

Background: Bacterial agarolytic systems are frequent and play an important role in algal biomass conversion.
Results: Structural and biochemical analyses of several agar-related enzymes reveal details on substrate recognition and complementary roles.
Conclusion: The diversity of agar-related enzymes within a bacterial organism reflects the complexity of the natural substrate.
Significance: Marine microbes employ complex systems to catalyze degradation of polysaccharides with unique structural characteristics.
Keywords: Bacterial Metabolism, Biodegradation, Carbohydrate Metabolism, Crystal Structure, Enzyme Catalysis, Agarolytic Enzyme System, Marine Biomass Degradation
Abstract
Zobellia galactanivorans is an emerging model bacterium for the bioconversion of algal biomass. Notably, this marine Bacteroidetes possesses a complex agarolytic system comprising four β-agarases and five β-porphyranases, all belonging to the glycoside hydrolase family 16. Although β-agarases are specific for the neutral agarobiose moieties, the recently discovered β-porphyranases degrade the sulfated polymers found in various quantities in natural agars. Here, we report the biochemical and structural comparison of five β-porphyranases and β-agarases from Z. galactanivorans. The respective degradation patterns of two β-porphyranases and three β-agarases are analyzed by their action on defined hybrid oligosaccharides. In light of the high resolution crystal structures, the biochemical results allowed a detailed mapping of substrate specificities along the active site groove of the enzymes. Although PorA displays a strict requirement for C6-sulfate in the −2- and +1-binding subsites, PorB tolerates the presence of 3–6-anhydro-l-galactose in subsite −2. Both enzymes do not accept methylation of the galactose unit in the −1 subsite. The β-agarase AgaD requires at least four consecutive agarose units (DP8) and is highly intolerant to modifications, whereas for AgaB oligosaccharides containing C6-sulfate groups at the −4, +1, and +3 positions are still degraded. Together with a transcriptional analysis of the expression of these enzymes, the structural and biochemical results allow proposition of a model scheme for the agarolytic system of Z. galactanivorans.
Introduction
Red seaweeds play a crucial role in the primary production of coastal ecosystems, and their biomass represents an important food and industrial resource. Like land plants, red macroalgae belong to the Archaeplastida phylum, but the cell walls of these eukaryotic lineages fundamentally differ (1). In red seaweeds, cellulose fibers interact with unusual hemicelluloses, such as glucomannans and sulfated mixed linked glucans (2). These semi-crystalline polysaccharides are embedded in a matrix of sulfated galactans (agars or carrageenans), which are the most abundant components of red algal cell walls (1). These polysaccharides consist of a linear backbone of galactose residues linked by alternating β-1,4 and α-1,3 glycosidic bonds. Although all the β-linked residues are in the d configuration (G monomer), the α-linked galactose units are in the l configuration in agars (L monomer) and in the d configuration in carrageenans (D monomer).
The regular structure of the agar backbones is often masked by various chemical modifications, such as ester sulfate groups, methyl groups, or pyruvic acid acetal groups (3, 4). The most common modification is the presence of a 3,6-anhydro bridge in the l-galactose monomer (LA monomer), and the main agar repeating moiety is the agarobiose (G-LA, the equivalent of DP2, see Fig. 1A) (5). Another frequent modification is the sulfation of the C6 of the l-galactose residues resulting in α-l-galactose 6-sulfate (L6S).6 This L6S monomer is twice more abundant than LA in the agar extracted from Porphyra species (6). This agar is commonly named porphyran, and its main repeating unit is the disaccharide G-L6S (Fig. 1A), which is here referred to as porphyranobiose. L6S units are found in various amounts in the agars of most agarophytes, including Gracillaria and Gelidium spp. (7). The structure of porphyran is further complicated by the frequent methylation of the C6 in the β-d-galactopyranose monomer (3, 8).
FIGURE 1.
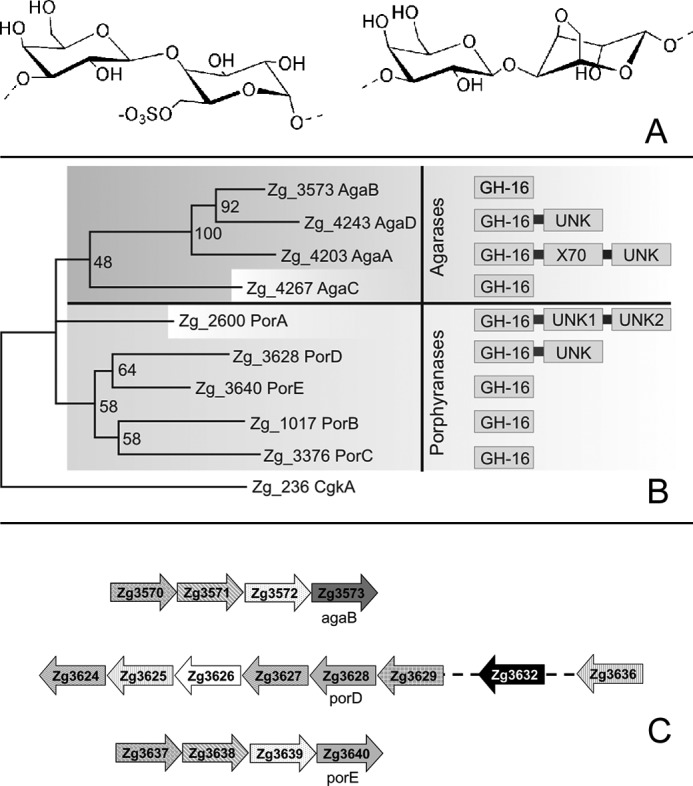
A, schematic representation of the chemical structure of porphyranobiose (left disaccharide) and agarobiose (right disaccharide). B, phylogenetic analysis of GH16 enzymes from Z. galactanivorans, active on agars, highlighting the separation in two clades. The modular structure of the enzymes is also represented. C, representation of the operon-like structure showing the genomic context of the genes agaB, porD, and porE. Top structure contains agaB. Zg3570, susC-like receptor; Zg3571, susD-like protein; Zg3572, lipoprotein with a CBM22-like domain; Zg3573, agaB. Middle structure contains porD. Zg3624 and Zg3627, GH74 glycoside hydrolases; Zg3625, putative carbohydrate esterase. CE4; Zg3626, hypothetical conserved protein; Zg3628, porD; Zg3629, sulfatase; Zg3632, GH28 glycoside hydrolase; Zg3636, one-component system sensor protein. Bottom structure contains porE. Zg3637, susC-like receptor; Zg3638, susD-like protein; Zg3639, lipoprotein with a CBM22-like domain; Zg3640, porE.
The amount of sulfated, anhydro, or otherwise modified sugar units in agars vary strongly between red algal species in their number and distribution within the molecular chain. This heterogeneous character of agars, their gelling properties, and their interactions with the other components of red algal cell walls challenge the microorganisms, which use these sulfated galactans as carbon sources. To break down these complex polysaccharides, marine bacteria produce specific glycoside hydrolases such as β-agarases (EC 3.2.1.81), which hydrolyze the β-1,4 glycosidic bond between two neoagarobiose units (LA-G), or α-agarases (EC 3.2.1.158), which cleave the α-1,3 linkage between two agarobiose units (9). These enzymes belong to different families of glycoside hydrolases as follows: GH96 family for α-agarases (10) and families GH16, GH50, GH86 (9), and GH118 (11) for β-agarases. However, little is known about the complementarities of the various enzymes needed for the complete assimilation of complex marine polysaccharides such as agars. Indeed, the only agarolytic system that has been studied extensively to date is the one of Saccharophagus degradans 2-40, a salt marsh Gammaproteobacterium, which possesses five β-agarases belonging to the families GH16, GH50, and GH86 (12). But this study focused solely on the neutral fractions of agars and not on the complex character of these polysaccharides in nature. Moreover, agarases catalyze only the initial step of agar degradation and are not sufficient alone to lead to complete substrate assimilation.
