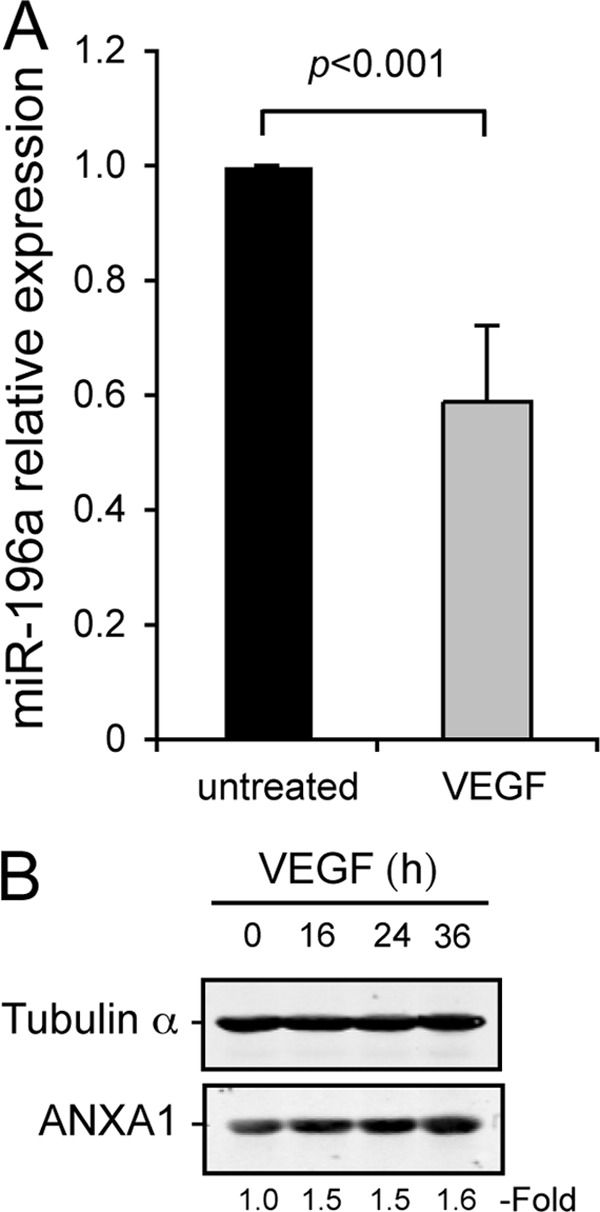
VEGF induced miR-196a decrease and ANXA1 increase in endothelial cells. A, quiescent HUVECs were treated or not with VEGF (10 ng/ml) for 24 h. After treatments, total RNA was extracted and miR-196a expression level was evaluated by TaqMan RT-qPCR (n = 4). B, quiescent HUVECs were treated or not with VEGF (10 ng/ml) for increasing periods of time. After treatments, proteins were extracted, were separated by SDS-PAGE and transferred onto nitrocellulose membrane. ANXA1 and tubulin α were revealed by Western blot using specific antibodies.
miR-196a Inhibits ANXA1-dependent Actin Remodeling and Endothelial Cell Migration Induced by VEGF
In accordance with a role of miR-196a in mediating endothelial cell migration, we obtained results indicating that the expression of a miR-196a mimic in HUVECs was associated with an inhibition of endothelial cell migration in response to VEGF in wound closure and Boyden Chamber assays (Fig. 3 and supplemental Fig. S1). Moreover, miR-196a-dependent inhibition of cell migration was rescued by overexpressing ANXA1 by means of a plasmid that lacks the 3′-UTR region (Fig. 3, F and G). This indicated that miR-196a inhibited cell migration by targeting ANXA1.
FIGURE 3.