Alongside Gammaproteobacteria, members of the Bacteroidetes phylum are increasingly recognized as specialists for the degradation of polysaccharides and other polymers (13, 14). Within this phylum, Zobellia galactanivorans, which was isolated from the surface of the red alga Delesserria sanguinea, is remarkable for its extensive capacity to degrade algal biomass (15). Before the sequencing of its genome, Z. galactanivorans was already known to possess three β-agarases. AgaA and AgaB were expressed in Escherichia coli, and their capacity to degrade commercial agarose was characterized (16). The crystal structure of these recombinant enzymes was also determined (17, 18), which is the only structural data on β-agarases available to date. Wild type AgaC was also purified but not completely characterized (16). The recent genomic analysis of Z. galactanivorans7 has revealed one of the most expanded GH16 family among bacterial genomes with 16 genes.8 Interestingly, and in contrast to S. degradans (12), the genome of Z. galactanivorans does not encode any of the other known β-agarases, belonging to families GH50, GH86, or GH118. However, we have recently described the discovery of the first β-porphyranases (EC 3.2.1.178) among the Z. galactanivorans GH16 gene family. These enzymes, PorA and PorB, specifically cleave the β-1,4 linkage between two neoporphyranobiose units (L6S-G) in agar chains (7). We also used these enzymes to produce defined porphyran oligosaccharides (8). Thus, we hypothesize that the observed expansion of GH16 enzymes in Z. galactanivorans reflects an adaptation to the naturally occurring heterogeneity of agars. Recently, we have also described a new glycoside hydrolase family of marine origin (GH117 family), which includes an enzyme involved in the terminal steps of agar catabolism, the α-1,3-(3,6-anhydro)-l-galactosidase AhgA (19). Together with the previously described β-agarases and β-porphyranases, AhgA is thus part of a sophisticated degradation system present in Z. galactanivorans.
In this context, this study focuses on the expression and substrate specificity of a subgroup of β-porphyranases and β-agarases present in Z. galactanivorans by combining biochemical and three-dimensional structural and transcriptional data. In particular, we describe the crystal structure of the native PorA, of AgaB in complex with a neoagarooctaose, and of a third β-agarase, namely the catalytic domain of AgaD that displays a substantially longer binding cleft, resulting in a higher specificity for nonsubstituted agarobiose repetitions. By using pure porphyran oligosaccharides, as well as defined hybrid oligosaccharides containing both neoporphyranobiose and neoagarobiose moieties, we show that PorA is a strict β-porphyranase specifically cleaving between adjacent neoporphyranobiose moieties, whereas PorB can degrade hybrid molecules. Combined, these results support that the expansion of GH16 in Z. galactanivorans is an adaptation to the natural heterogeneity found in agars.
MATERIALS AND METHODS
Sequence Analysis and Phylogeny
The putative β-agarase and β-porphyranase genes of Z. galactanivorans were annotated in the Z. galactanivorans genome (sequenced by Genoscope)7 using the program GenDB (20). The conserved protein modules were queried against the Pfam database (21) and by BlastP searches against the GenBankTM nr database. Signal peptide and transmembrane helices were predicted using SignalP version 2.0 (22) and TMHMM (23), respectively. For the further phylogenetic analyses together with GH16 enzymes of other bacteria, the sequences were selected using the CAZY database (24). The selected proteins were subjected to multiple sequence alignments using MAFFT (25), with the iterative refinement method and the scoring matrix Blosum62. These alignments were manually edited using Bioedit (©Tom Hall), on the basis of the superposition of the structure of the κ-carrageenase of Pseudoalteromonas carrageenovora (26), the β-agarase AgaA (18), and the β-porphyranases PorA and PorB from Z. galactanivorans (7). The different phylogenetic trees were derived from these refined alignments using the Maximum Likelihood method with the program PhyML (27). The reliability of the trees was tested by bootstrap analysis using 100 resamplings of the dataset. The final tree was displayed with MEGA5 (28).
Cloning, Expression, and Purification of Recombinant Agarolytic Enzymes
Six of the nine genes encoding putative porphyranases and agarases (agaD and porA through porE) were selected for a medium throughput cloning strategy performed as in Groisillier et al. (29) (Fig. 1B). The gene agaC was excluded because the coding sequence features restriction sites noncompatible with the medium throughput cloning strategy. Primers were designed to amplify the coding region corresponding to the mature proteins, except for agaD, porA, and porD, for which only the catalytic modules were cloned and are hereafter called AgaDcat, PorAcat, and PorDcat. Briefly, the genes were amplified by PCR from Z. galactanivorans genomic DNA using the oligonucleotide primers reported in supplemental Table S1. For one of the targeted genes, namely porC, no positive clones were obtained, and no further steps were performed. Using the restriction sites indicated in supplemental Table S1, the PCR products were then ligated into the expression vector pFO4, resulting in a recombinant protein with an N-terminal hexa-histidine tag. The plasmids were transformed into E. coli BL21(DE3) expression strains. Recombinant strains were cultured at 20 °C during 3 days in 1 liter of ZYP medium (30) supplemented with 100 mg/ml ampicillin. No significant heterologous expression was detected for the cells containing the plasmids encoding for porD and porE. The cells of the three remaining targets were resuspended in 25 mm Tris-HCl, pH 7.5, 200 mm NaCl, and 5 mm imidazole buffer, supplemented with one dose of an anti-protease mixture (Complete EDTA-free, Roche Applied Science) and 0.1 mg/ml of DNase. The cells were lysed with lysis buffer composed of 50 mm Tris, pH 8, 300 mm NaCl, 1 mg/ml lysozyme, and DNase. The samples were centrifuged for 2 h at 20,000 × g and at 4 °C. The supernatant was loaded onto a HyperCell PAL column charged with NiSO4 (0.1 m) and pre-equilibrated with resuspension buffer, composed of 50 mm Tris, pH 8, 200 mm NaCl, 20 mm imidazole. The His-tagged proteins were eluted with a linear imidazole gradient (0.005–0.5 m imidazole) at a flow rate of 1 ml·min−1. Fractions that corresponded to the major eluted peak were analyzed by SDS-PAGE. Fractions that contained a protein band of right molecular weight were pooled and concentrated to a final volume of ∼5 ml for AgaDcat, PorAcat, and PorB, by ultrafiltration on an Amicon membrane (polyethersulfone, 10-kDa cutoff). For final protein purification, a Sephacryl S-200 column (GE Healthcare) was pre-equilibrated with buffer C at a flow rate of 1 ml/min. The purified enzymes were concentrated to ∼3 mg/ml for PorAcat, ∼6 mg/ml for PorB, and ∼8 mg/ml for AgaDcat by ultrafiltration on an Amicon membrane (10-kDa cutoff). All chromatographic steps were carried out on an ÄKTA Explorer Chromatography system (GE Healthcare) at 20 °C.
Mutagenesis of AgaB
The site-directed mutagenesis kit QuikChange (Stratagene) was used to introduce a mutation in the agaB gene. To maintain the charge balance within the active site, the conservative mutation, shortening the nucleophile by one carbon, Glu → Asp was chosen. Briefly, the pET20b/AgaB was entirely amplified by the Pfu Turbo DNA polymerase by using two complementary mutated primers as follows: 5′ GCC GAT GAC ACC CAA GAT ATA GAT ATT CTA GAG GCA 3′ and 3′ CGG CTA CTG TGG GTT CTA TAT CTA TAA GAT CTC CGT 5′ in which a Glu codon (GAG) was replaced by a Asp codon (GAT) at position 189. The parental plasmid was digested by DnpI, and the vector containing the mutation was transformed in XL1-Blue cells. Transformants were selected on LB ampicillin agar plates. Mutations were verified by automated DNA sequencing (Genome Express, Meylan, France), and the resulting construct was referred to as pET20b/ΑgaB-E189D.
Enzymatic Degradation of Polysaccharides
The natural agar samples were extracted and prepared as described in Correc et al. (8). Porphyran (1% (w/v) in deionized water) extracted from Porphyra umbilicalis, natural agar from Gracilaria sp., or agarose (Eurogentec) were incubated for 12 h at 30 °C with PorAcat, PorB, AgaB, and AgaDcat, at an enzyme concentration of 0.15 μg/ml each. During the hydrolysis, aliquots were taken to monitor enzyme activity by reducing sugar assay and size exclusion chromatography. At completion of the enzymatic reaction, which was tested by adding fresh enzyme solution again to verify that no further degradation was induced, the enzyme was inactivated by boiling during 1 h. In the case of further analyses, the solution was centrifuged to remove a small amount of white precipitate before freeze-drying of the hydrolysis products.
Purification of the Oligosaccharides by Preparative Size Exclusion Chromatography
The freeze-dried hydrolysis product was dissolved in deionized water at a concentration of 4% (w/v). After filtration through a 0.45-μm syringe filter (Millipore), 4 ml of sample were injected. The purification of the porphyran oligosaccharides was carried out by preparative size exclusion chromatography with three columns of Superdex 30 (26/60) (GE Healthcare) in series and integrated on an HPLC system liquid injector/collector (Gilson). Detection was achieved by a refractive index detector (Spectra System RI-50). The Gilson system and the detector data were monitored by Unipoint software (Gilson). Fifty mm ammonium carbonate ((NH4)2CO3) was used as elution buffer, at a flow rate of 1.5 ml/min during 650 min. Fractions of oligosaccharides were collected and subjected to further analysis by anionic exchange chromatography. Then the fractions were freeze-dried before being analyzed by electrophoresis techniques and nuclear magnetic resonance spectroscopy (NMR; supplemental Fig. S5) (8).
Enzymatic Assay
The amount of reducing sugars produced during the enzymatic digestion of extracted and purified porphyran and agarose (Eurogentec) was determined using a method adapted from Kidby and Davidson (31). Aliquots (100 μl) of the reaction medium diluted 5× were mixed with 1 ml of ferricyanide solution (300 mg of potassium hexocyanoferrate III, 29 g of Na2CO3, 1 ml of 5 m NaOH, completed to 1 liter with water). The mixture was boiled for 15 min and cooled to room temperature, and its absorbance was measured at 420 nm.
Fluorophore-assisted Carbohydrate PAGE
Fractions obtained after gel chromatographic separation of oligosaccharides were analyzed by fluorophore-assisted carbohydrate-PAGE. Aliquots of 100 μl (∼25 μg of material) were dried in a speed-vacuum centrifuge, and the oligosaccharides were labeled with 8-aminonaphthalene-1,3,6-trisulfonate (ANTS) and 2-aminoacridone (AMAC) adapted from Starr and Masada (32). For derivatization with ANTS, the pellet was redissolved in 2 μl of ANTS solution (0.15 m ANTS in acetic acid/water (3:17, v/v). Once the pellet was redissolved, 5 μl of 1 m sodium cyanoborohydride in dimethyl sulfoxide (DMSO) was added, and the mixture was incubated for 16 h at 37 °C. For the derivatization with AMAC, the pellet was redissolved in 2 μl of AMAC solution (0.1 m AMAC in acetic acid/DMSO (3:17, v/v)) and 5 μl of 1 m sodium cyanoborohydride in water also followed by 16 h of incubation at 37 °C. The samples were analyzed on a 30% polyacrylamide running gel with a 4% stacking gel and a Bio-Rad gel system. Gels were run for half an hour at 15 mA followed by 40 mA for 3 h at 4 °C.
Determination of Kinetic Constants of AgaDcat
Michaelis constant (Km) and turnover number (kcat) were experimentally determined with dissolved agarose at a temperature of 44 °C. Substrate concentrations were varied from 0.0125 to 0.75% agarose (corresponding to 0.4–22 nm of cleavable glycosidic bonds calculated with the molecular weight of neoagarobiose as final reaction product) in 300 nm NaCl and 50 mm MOPS buffer as reported in Jam et al. (16). All values were determined in triplicate, and the amount of released sugars was estimated by the ferricyanide reducing sugar assay (31). For each concentration, five time points were analyzed during a total reaction time of 10 min. All experiments were performed in quadruplicate. The calibration and estimation of cleavage events were made with glucose as standard. The Michaelis constant and the turnover number were determined by a nonlinear regression analysis.
Bacterial Strain, Culture Conditions, and RT-PCR
The type strain DsijT of Z. galactanivorans was routinely grown at 20 °C and 150 rpm in Zobell medium 2216E (5 g/liter tryptone, 1 g/liter yeast extract, sea water (33)) until mid-exponential phase (A600 nm around 0.3). For RNA extraction, cells were transferred in the Marine Mineral Medium supplemented with 2 g·liter−1 agar from Gelidium spinosum, porphyran from P. umbilicalis or laminarin as sole carbon sources. Briefly, 1 liter of Marine Mineral Medium was composed of 24.7 g of NaCl, 6.3 g of MgSO4·7H2O, 4.6 g of MgCl2·H2O, 2 g of NH4Cl, 0.7 g of KCl, 0.6 g of CaCl2, 200 mg of NaHCO3, 100 mg of K2HPO4, 50 mg of yeast extract, 20 mg of FeSO4·7H2O, in 50 mm Tris-HCl, pH 8.0 (34). Total RNA isolation and cDNA synthesis were performed as described previously (34). Briefly, RNA was isolated from 2 ml of Marine Mineral Medium in mid-log phase (A600 nm 0.3–0.5) using RNA mini kit (Qiagen) and treated with Turbo DNase (Ambion). Total elimination of genomic DNA was checked by PCR on RNA samples. Eight hundred ng of RNA were retro-transcribed using the Phusion RT-PCR kit (Finnzymes) with random hexamer primers. cDNA samples were stored at −20 °C and used in PCRs within a month. PCRs were performed using the Phusion RT-PCR Kit (Finnzymes) with 0.5 μm of each primer (supplemental Table S1) using the following cycling parameters: initial denaturation at 98 °C for 30 s, cycles of 10 s at 98 °C, 10 s at 55 °C, and 70 s at 72 °C, followed by a final step at 72 °C for 5 min. The number of cycles used for each gene is reported in Fig. 6. Samples were analyzed by electrophoresis on 0.8% agarose gel in TAE buffer and visualized by ethidium bromide.
FIGURE 6.

Differential gene expression depending on the carbon source. A, gel electrophoresis of the RT-PCR amplification products. Total RNA from cells grown with laminarin, agar, or porphyran as sole carbon source was used as a template for reverse transcription. PCRs were performed on the resulting cDNA using oligonucleotides primers for the genes coding the four agarases and five porphyranases. Experiments were performed on biological triplicates. B, Venn diagram of the genes expressed in the three conditions.
Crystal Structure Determination of AgaDcat, AgaB_E189D/Neoagarooctaose, and PorA at High Resolution
Crystal Structure of AgaDcat
AgaD is a multimodular enzyme composed of an N-terminal GH16 catalytic domain and a C-terminal domain of ∼10 kDa with unknown function. For biochemical and structural determination, only the catalytic domain, AgaDcat, was cloned and further analyzed in this study. This domain, consisting of 357 residues with a molecular mass of 40.2 kDa and a theoretical isoelectric point of 8.35, was expressed in E. coli, as described above and purified and crystallized as reported previously (35). Single crystals were soaked in crystallization solutions supplemented with 20% glycerol prior to flash-freezing in a nitrogen stream at 100 K.