miR-196a inhibits VEGF-mediated endothelial cell migration. HUVECs were transfected with miR-196a (B, C, E, and F) or control (A and D) mimics (100 nm, for a total of 48 h) together with an empty vector (pIRES; A, B, D, and E) or pIRES-ANXA1-HA (C and F) for 48 h. Cells were processed for wound healing assay in response to VEGF (10 ng/ml). The black lines represents the edge of the wound and pictures were taken after 0 (A–C) and 16 (D–F) hours of treatment. Images were captured using an inverted microscope (10×). G, cells that have crossed the edge of the wound have been counted and the results from 2 separate experiments are shown. H, HUVECs were transfected with a miR-196a mimic or a control mimic (100 nm, for a total of 48 h) together with pEGFP to visualize and count transfected cells. Overnight-serum-starved cells were evaluated for cell migration in a Boyden chamber assay using VEGF (10 ng/ml for 4 h) as chemoattractant. I, HUVECs were transduced with miR-196a precursor (pmiR-196a) or with an empty vector (pmiR-empty) both expressing GFP using lentiviral-mediated infection (MOI of 65) for 96 h. Overnight-serum-starved cells were processed for cell migration assay as in H.
The VEGF-induced endothelial cell migration requires actin cytoskeleton remodeling into lamellipodia, and ANXA1 has been reported to be recruited in lamellipodia following its phosphorylation in response to EGF (6, 23). Along these lines, we observed, in Western blot, that actin co-precipitates with ANXA1 and that the association was increased by VEGF (Fig. 4). Similar results were observed in mass spectrometry analysis where we found, in an immunoprecipitate of ANXA1 obtained from cells treated with VEGF, a band of 46 kDa whose analysis revealed 69 peptides that corresponded to actin. More interestingly, the decreased migratory potential of HUVECs overexpressing miR-196a, and thus showing decreased level of ANXA1, was associated with an inhibition in the formation of lamellipodia in response to VEGF. In particular, the mean number of lamellipodia per cell was increased by 1.5∼fold when it peaks at 30 min in response to VEGF treatment in cells expressing a control mimic. In contrast, the number of lamellipodia remained to basal level in cells expressing miR-196a mimic (Fig. 5, A and B).
FIGURE 4.
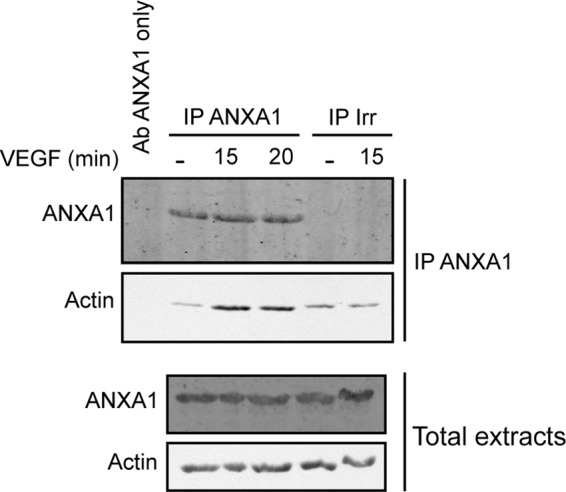
Actin associates with ANXA1 in response to VEGF. Quiescent HUVECs were treated with VEGF (10 ng/ml for 15 or 20 min). After treatments, cells were lysed, and ANXA1 was immunoprecipitated using a mouse monoclonal antibody or a control irrelevant antibody. Proteins were separated by SDS-PAGE and were transferred to a nitrocellulose membrane. Only the ANXA1 antibody used for immunoprecipitation was loaded as control. Actin co-immunoprecipitation with ANXA1 was confirmed by Western blotting using a goat polyclonal anti-actin antibody and the presence of ANXA1 was revealed using a mouse monoclonal antibody. Total cell extracts were identically assessed for loading control.
FIGURE 5.

miR-196a represses VEGF-induced lamellipodia. HUVECs were transfected with miR-196a or control mimics (100 nm, for a total of 72 h). Cells were serum-starved overnight before being treated with VEGF (10 ng/ml) for increasing periods of time. Thereafter cells were fixed, permeabilized, and stained for F-actin and ANXA1 using phalloidin and ANXA1 specific mouse antibody, respectively. A, minimum of 150 cells per condition were evaluated to quantified lamellipodia per cell in two separated experiments. The mean number of ruffles per cell is represented in each condition ± S.D. B, representative field for each condition was captured using a fluorescence microscope (40×).