Diffraction data for AgaDcat at 1.5 Å resolution were collected on beamline ID14-EH1 (ESRF, Grenoble, France). The crystals (P212121) contained one molecule in the asymmetric unit, giving a Matthews coefficient VM of 2,14 Å3/Da and a solvent content of 43%. The three-dimensional protein structure was solved by molecular replacement with AMORE (36) using AgaB as a search model within the resolution range from 10 to 4.0 Å. The unambiguous result gave one contrasted solution with a correlation factor of 25.4 and an initial R-factor of 50.9%.
The model construction and adjustments of residues were performed manually using COOT (37). The refinement was performed with REFMAC 5 (38) as part of the CCP4 suite (39). Water molecules were added using CCP4/wARP (40).
AgaB-E189D in Complex with Neoagarose-Octaoligosaccharides
The mutant protein AgaB-E189D was purified by the same procedure as described for the native protein (18), and activity was monitored by the reducing sugar assay (data not shown). No significant residual activity was detected, and the mutant enzyme was therefore crystallized in the presence of neoagarooctaose. The production and purification of pure oligosaccharides of defined length have been described previously (17).
AgaB-E89D was concentrated to 5 mg/ml and stored in 50 mm Tris buffer, pH 7.5, with 25 mm NaCl. Crystallization conditions were established using two sparse-matrix sampling kits (Molecular Dimensions and Stura Footprint). Further optimization allowed growing monoclinic crystals of the enzyme-substrate complex. The best conditions for co-crystallization of AgaB-E189D with neoagarooctaose contained 32% PEG 8000, 400 mm ammonium acetate, and 100 mm sodium cacodylate, pH 6.5. Crystals were grown by mixing 2 volumes of protein solution, equally containing about 4 mm neoagarooctaose, with 1 volume of precipitant solution in a hanging-drop vapor diffusion setup at 20.0 °C. Crystals grew within 1 week and belong to the space group P21, having unit cell parameters a = 74.10 Å, b = 105.90 Å, c = 96.93 Å, and β = 93.44°. Thereafter, a single crystal was soaked for 30 s in successive solutions with increasing glycerol concentrations up to a maximum of 15%.
Diffraction data for AgaB-E189D/neoagarooctaose at 1.9 Å resolution were collected on beamline ID14-EH2 (ESRF, Grenoble, France). The complex co-crystals (P21) contained four molecules in the asymmetric unit, giving a Matthews coefficient VM of 2.66 Å3/Da and a solvent content of 54%. The structure was solved by molecular replacement, using AMoRe (36) and native AgaB as a search model. The correlation and R-factor of the molecular replacement are 63.9 and 17.6% for the four solutions present in the asymmetric unit of AgaB-E189D/agaro-octaose crystals.
The corrections and adjustments of the residues and the addition of the agarose sugar units for the complex structure were performed manually using COOT (37). The refinement was performed with REFMAC 5 (38) as part of the CCP4 suite (39). Water molecules were added using CCP4/wARP and manually inspected (40).
PorA High Resolution
PorAcat was crystallized by the hanging drop method as described previously (7). Drops of 2-μl volume of protein solution (2.6 mg/ml) were mixed with 1 μl of crystallization solution and equilibrated against a reservoir containing 500 μl. The native crystals were flash-frozen at 100 K in a crystallization solution supplemented with 10% glycerol. Native data were collected to 1.1 Å resolution on beamline ID23–1 equipped with an ADSC Quantum Q315r detector (ESRF, Grenoble, France) and a wavelength of 0.827 Å.
The three-dimensional structure was solved by the MAD method (7), and starting phases were used for the initial model that was built with ARP/wARP (41) and REFMAC 5 (38) as part of the CCP4 suite (39). The refinement was carried out with REFMAC 5 and model building with COOT (37). Water molecules were added automatically with REFMAC-ARP/wARP and visually verified.
In all cases, the stereochemistry of the final structure was evaluated using PROCHECK (42). All further data collection and refinement statistics are summarized in Table 1.
TABLE 1.
Data collection and refinement statistics of AgaDcat, AgaB-E189D/agarooctaose, and PorA at 1.1 Å resolution
Data collection and refinement statistics are for the crystal structure of AgaB-E189D in complex with agarooctaose, AgaDcat, and PorA at very high resolution. r.m.s.d., root mean square deviation.
| AgaDcat | AgaB-E189D with agarooctaose | PorA high resolution | |
|---|---|---|---|
| Data collection | |||
| ESRF beamline | ID14-EH1 | ID14-EH2 | ID23–1 |
| Wavelength | 0.934 | 0.933 | 0.827 |
| Space group | P212121 | P21 | P3121 |
| Unit cell parameters | a = 53.25, b = 77.27, c = 83.7 | a = 74.10, b = 105.90, c = 96.93, β = 93.44° | a = b = 70.27, c = 92.47 |
| Resolution range | 56.7 to 1.5 (1.58–1.50) | 39.8 to 1.8 (1.868–1.80) | 25.86 to 1.10 (1.129–1.10) |
| No. of observations (F >0) | 385,296 (55,310) | 357,984 | 1,022,069 (48,384) |
| No. of unique reflections | 55,977 (8035) | 133,189 (11,867) | 181,505 (8191) |
| Completeness | 99.9% (99.9%) | 96.4% (87.8%) | 87.5% (53.2%) |
| Mean I/σ(I) | 21.3 (9.1) | 9.8 (2.2) | 9.3 (5.4) |
| Rmerge | 5.3% (24.9%) | 8.6% (37.8%) | 9.3% (22.8%) |
| Redundancy | 6.9 (6.9) | 2.7 (2.5) | 5.6 (5.9) |
| Refinement | |||
| Resolution range | 43.9 to 1.8 | 39.8 to 1.9 | 25.8 to 1.1 |
| No. of unique reflections | 32,693 | 107,508 | 89,663 |
| R-factor (Rfree on 5%) | 14.01 (18.51) | 15.8 (19.8) | 12.3 (15.2) |
| No of protein atoms (mean B-factor in Å2) | 2874 (9.36) | 9840 (16.99) | 2185 (9.59) |
| No. of solvent atoms (mean B-factor in Å2) | 600 (11.35) | 324 (24.98) | 450 (23.33) |
| No. of ions/ligands (mean B-factor in Å2) | 1 (8.98) | 945 (27.14) | 7 (8.15) |
| r.m.s.d. bond lengths | 0.022 Å | 0.026 Å | 0.027 Å |
| r.m.s.d. bond angles | 1.92° | 2.12° | 2.28° |
| Mean overall Bfact | 13.14 Å2 | 18.05 Å2 | 11.89 Å2 |
| Ramachandran plot, most favored | 98.0% | 98.5% | 97.2% |
| Ramachandran plot, additional allowed | 2.0% | 1.5% | 2.8% |
The coordinates and structure factors of the three structures reported here have been deposited in the Protein Data Bank. The accession codes are 4asm for AgaDcat, 4atf for AgaB-E189D, and 4ate for PorA at high resolution.
RESULTS
Z. galactanivorans Genome Encodes Nine GH16 Porphyranases and Agarases
Alongside the characterized genes agaA, agaB, agaC (16), porA, and porB (7), four additional genes were predicted in the genome of Z. galactanivorans7 to encode putative β-agarases or β-porphyranases: agaD, Zg_4243; porC, Zg_3376; porD, Zg_3628; and porE, Zg_3640. In a partial phylogenetic tree of the GH16 family, the corresponding proteins consistently cluster into two distinct clades corresponding to the β-agarases (clade 1) and the β-porphyranases (clade 2) (Fig. 1B). AgaA and AgaD are modular proteins, containing N-terminal signal peptides targeting the periplasmic space and a C-terminal domain only conserved in Bacteroidetes (43) and likely acting as a sorting signal for a secretion system unique to this phylum (44, 45). AgaB and AgaC both contain a lipoprotein-type signal peptide that targets the outer membrane. The wild type AgaA and AgaC were previously purified from the extracellular medium of Z. galactanivorans cultivated in Zobell medium supplemented with agar (16). These purified proteins corresponded only to the GH16 family modules, suggesting that AgaA and AgaC were further processed by proteolysis to be released in the culture medium. In contrast, AgaB and AgaD were not found secreted in the medium in this culture condition (16). All the β-porphyranases are predicted to be targeted to the periplasm, with the exception of PorC, which displays a lipoprotein signal peptide and is likely localized in the outer membrane. Like AgaA and AgaD, PorA comprises a potential Bacteroidetes-specific secretion domain in its C terminus.
The genes agaA, agaC, agaD, porA, porB, and porC are isolated in the genome without any neighboring gene obviously related to agar utilization. In contrast, agaB and porE are part of operon-like structures, which both include genes encoding a TonB-dependent receptor (TBDR, SusC-like protein), its associated SusD-like lipoprotein, and a lipoprotein containing a putative carbohydrate-binding module distantly related to family CBM22 (Fig. 1C). The conserved synteny of these two clusters suggests that they likely evolved by gene duplication. This organization is reminiscent of the alginolytic operon that we have recently characterized in Z. galactanivorans (46) and of the Starch Utilization System (Sus) of Bacteroides thetaiotaomicron. In this latter bacterium, SusC and SusD form a complex responsible for the uptake of starch degradation products (47, 48). Therefore, the homologs of these genes in the agaB and porE gene clusters may be similarly involved in the import of agaro- and porphyrano-oligosaccharides, respectively. porD is localized in a large cluster of 24 genes comprising numerous putative carbohydrate-related genes as follows: two GH2 β-galactosidases, two GH28 polygalacturonases, three GH29 fucosidases, one GH117, one CE4 carbohydrate esterase, two genes distantly related to GH74 enzymes, and three sulfatases. Considering the length of this cluster and the functional diversity of its predicted genes, it is unclear whether this group constitutes one operon or several unrelated transcriptional units.
Determination of Kinetic Parameters of AgaDcat
Among the four new putative agarases and porphyranases (AgaD, PorC, PorD, and PorE) that we attempted to express in E. coli, only the catalytic module of AgaD was expressed in soluble form. This recombinant protein, hereafter called AgaDcat, consists of 357 residues (40.2 kDa) and has an isoelectric point of 8.35. AgaDcat shares 32% identity with the catalytic module of AgaA (AgaAcat) and 39% identity with AgaB (supplemental Table S2). Sequence analysis reveals three major insertions in comparison with the two latter enzymes (35). When the three β-agarases were used to degrade agarose solutions (44 °C), the initial velocity of degradation was almost identical between AgaDcat, AgaAcat, and AgaB (supplemental Fig. S1c). Nevertheless, a plateau was reached more rapidly with AgaDcat, and therefore only ∼50% of oligosaccharides in respect to AgaAcat and AgaB were released at the final stage of hydrolysis under these conditions. Further addition of AgaDcat to the agarose sample resulted in additional amounts of released oligosaccharides, suggesting that the previous end of reaction was due to a low stability of the enzyme. Therefore, the melting point of AgaDcat was experimentally determined by dynamic light scattering to be 32 °C. Because experiments with agarose solutions are carried out above 40 °C to hinder agarose from gelation, this value well explains the lower efficiency of AgaDcat on agarose solutions. However, agars are not in liquid phase in nature and form gel in the cell wall of red seaweeds. In gel phase at 30 °C, AgaDcat degraded 70% of the agarose gel compared with AgaAcat. Addition of a new sample of AgaDcat after 12 h of digestion did not change the amount of released oligosaccharide this time. The lower thermostability of AgaDcat is thus not the only factor influencing the difference in amount of released sugars produced on neutral agarose (supplemental Fig. S1a). These findings were subsequently taken into account for the experimental determination of the kinetic constants of AgaDcat, for which the initial velocity of degradation on agarose in solution was recorded. The kinetic parameters were as follows: Km = 6.5 mm, kcat = 248 s−1, and kcat/Km = 48 mm−1s−1. Because these values were obtained as described previously for AgaAcat and AgaB (16), they can directly be compared as follows: AgaAcat, Km = 2 mm, kcat = 150 s−1, and kcat/Km = 75 mm−1 s−1; AgaB, Km = 1 mm, kcat = 100 s−1, and kcat/Km = 105 mm−1 s−1. Even though the values for AgaDcat were obtained from the initial rate of degradation, they may be underestimated due to the lower thermostability of AgaDcat. Nevertheless, they range in the same order of magnitude as measured for the AgaAcat and AgaB.
AgaDcat Is an Endo β-Agarase Cleaving β-1,4 Linkages in Agarose
AgaDcat has an endo-like degradation behavior (where endo means the enzyme attacks anywhere along the polysaccharide chain) (16) as shown by analysis of the oligosaccharide reaction products. AgaDcat produces randomly sized oligosaccharides at the onset of the reaction (supplemental Fig. S1a, lanes 15, 30, 60 min), which are subsequently degraded to obtain neoagarohexose (DP6) and neoagarotetraose (DP4) as final reaction products (supplemental Fig. S1a, lane 1440 min). Prolonged incubation of DP6 or DP4 agaro-oligosaccharides with high concentrations of AgaDcat did not yield smaller units (supplemental Fig. S1b), indicating that these oligosaccharides are the final reaction products of AgaDcat. This is in contrast to AgaB, which was shown to release DP2 and DP4 by degradation of DP6 (supplemental Fig. S1b) (18), and to a minor extent AgaAcat, which only releases very small amounts of DP2 from DP6 (18).
Comparison of the Agarolytic Activity of the Three β-Agarases on Naturally Extracted Agars from Different Agarophytes
Despite the large structural differences between AgaDcat, AgaAcat, and AgaB, the differences in activity on neutral, industrially available agarose, were rather subtle. To further investigate polysaccharide degradation by these enzymes, we analyzed their activity on natural substrates, i.e. on simple hot water extractions from marine red algae, without any further chemical treatment of the extracted fractions. The activity of the three enzymes was compared for agars extracted from two red algal species: (i) the economically exploited agarophyte G. spinosum, because these species contain agars forming strong gels due to the low amounts of substitutions, and for comparison (ii) P. umbilicalis, which in contrast contains the highly sulfated agars, commonly referred to as porphyran.
Expectedly, all three enzymes had highest relative activity on extracts from G. spinosum (Fig. 2A), which is explained by the presence of high amounts of unmodified neoagarobiose moieties (LA-G) within the polysaccharide chain. Consistently, the amount of oligosaccharides produced by the three β-agarases on the extracted agar from P. umbilicalis is significantly lower. Interestingly, for AgaDcat the released quantity of oligosaccharides is diminished by only about one-third, whereas for AgaAcat and AgaB the difference is more than one-half of the quantity measured on the Gelidium agar. This is reminiscent of the activity on agarose gel on which AgaDcat typically releases ∼70% of reducing-end equivalents, as compared with AgaAcat. On porphyran this difference is even accentuated, because AgaDcat releases only 50% of oligosaccharides with respect to AgaAcat and AgaB (Fig. 2C). This result suggests that AgaAcat and AgaB seem to tolerate modifications of the ideal neoagarobiose moieties to some extent, such as the presence of sulfations as in porphyran. In contrast, AgaDcat may be less promiscuous and have rigid specificity for nonsubstituted stretches in the polysaccharide chain. To test this hypothesis, purified hybrid reaction products that were released by porphyran hydrolysis were further analyzed by fluorophore-assisted oligosaccharide gel electrophoresis.