Altogether, these results indicate that ANXA1 by associating with actin is a major determinant of cell migration. Moreover, they suggest that the repression of ANXA1 expression by miR-196a is an important negative modulator of actin remodeling into lamellipodia and endothelial cell migration in response to VEGF.
miR-196a Inhibits the Formation of Capillary-like Structures in a Reconstituted Tissue Model of Angiogenesis
Endothelial cell migration is an essential step of angiogenesis. As miR-196a blocks endothelial cell migration, we investigated next, whether it repressed angiogenesis using a simplified human reconstructed tissue culture model of neovascularization in vitro (42). Briefly, HUVECs expressing miR-196a precursor or an empty vector were seeded on a monolayer of Normal Human Dermal Fibroblasts (NHDFs) and were treated or not with VEGF. In this model, the addition of VEGF induced a well-developed network of capillary-like structures. These structures were already evident after 3 days of VEGF treatment and further gain in organization after 5 days (Fig. 6, A–D). As expected, miR-196a impaired the formation of these capillary-like structures induced by VEGF treatment (Fig. 6, E–H). In fact, in HUVECs overexpressing miR-196a and treated with VEGF, we noted a marked decrease in the mean number and length of capillary-like structures, as well as in their branching (Fig. 6, I–K). The dynamic of the capillary-structure formation in response to VEGF and its impairment by miR-196a is more clearly seen in movies presented in supplemental Figs. S2, A and B. Additionally, supplemental Fig. S2C further shows that the process is ANXA1-dependent given that the inhibition of capillary-structure formation associated with miR-196a was rescued by overexpression of ANXA1.
FIGURE 6.

miR-196a modulates VEGF-mediated formation of capillary-like structures. A–H, HUVECs were transduced with miR-196a precursor (pmiR-196a) or pmiR-empty (MOI of 65), both co-expressing GFP, using lentiviral-mediated infection. After 96 h, 50 000 transduced cells were plated on a 12 days normal human dermal fibroblasts (NHDF) monolayer. Co-cultures were maintained for 6 days, and media were replaced every 48 h with or without VEGF treatment (10 ng/ml) for a total of 3 treatments (n = 4). Fluorescent capillary-like structures were observed at day 3 and 5 using an inverted microscope (4×). Representative fields are shown for each condition (A–H). Fifteen pictures per condition from different fields were captured to measure the number of capillary-like structures (I) and the number of junctions (J) using AnalyzeSkeleton plugin from ImageJ. K, using the same pictures, the length of the capillaries was measured using ImageJ software.
miR-196a Inhibits in Vivo Angiogenesis
We next ascertained whether miR-196a regulates VEGF-induced angiogenesis in vivo. To this end, we used the mouse Matrigel Plug assay. Briefly, Matrigel containing or not VEGF in the presence of the miR-196a mimic, or a control mimic, was injected in mice. Twelve days later, the plugs were harvested and the fomation of vessels was evaluated in situ and their functionality was evaluated by determining the hemoglobin content in the plug. The results showed that VEGF induces a massive neovascularization in the plug as evidenced by the redness and the number of capillaries. As expected, this is impaired in the presence of miR-196a (Fig. 7, A, B, and D). Similarly, the amount of hemoglobin was markedly reduced in the presence of miR-196a (Fig. 7C). Overall, these results indicate that miR-196a, by impairing the expression of ANXA1, is a major repressor of angiogenesis induced by VEGF.
FIGURE 7.

miR-196a inhibits VEGF-induced angiogenesis in the Matrigel plug assay. A, Matrigel was mixed with miR-196a mimic or control mimic together with VEGF or PBS as control. The Matrigel plugs were injected subcutaneously in the mid-ventral region of FVB/n mice. Twelve days later, the plugs were harvested. Representative images taken from at least 5 mice per condition are shown. B, plugs have been embedded in paraffin and stained with H&E to visualize the blood vessels that have been formed. C, hemoglobin content was measured and normalized to the weight of Matrigel in each plugs. D, new blood vessels stained in H&E were quantified and the mean number of capillaries per ROI was calculated.
DISCUSSION
We recently reported that the VEGF-induced activation of the p38/MAPKAP kinase 2 axis leads to activation of LIM kinase 1 (LMK1) and phosphorylation of ANXA1. In turn, this contributes to trigger endothelial cell migration and tube formation in Matrigel (7). More recently, we found that miRNAs are also important regulator of the p38 pathway by showing that miR-20a inhibits the activation of p38 by repressing MKK3 (43). In this report, we reported that, in addition to phosphorylation, the migratory functions of ANXA1 are also tightly regulated by miR-196a-dependent regulation of ANXA1 expression.
The fact that miR-196a is a major regulator of endothelial cell migration and angiogenesis in response to VEGF is supported by three complementary lines of evidence: 1) the expression of a miR-196a mimic in HUVECs almost completely impairs their migration; 2) the overexpression of miR-196a dysregulates the formation and branching of capillary-like structures induced by VEGF in vitro and 3) miR-196a inhibits the formation of functional blood vessel induced by VEGF in vivo in the Matrigel plug assay.
In accordance with a role of miR-196a in cell migration, a previous study reported that a low level of miR-196a expression is associated with an enhanced migratory potential of melanoma, presumably as a result of a raised ETS-1 and BMP4 activity (44). In contrast, high levels of miR-196a promote cancer cell detachment, migration and invasion of other cancer cell types including colon cancer cells (39, 45). Together with our present findings, these results suggest that the effect of miR-196a on cell migration may vary from cell type to cell type and warrants further investigations.
MicroRNAs repress protein expression by binding to the 3′-UTR region of mRNAs, which results in translation inhibition or mRNA degradation (32). They have different targets and thereby may affect a same cellular process via different pathways. In the case of miR-196a, it was shown that the human HOXB8 3′-UTR has perfect base-pairing for this miRNA, which suggests that HOXB8 mRNA is targeted by miR-196a for cleavage. In fact, miR-196a directs the cleavage of HOXB8 mRNA in mouse embryos (46). A major finding of our study is to have demonstrated that the miR-196a-dependent inhibition of endothelial cell migration and angiogenesis relies on the repression of ANXA1. Firstly, this is supported by the finding that miR-196a decreases the expression of ANXA1 in response to VEGF by targeting the 3′-UTR of ANXA1 mRNA in endothelial cells. Reciprocally, the expression of an antago-miR-196a increases the ANXA1 level. In fact, this finding is consistent with a previous study that showed that increased levels of miR-196a have a significant inverse correlation with the level of ANXA1 mRNA in cell lines from esophageal, breast and endometrial cancer origin (40). Secondly, this is also supported by our results indicating that the miR-196a-mediated inhibition of cell migration and capillary-like structures in vitro is rescued by overexpressing ANXA1 in the presence of a miR-196a mimic. Thirdly, ANXA1 level is increased by VEGF.
ANXA1 is a calcium- and phospholipid-binding protein involved in regulating a broad range of cellular events, including migration in different types of cells such as myoblasts, leukemia, and breast cancer lines and endothelial cells (7, 26, 47, 48). Moreover, a recent study showed that ANXA1−/− mice are unable to heal the damage associated with indomethacin-induced gastric ulcers (30). Similary, aortic ring assays reveal an increased ANXA1 expression in sprouting endothelial cells of normal mice whereas aortas from ANXA1 KO mice exhibit impaired endothelial cell sprouting that is rescued by adenoviral expression of ANXA1 (29). The mechanisms that underly the role of ANXA1 in regulating cell migration and angiogenesis are still ill defined. In endothelial cells, we found that phosphorylation of ANXA1 is required to trigger cell migration (7). Moreover, EGF-induced phosphorylation of ANXA1, triggers its colocalization with F-actin in the lamellipodia (23). Given the important role that lamellipodia played in conferring forward movements to endothelial cells during their migration, this suggests that ANXA1 mediates VEGF-induced endothelial cell migration by regulating the formation of lamellipodia. In this context, ANXA1, both in basal and VEGF-induced conditions, colocalizes with F-actin in the lamellipodia of endothelial cells (7). Moreover, here we found that VEGF induces the ruffling activity of endothelial cells and that this is impaired by knocking down ANXA1 by overexpressing miR-196a. We also found that VEGF induces the association between ANXA1 and actin (Fig. 4). Overall, these findings suggest that ANXA1 should be present in the lamellipodia to sustain cell migration but that the triggering event is induced by its phosphorylation. Based on our present and previous findings, VEGF seems to be involved in regulating both events: 1) it decreases the expression level of miR-196a, and thereby favors the expression of ANXA1 to levels allowing cell migration (see Fig. 2) and; 2) it triggers the phosphorylation of ANXA1 downstream of p38 pathway (7).