FIGURE 2.

A, activity of AgaAcat, AgaB, and AgaDcat on the agars from G. spinosum and P. umbilicalis. The polysaccharides were obtained by hot water extraction. The activity was monitored using the reducing sugar assay and normalized to glucose equivalents. B, β-agarases AgaA and AgaB produce hybrid hexasaccharides with different neoporphyranobiose/neoagarobiose structures. AMAC fluorophore-assisted carbohydrate-polyacrylamide gel shows the degradation products of AgaA (lane 1) and AgaB (lane 2) as compared with those of PorA (lanes 4 and 5) on hot water-extracted and AgaB pre-digested porphyran. AgaD (lane 3) only produces a limited number of high molecular weight hybrid oligosaccharides. C, high performance anion exchange chromatography of the reaction products produced by AgaB, AgaD, and PorA corresponding to experiments of the AMAC-labeled oligosaccharides in B.
Subsite Specificity Mapping of AgaB, PorA, and PorB through the Degradation of Characterized (Hybrid) Oligosaccharides
Subtle differences in substrate specificity can be identified for β-agarases and β-porphyranases acting on the heterogeneous natural agars. Here, the reaction products of the three β-agarases were compared with those produced by PorA (Fig. 2, B and C). Both AgaAcat and AgaB produce a number of discrete bands, of which the lowest molecular weight product, present in a double band, had the same velocity as hybrid hexasaccharides produced by PorA. The degradation pattern of AgaDcat clearly differs from other β-agarases. In particular, no bands of low molecular weight oligosaccharides are visible after degradation with AgaDcat, even when adding extensive amounts of enzyme. In contrast, AgaDcat produces a smear of bands, which migrate with lower velocity.
We further investigated the degradation of defined hybrid hexasaccharides (supplemental Fig. S2) by PorAcat and PorB. Both enzymes degraded the neoporphyranotetraose only in the unmethylated form, showing that the shortest substrate accepted is the tetrasaccharide but also that both enzymes do not tolerate a methylation of the galactose in the −1-binding subsite (supplemental Fig. S3). Interestingly, PorAcat did not degrade any of the hybrid hexasaccharides, indicating that this enzyme strictly requires the presence of two consecutive neoporphyranobiose units (L6S-G-L6S-G) for hydrolysis. In contrast, PorB was able to degrade the hybrid hexasaccharide L6S-G-LA-G-L6S-G (supplemental Fig. S2c), as well as its methylated variant (L6S-G(Met)-LA-G-L6S-G; see supplemental Fig. S3b). Because PorB does not accept methylation of the tetrasaccharide at the −1 subsite, we can conclude that the cleavage point of the methylated hexasaccharide is such that the LA unit is bound to the −2-binding subsite.
Crystal Structure of AgaDcat
The crystal structure of AgaDcat confirms that this protein adopts the jelly roll fold typical of GH16 enzymes (Fig. 3 and supplemental Fig. S4). The respective root mean square deviation values of AgaDcat are 1.26 Å with AgaAcat and 1.2 Å with AgaB (Table S2) (18). The core of the jelly roll fold is composed of two sandwich-like stacked β-sheets, each composed of 5–7 β-strands, which are twisted to form the substrate binding cleft that is further accentuated by several extending surface loops. Most variations determining the substrate specificities are located within these regions. AgaDcat contains three rather large insertions that do not exist in AgaAcat and in AgaB (Fig. 3A) (Protein Data Bank codes 1o4y and 1o4z, respectively). These insertions are arranged around the substrate binding cleft and include residues Asn57–Asn63 (loop 1), Ala96–Pro110 (loop 2), and Asp244–Val296 (loop 3; Fig. 3B). Loop 1 extends the groove on the negative binding sites, after the naming convention where −n represents binding sites on the nonreducing end, and +n represents the binding sites on the reducing end of the substrate (49). Loop 2 extends above the substrate binding cleft and leads to a more tunnel-like topology when compared with the structures of AgaAcat and AgaB. The loop insertion 3 is best described as a new subdomain composed of three β-strands and one α-helix. These three β-strands extend the convex outer β-sheet at the positive binding subsites of the enzyme. All together, these three loop insertions significantly extend the substrate binding cleft of AgaDcat to roughly 42 Å, as compared with that of AgaAcat (∼34 Å) or AgaB (∼33 Å). Moreover, the additional loop regions result in a deeper and steeper active site cleft for AgaDcat, as compared with the open cleft of AgaAcat and AgaB.
FIGURE 3.

A, ribbon representation of AgaDcat highlighting the localization of the three major loop insertions surrounding the active site cleft of the enzyme. B, extractions of multiple sequence alignments of AgaA, AgaB, putative agarase from MS116, and AgaD showing the large insertions present in AgaD and the agarase MS116 forming a large domain extension at the reducing end of the catalytic binding cleft.
Crystal Structure of the Inactivated Mutant AgaB-E189D in Complex with Neoagarooctaose
The crystals of AgaB-E189D in the presence of neoagarooctaose (AgaB-E189Dco) displayed the same molecular packing of four molecules in the asymmetric unit and space group (P21) as the native protein (18), but interestingly, in the presence of the oligosaccharide they diffracted to a higher resolution (1.9 Å versus 2.3 Å in 1o4z). Overall, the molecular structures are identical, with an average root mean square deviation in the range of 0.21 to 0.30, when superimposing one of the four molecules of AgaB in the native structure with one of AgaB-E189Dco. Between the apo and the complex structure of AgaB, no major shift or structural rearrangement of loops or specific residues were observed, except for one loop, including residues from Ile218 to Gln223, that in average shifts by roughly 1.4 Å. This loop contains Arg219 and Phe222 that shift by about 1.1 Å toward the 3,6-anhydro-group of the agarose unit to bind this sugar into subsite −2.
The electron density in the active site clefts of all four molecules clearly indicates the presence of neoagarose units on the nonreducing end (subsites −4 to −1) that were easily modeled into the structure and refined. Some discontinuous density was also seen in the positive binding sites that appeared to be more than ordered water molecules but was not easily identified as neoagarobiose units. Knowing that purified neoagarooctaose was used for co-crystallization, supplementary neoagarobiose units were expected to be present in the crystal structure. The ncs-averaged map of all four molecules in the asymmetric unit of the crystal packing indeed displayed residual density that seemed to trace the presence of the missing neoagarobiose units (supplemental Fig. S5). Moreover, when building the continuous neoagarooctaose chain, following the ncs-averaged density in the active site cleft, typical sugar-binding residues became obvious and displayed reasonable binding distances to the modeled sugar chain. However, these neoagarobiose units on the positive binding sites were not stable upon crystallographic refinement. Our assumption is that the binding affinity for the subsites +1 to +4 is not strong enough to fix these sugar units into a tight binding and that these units are rather disordered. In addition, different chain lengths of the trapped oligosaccharides, due to the presence of traces of shorter ones in the purified octasaccharide, might also produce lower occupancies in these binding sites, making the electron density blurry. Nevertheless, the residual electron density allowed to model one possible sugar chain orientation that helps to identify important residues that define the subsites +1 to +4. Although the 3,6-anhydro-l-galactose in the +1 site is flanked by two glutamine residues, Gln310 and Gln226, the binding subsites +2 to +4 are formed by a triad of aromatic hydrophobic “platforms,” Trp312, Trp233, and Tyr205, that are optimally positioned and oriented to create hydrophobic and carbon-π interactions (50) with the three successive sugar units to be bound (Fig. 4).