By decreasing the expression of miR-196a to maintain the expression of ANXA1 to physiological levels, we now propose that VEGF activates a feedback loop to enable p38-dependent cell migration. This conclusion is in line with clinical findings showing that the level of miR-196a is increased in pathologies such as diabetes mellitus that are characterized by the disruption of normal wound repair process associated with an insufficient production of growth factors (49). Moreover, the mRNA levels of VEGF are severely decreased during the first phase of the healing process in the wounds of genetically diabetic db/db mice, and only low VEGF mRNA expression is still detectable at day 12 (49, 50).
By showing that miR-196a modulates angiogenesis by targeting the expression of annexin-1, our results bring important new insights in understanding the mechanisms underlying pathologies such as diabetes. In corollary, our manuscript establishes miR-196a and annexin-1 as putative targets for the development of drugs designed to modulate wound healing and VEGF-driven angiogenesis.
Supplementary Material
Acknowledgments
We thank Dr. Karim Ghani and Dr. Manuel Caruso for help in preparing the lentiviral vectors. We also thank Dr. Hiroyuki Miyoshi (Riken Institute Tsukuba, Japan) for providing the Venus construct. We also thank Dr. Éric Bonneil from IRIC (Montreal) for performing MS analysis.
This study was supported by grants (to J. H.) from the Canadian Institutes for Health Research (CIHR), The Heart Stroke Foundation of Canada (HSFC), and The Natural Sciences and Engineering Research Council of Canada (NSERC) and by CIHR grants (to M. J. S. and I. R.).

This article contains supplemental Figs. S1 and S2.
- VEGF
- vascular endothelial growth factor
- HSP27
- heat shock protein-27
- HUVEC
- human umbilical vein endothelial cells
- MAPK
- mitogen-activated protein kinase
- MAPKAP K2
- MAP kinase-activated protein kinase-2
- MKK3
- mitogen-activated protein kinase kinase 3
- NHDF
- normal human dermal fibroblast
- PI3K
- phosphatidylinositol 3-kinase
- RISC
- RNA-induced silencing complex
- 3′-UTR
- 3′-untranslated region
- VEGFR
- vascular endothelial growth factor receptor.
REFERENCES
- 1. Filho A. L., Lopes J. M., Schmitt F. C. (2010) Angiogenesis and breast cancer. J. Oncol. 2010, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Breier G., Risau W. (1996) The role of vascular endothelial growth factor in blood vessel formation. Trends Cell Biol. 6, 454–456 [DOI] [PubMed] [Google Scholar]
- 3. Carmeliet P. (2005) Angiogenesis in life, disease and medicine. Nature 438, 932–936 [DOI] [PubMed] [Google Scholar]
- 4. Oklu R., Walker T. G., Wicky S., Hesketh R. (2010) Angiogenesis and current antiangiogenic strategies for the treatment of cancer. J. Vasc. Interv. Radiol. 21, 1791–1805 [DOI] [PubMed] [Google Scholar]
- 5. Garnier D., Rak J. (2010) in Cancer Metastasis-Biology and Treatment. Metastasis of Colorectal Cancer (Beauchemin N, Huot J., eds), Vol. 14, pp. 243–286, Springer [Google Scholar]
- 6. Lamalice L., Le Boeuf F., Huot J. (2007) Endothelial cell migration during angiogenesis. Circ. Res. 100, 782–794 [DOI] [PubMed] [Google Scholar]
- 7. Côté M. C., Lavoie J. R., Houle F., Poirier A., Rousseau S., Huot J. (2010) Regulation of vascular endothelial growth factor-induced endothelial cell migration by LIM kinase 1-mediated phosphorylation of annexin 1. J. Biol. Chem. 285, 8013–8021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feige J. J. (2010) Tumor angiogenesis: recent progress and remaining challenges. Bull Cancer 97, 1305–1310 [DOI] [PubMed] [Google Scholar]
- 9. Shalaby F., Rossant J., Yamaguchi T. P., Gertsenstein M., Wu X. F., Breitman M. L., Schuh A. C. (1995) Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66 [DOI] [PubMed] [Google Scholar]
- 10. Fong G. H., Rossant J., Gertsenstein M., Breitman M. L. (1995) Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376, 66–70 [DOI] [PubMed] [Google Scholar]
- 11. Lamalice L., Houle F., Huot J. (2006) Phosphorylation of Tyr1214 within VEGFR-2 triggers the recruitment of Nck and activation of Fyn leading to SAPK2/p38 activation and endothelial cell migration in response to VEGF. J. Biol. Chem. 281, 34009–34020 [DOI] [PubMed] [Google Scholar]
- 12. Lamalice L., Houle F., Jourdan G., Huot J. (2004) Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene 23, 434–445 [DOI] [PubMed] [Google Scholar]
- 13. Dance M., Montagner A., Yart A., Masri B., Audigier Y., Perret B., Salles J. P., Raynal P. (2006) The adaptor protein Gab1 couples the stimulation of vascular endothelial growth factor receptor-2 to the activation of phosphoinositide 3-kinase. J. Biol. Chem. 281, 23285–23295 [DOI] [PubMed] [Google Scholar]
- 14. Laramée M., Chabot C., Cloutier M., Stenne R., Holgado-Madruga M., Wong A. J., Royal I. (2007) The scaffolding adapter Gab1 mediates vascular endothelial growth factor signaling and is required for endothelial cell migration and capillary formation. J. Biol. Chem. 282, 7758–7769 [DOI] [PubMed] [Google Scholar]
- 15. Lim L. H., Pervaiz S. (2007) Annexin 1: the new face of an old molecule. Faseb J. 21, 968–975 [DOI] [PubMed] [Google Scholar]
- 16. Flower R. J., Rothwell N. J. (1994) Lipocortin-1: cellular mechanisms and clinical relevance. Trends Pharmacol. Sci. 15, 71–76 [DOI] [PubMed] [Google Scholar]
- 17. Roviezzo F., Getting S. J., Paul-Clark M. J., Yona S., Gavins F. N., Perretti M., Hannon R., Croxtall J. D., Buckingham J. C., Flower R. J. (2002) The annexin-1 knockout mouse: what it tells us about the inflammatory response. J. Physiol. Pharmacol. 53, 541–553 [PubMed] [Google Scholar]
- 18. Rescher U., Danielczyk A., Markoff A., Gerke V. (2002) Functional activation of the formyl peptide receptor by a new endogenous ligand in human lung A549 cells. J. Immunol. 169, 1500–1504 [DOI] [PubMed] [Google Scholar]