FIGURE 4.

Close-up view of AgaB-E189D in complex with the modeled continuous neoagarooctaose, the hydrophobic residues (Trp344, Trp221, and Tyr192 in AgaB) present in the +2-, +3-, and +4-binding sites, respectively, are highlighted.
High Resolution (1.1 Å) Structure of Native PorA Loop Obstruction of Binding Subsite +1
The crystals of native PorAcat belong to the space group P3121 and diffract to 1.1 Å resolution. The structure was solved by the MAD method as described in Ref. 7. A native data set collected at maximum resolution was refined to a final R- and Rfree-factors of 12.5 and 15.2%, respectively, applying anisotropic B-factors to all the atomic coordinates present in the structure. The root mean square deviation of the coordinates is calculated to be 0.86 for 199 matching residues, when superimposing PorAcat onto the coordinates of the closest β-agarase (AgaAcat) with known structure. The largest differences between the overall structures of these both enzymes concern eight loops connecting the different β-strands of the jelly roll fold, most of which are surrounding the active site groove. These loops contain the crucial residues that determine the differences in the substrate recognition of β-agarases versus β-porphyranases on the nonreducing end of the active site groove (the negative binding subsites), which were described in detail in Ref. 7. A major difference concerns the central, and active site groove-forming, concave β-sheet. This β-sheet is composed of six β-strands in PorAcat, whereas it contains only five β-strands in all three-dimensional structures of β-agarases so far; the sixth terminal β-strand at the reducing end of the active site cleft (residues 178–182 in PorAcat) is replaced by a short α-helix containing stretch in agarases (Fig. 5A).
FIGURE 5.
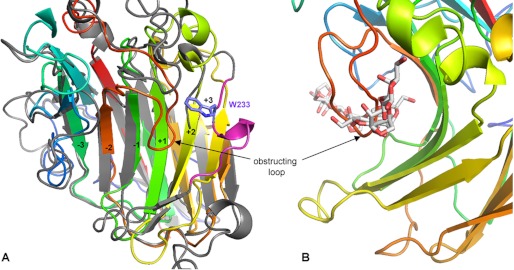
A, ribbon representation of PorA (colored strands) superimposed on AgaB (gray strands and pink helix carrying Trp233) highlighting the differences of the concave β-sheet on the + binding subsites. B, close-up view of native PorA showing the loop insertion partially obstructing the catalytic active site groove at the +1-binding site.
The most intriguing feature in native PorAcat is the presence of a short loop insertion (residues 238–245), absent in AgaAcat, AgaB, or AgaDcat that protrudes into the active site cleft and binds at place of the +1-binding site (Fig. 5B). Interestingly, this loop is highly disordered in the complex mutant structure, containing a neoporphyranotetraose bound to the binding subsites −4 to −1. The order-disorder transition of this loop is also demonstrated when comparing the thermal B-factors of this region between the bound and unbound β-porphyranase structures; the mean B-factors of the residues from Phe238 to Asn245 range between 7 and 8 Å2 in the native PorAcat (overall B-factor of the protein is 11.90 Å2), although they range from 15 to 27 Å2 in complexed PorA (overall B-factor of the protein is 13.34 Å2).
Transcriptomic Analysis of the β-Agarase and β-Porphyranase Genes
We analyzed the transcription of the nine β-agarase and β-porphyranase genes with either agar, porphyran, or laminarin as the sole carbon source (Fig. 6). As expected, the four β-agarases AgaA, AgaB, AgaC, and AgaD were expressed when cells were grown with the low sulfated agar from G. spinosum. These genes were not transcribed when laminarin was used as the carbon source. Interestingly, cells grown with the porphyran from P. umbilicalis expressed the β-agarases genes agaA, agaB, and agaC but not agaD. Indeed, the expression of β-agarases when growing on porphyran can be explained by the hybrid character of this highly sulfated agar, which always contains both motifs, in general up to 60% neoporphyranobiose (L6S-G) units and 40% neoagarobiose (LA-G) units. The pattern of expression for the five β-porphyranases differed from one enzyme to the other. The gene PorA was expressed under the three conditions tested, which might indicate a constitutive expression. The gene porC was not expressed in the agar and laminarin conditions, whereas transcription was detected when cells used porphyran. A similar profile was found for porD, although only a weak expression was found with porphyran. The expression of porE was drastically decreased in cells using agar, compared with the laminarin and porphyran conditions. Finally, porB was expressed only in cells growing in the laminarin-supplemented medium.
DISCUSSION
Global Structural Comparison of Agarases and Porphyranases
The crystal structure of the inactivated mutant AgaB-E189D allowed modeling of a neoagarooctaose spanning the entire active site groove of the enzyme. With the help of this model, the pattern of the specific residues, defining the eight binding subsites, was identified in detail, which can also be deduced for the other β-agarases and the two β-porphyranases, by superimposition of the structures of the different GH16 enzymes and enzyme complexes (Table 2). These comparisons provide a straightforward explanation for the substrate degradation pattern observed in the biochemical product analyses.
TABLE 2.
Residues involved in the different binding subsites of the catalytic active site groove of β-agarases and β-porphyranases
| Enzymes | −4 | −3 | −2 | −1 | +1 | +2 | +3 | +4 |
|---|---|---|---|---|---|---|---|---|
| AgaA | Asn71 | Tyr69 | Trp138 | Glu152 | His168 | Trp190 | Trp162 | |
| Trp73 | Arg176 | His172 | Tyr154 | Gln183 | ||||
| Glu144 | Phe179 | Glu254 | Gln256 | Trp259 | ||||
| AgaB | Asn107 | Tyr105 | Asp173 | Asp189 | His211 | Trp233 | Tyr205 | |
| Trp109 | Arg219 | Trp175 | Tyr191 | Gln226 | ||||
| Phe217 | His215 | Gln310 | Trp312 | |||||
| Glu308 | ||||||||
| AgaD | Arg77 | Asn83 | Tyr74 | Asp163 | Glu179 | His198 | Trp221 | Arg350 |
| Asn84 | Phe85 | Arg206 | Trp165 | Glu181 | Gln214 | Tyr296 | Tyr192 | |
| Phe204 | His202 | Gln342 | Trp344 | |||||
| Glu340 | ||||||||
| PorA | Trp56 | His53 | Arg59 | Glu144 | Sterical clash with loop 240–243 | |||
| Arg133 | Trp56 | Trp131 | Tyr191 | |||||
| Arg59 | His167 | Phe236 | ||||||
| Arg133 | Glu234 | |||||||
| PorB | Arg252 | Trp67 | Arg70 | Glu161 | Trp260 | Sterical clash with loop 170–176 | ||
| Arg70 | Trp139 | Tyr258 | ||||||
| Arg187 | His185 | |||||||
| Glu256 |
Both AgaA and AgaB do not specifically bind to the 3,6-anhydro-l-galactose bound in the −4 sub-binding site leaving space to accommodate the L6S of a neoporphyranobiose unit. In contrast, in AgaD the additional loop not only would provoke sterical clash with an L6S unit but would provide an arginine that is in binding position (2.2 Å) to the O3 oxygen of a 3,6-anhydro-bridge of a nonsulfated unit bound in −4. This is in agreement with the fact that AgaDcat requires at least two repeats of consecutive neoagarobiose units on the nonreducing end to show activity, whereas the AgaAcat and AgaB produce L6S-G-L6S-G-LA-G hybrid oligosaccharides (supplemental Fig. S2b). Thus, only the binding subsite −2 strictly requires the presence of an LA unit for these two enzymes to be active.