- 19. Skouteris G. G., Schröder C. H. (1996) The hepatocyte growth factor receptor kinase-mediated phosphorylation of lipocortin-1 transduces the proliferating signal of the hepatocyte growth factor. J. Biol. Chem. 271, 27266–27273 [DOI] [PubMed] [Google Scholar]
- 20. Solito E., Mulla A., Morris J. F., Christian H. C., Flower R. J., Buckingham J. C. (2003) Dexamethasone induces rapid serine-phosphorylation and membrane translocation of annexin 1 in a human folliculostellate cell line via a novel nongenomic mechanism involving the glucocorticoid receptor, protein kinase C, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase. Endocrinology 144, 1164–1174 [DOI] [PubMed] [Google Scholar]
- 21. Alldridge L. C., Bryant C. E. (2003) Annexin 1 regulates cell proliferation by disruption of cell morphology and inhibition of cyclin D1 expression through sustained activation of the ERK1/2 MAPK signal. Exp. Cell Res. 290, 93–107 [DOI] [PubMed] [Google Scholar]
- 22. Alldridge L. C., Harris H. J., Plevin R., Hannon R., Bryant C. E. (1999) The annexin protein lipocortin 1 regulates the MAPK/ERK pathway. J. Biol. Chem. 274, 37620–37628 [DOI] [PubMed] [Google Scholar]
- 23. Campos-Gonzalez R., Kanemitsu M., Boynton A. L. (1990) Epidermal growth factor induces the accumulation of calpactin II on the cell surface during membrane ruffling. Cell Motil. Cytoskeleton 15, 34–40 [DOI] [PubMed] [Google Scholar]
- 24. Traverso V., Morris J. F., Flower R. J., Buckingham J. (1998) Lipocortin 1 (annexin 1) in patches associated with the membrane of a lung adenocarcinoma cell line and in the cell cytoplasm. J. Cell Sci. 111, 1405–1418 [DOI] [PubMed] [Google Scholar]
- 25. Hayes M. J., Rescher U., Gerke V., Moss S. E. (2004) Annexin-actin interactions. Traffic 5, 571–576 [DOI] [PubMed] [Google Scholar]
- 26. Babbin B. A., Lee W. Y., Parkos C. A., Winfree L. M., Akyildiz A., Perretti M., Nusrat A. (2006) Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J. Biol. Chem. 281, 19588–19599 [DOI] [PubMed] [Google Scholar]
- 27. Cicek M., Samant R. S., Kinter M., Welch D. R., Casey G. (2004) Identification of metastasis-associated proteins through protein analysis of metastatic MDA-MB-435 and metastasis-suppressed BRMS1 transfected-MDA-MB-435 cells. Clin. Exp. Metastasis 21, 149–157 [DOI] [PubMed] [Google Scholar]
- 28. Wu W., Tang X., Hu W., Lotan R., Hong W. K., Mao L. (2002) Identification and validation of metastasis-associated proteins in head and neck cancer cell lines by two-dimensional electrophoresis and mass spectrometry. Clin. Exp. Metastasis 19, 319–326 [DOI] [PubMed] [Google Scholar]
- 29. Yi M., Schnitzer J. E. (2009) Impaired tumor growth, metastasis, angiogenesis and wound healing in annexin A1-null mice. Proc. Natl. Acad. Sci. U.S.A. 106, 17886–17891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin G. R., Perretti M., Flower R. J., Wallace J. L. (2008) Annexin-1 modulates repair of gastric mucosal injury. Am. J. Physiol. Gastrointest Liver Physiol. 294, G764–769 [DOI] [PubMed] [Google Scholar]
- 31. Aslam M. I., Taylor K., Pringle J. H., Jameson J. S. (2009) MicroRNAs are novel biomarkers of colorectal cancer. Br J. Surg. 96, 702–710 [DOI] [PubMed] [Google Scholar]
- 32. Suárez Y., Sessa W. C. (2009) MicroRNAs as novel regulators of angiogenesis. Circ. Res. 104, 442–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kwak P. B., Iwasaki S., Tomari Y. (2010) The microRNA pathway and cancer. Cancer Sci 101, 2309–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee R., Feinbaum R., Ambros V. (2004) A short history of a short RNA. Cell 116, 89–92 [DOI] [PubMed] [Google Scholar]
- 35. Hutvagner G., Simard M. J. (2008) Argonaute proteins: key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 9, 22–32 [DOI] [PubMed] [Google Scholar]
- 36. Ozen M., Creighton C. J., Ozdemir M., Ittmann M. (2008) Widespread deregulation of microRNA expression in human prostate cancer. Oncogene 27, 1788–1793 [DOI] [PubMed] [Google Scholar]
- 37. Lü J., Qian J., Chen F., Tang X., Li C., Cardoso W. V. (2005) Differential expression of components of the microRNA machinery during mouse organogenesis. Biochem. Biophys. Res. Commun. 334, 319–323 [DOI] [PubMed] [Google Scholar]
- 38. Kumar M. S., Lu J., Mercer K. L., Golub T. R., Jacks T. (2007) Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 39, 673–677 [DOI] [PubMed] [Google Scholar]
- 39. Chen C., Zhang Y., Zhang L., Weakley S. M., Yao Q. (2011) MicroRNA-196: critical roles and clinical applications in development and cancer. J. Cell Mol. Med. 15, 14–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luthra R., Singh R. R., Luthra M. G., Li Y. X., Hannah C., Romans A. M., Barkoh B. A., Chen S. S., Ensor J., Maru D. M., Broaddus R. R., Rashid A., Albarracin C. T. (2008) MicroRNA-196a targets annexin A1: a microRNA-mediated mechanism of annexin A1 downregulation in cancers. Oncogene 27, 6667–6678 [DOI] [PubMed] [Google Scholar]
- 41. Huot J., Houle F., Marceau F., Landry J. (1997) Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ. Res. 80, 383–392 [DOI] [PubMed] [Google Scholar]
- 42. Gibot L., Galbraith T., Huot J., Auger F. A. (2010) A preexisting microvascular network benefits in vivo revascularization of a microvascularized tissue-engineered skin substitute. Tissue Eng. Part A 16, 3199–3206 [DOI] [PubMed] [Google Scholar]
- 43. Pin A., Houle F., Guillonneau M., Paquet E. R., Simard M. J., Huot J. (2012) miR-20a represses endothelial cell migration by targeting MKK3 and inhibiting p38 MAP kinase activation in response to VEGF. Angiogenesis, 10.1007/s10456-012-9283-z [DOI] [PubMed] [Google Scholar]
- 44. Braig S., Mueller D. W., Rothhammer T., Bosserhoff A. K. (2010) MicroRNA miR-196a is a central regulator of HOX-B7 and BMP4 expression in malignant melanoma. Cell Mol Life Sci 67, 3535–3548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schimanski C. C., Frerichs K., Rahman F., Berger M., Lang H., Galle P. R., Moehler M., Gockel I. (2009) High miR-196a levels promote the oncogenic phenotype of colorectal cancer cells. World J. Gastroenterol 15, 2089–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yekta S., Shih I. H., Bartel D. P. (2004) MicroRNA-directed cleavage of HOXB8 mRNA. Science 304, 594–596 [DOI] [PubMed] [Google Scholar]
- 47. Liao S. H., Zhao X. Y., Han Y. H., Zhang J., Wang L. S., Xia L., Zhao K. W., Zheng Y., Guo M., Chen G. Q. (2009) Proteomics-based identification of two novel direct targets of hypoxia-inducible factor-1 and their potential roles in migration/invasion of cancer cells. Proteomics 9, 3901–3912 [DOI] [PubMed] [Google Scholar]
- 48. Bizzarro V., Fontanella B., Franceschelli S., Pirozzi M., Christian H., Parente L., Petrella A. (2010) Role of Annexin A1 in mouse myoblast cell differentiation. J. Cell Physiol. 224, 757–765 [DOI] [PubMed] [Google Scholar]
- 49. Madhyastha R., Madhyastha H., Nakajima Y., Omura S., Maruyama M. (2012) MicroRNA signature in diabetic wound healing: promotive role of miR-21 in fibroblast migration. Int Wound J 9, 355–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Altavilla D., Saitta A., Cucinotta D., Galeano M., Deodato B., Colonna M., Torre V., Russo G., Sardella A., Urna G., Campo G. M., Cavallari V., Squadrito G., Squadrito F. (2001) Inhibition of lipid peroxidation restores impaired vascular endothelial growth factor expression and stimulates wound healing and angiogenesis in the genetically diabetic mouse. Diabetes 50, 667–674 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}