The more stringent specificity of AgaDcat is also true for the binding subsites on the reducing end (+ binding sites). Indeed, binding subsites +3 and +4 in AgaDcat are also more tailored for accommodating neoagarobiose units than the equivalent sites in AgaB or AgaAcat. However, the binding subsite −3 is lined by Trp109 in AgaB and by Phe85 in AgaD, which may account for weaker binding by the latter enzyme and explain its lack of activity on agarose hexasaccharides that are cleaved by AgaB. Although all three enzymes share the three aromatic residues (2 Trp and 1 Tyr or 3 Trp) constituting the hydrophobic binding-site platforms, two additional residues (Tyr296 and Arg350 in AgaDcat, Table 2) provide hydrogen bonds that specifically recognize an LA unit rather than the L6S sugar. This explains the requirement for neoagarobiose rather than neoporphyranobiose units on this side of the cleavage point for AgaDcat. These constraints on all binding subsites of AgaDcat easily account for the concomitant occurrence of medium sized oligosaccharides (DP4 to DP8; supplemental Fig. S1) with no production of DP2 and the lack of production of small oligosaccharides from the hybrid oligosaccharides (Fig. 2B, lane 3). Taking together the constraints of all binding sites in the catalytic groove of AgaDcat, it appears that this enzyme requires at least a stretch of four adjacent neoagarobiose units (eight sugar units) to be active.
Subtle differences of the residues present in the substrate binding groove of the two β-porphyranases also explain their diverging substrate specificity. Although the presence of an L6S unit in the −2-binding subsite is crucial for the activity of PorA, PorB also accepts LA units at this position for cleavage to occur. Indeed, the binding determinants of PorA at this position, formed by a histidine (His53) and an arginine (Arg133) residue that are at the border of a small pocket accommodating the sulfate group, have no equivalents in PorB (Table 2). Instead, in PorB a different arginine (Arg187) is positioned at the bottom of a much larger space, providing binding potential both for a sulfate group or the ring-oxygen (O3) of the 3,6-anhydro-bridge, as is typical in the binding pattern of neoagarobiose units (17).
The role or importance of the presence of a short loop obstructing the positive binding subsites in PorA can to date only be hypothesized. The loop could be involved in specific substrate recognition and only be displaced by the presence of neoporphyranobiose units. However, the absence of any positively charged residue in this loop is not particularly in favor of this hypothesis. Another explanation could be that the presence of the loop prevents the back-binding of short oligosaccharides that would only occupy the positive binding sites, thus hindering the reverse transglycosylation reaction that is always possible for enzymes displaying the retaining mechanism (51). Finally, the flexible loop could be involved in the dynamic turnover of the reaction, either favoring the departure of the reaction product or, on the contrary, be involved in processivity of the enzyme, maintaining the substrate close to the enzyme while diffusing to the next cleavage site. Further analyses combining enzymology with site-directed mutagenesis of this specific loop are needed to settle these questions.
Integrated View of the Agarolytic System of Z. galactanivorans
The data presented here show that significant differences, although subtle, in the substrate binding pattern of the various enzymes expressed by Z. galactanivorans can be correlated with substrate induction and the potential cellular location. Taken together, these results allow building a first global view and therefore help formulate working hypotheses about the assignment of specific functional roles to the different enzymes of this complex agarolytic system (Fig. 7). In this picture AgaD, which displays a C-terminal Bacteroidetes-specific domain (43, 45), could be secreted in the external medium or displayed at the surface of Z. galactanivorans only in the presence of low sulfated agars (Fig. 6). As the most stringent and therefore highly specific β-agarase, AgaD would prepare long oligosaccharide chains for other enzymes that complete the degradation. In contrast, the more tolerant β-agarases are not only induced by low sulfated agars but are also up-regulated in the presence of the highly sulfated agars, such as porphyran (Fig. 6). AgaA was shown to be a secreted enzyme, specialized for the degradation of regular neoagarobiose fibers forming the core of agar gels, additionally having a surface agarose-binding site that might be involved in the efficient degradation of solid substrate (16, 17). In this study, we demonstrate that AgaA is also tolerant for the sulfated moieties of agar chains, which are usually found in the junction zones separating the semi-crystalline neoagarobiose fibers. AgaB is an outer membrane-bound enzyme, which specializes in the degradation of soluble agaro-oligosaccharides (16). We have shown here that AgaB is also tolerant to hybrid substrates. Moreover, its gene is part of a cluster reminiscent of the Sus operon. Similarly to the role of the membrane-bound α-amylase SusG (52), AgaB may be involved in sugar uptake in connection with the associated SusC-like (Zg_3570) and SusD-like (Zg_3571) membrane proteins. The catabolic reaction in the periplasm would be further accomplished by the recently described 1,3-α-3,6-anhydro-l-galactosidase (AhgA) that specifically releases 3,6-anhydro-l-galactose monosaccharides from short oligosaccharides (19). Finally, AgaC is also predicted as an outer membrane protein, although the wild type enzyme was previously found secreted (16). The precise roles of this fourth β-agarase remain to be established.
FIGURE 7.

Schematic representation of the catabolic pathway of agar degradation and uptake in Z. galactanivorans. A, schematic model of the detailed subsite specificities of PorA, PorB, AgaB, and AgaD. B, respective roles of the different enzymes and their potential cellular localizations are illustrated. AgaA, modular enzyme was secreted in the exterior medium, and the biochemical properties of the enzyme and one of its attached modules (Footnote 7) have been determined. AgaB, membrane-attached lipoprotein with hypothetical external localization was biochemically characterized. AgaC, secreted in the exterior medium (Footnote 7), was not further characterized yet. AgaD, modular enzyme was secreted in the exterior medium, and the catalytic module is biochemically characterized; the attached module is a hypothetical CBM of unknown family. PorA, modular enzyme was most probably secreted in the exterior medium, and only the catalytic module is biochemically characterized. PorB, possibly secreted enzyme, was biochemically characterized. PorC and PorE are nonmodular, hypothetical membrane-associated enzymes, most likely located in the outer cell membrane. PorD is modular and contains a signal peptide targeting the outer membrane. AhgA (19) contains a lipoprotein-type signal peptide, and its biochemical function of terminal oligosaccharide degradation points toward a periplasmic location, associated with the membrane. Three of the enzymes, namely AgaB, PorD, and PorE, are found within operon-like gene clusters that also contain homologs of the TBDR SusC family porins.
Alongside the β-agarases, the β-porphyranases would act in synergy to more completely degrade the natural heterogeneous agars. The highly selective PorA might play the role of a secreted constitutive enzyme (Fig. 6) that provides a basal level of porphyran oligosaccharides. The role of PorB, which is only expressed in cells growing in the laminarin-supplemented medium, could be that of a secreted enzyme sensing the presence of porphyran and releasing the oligosaccharide signal for the induction of the other porphyranolytic enzymes to fully degrade the substrate. This is in line with its broader acceptance of hybrid substrates to be able to detect porphyranobiose moieties in various agar compositions. To complete the degradation and uptake, PorC, PorD, and PorE would be inducible enzymes (Fig. 6) predicted to be bound to the external membrane. Finally, like agaB, the gene porE is part of a Sus-like gene cluster and PorE may act in synergy with its associated SusC-like (Zg_3637) and SusD-like (Zg_3638) membrane proteins to uptake porphyrano-oligosaccharides. Genetic analyses are necessary to confirm these functional hypotheses. Trials to transform Z. galactanivorans are currently ongoing, and the success will give us the tool to investigate further the functional implementation of these enzymes within this model agarolytic system.
Supplementary Material
Acknowledgments
We are indebted to the staff of the European Synchrotron Radiation Facilities (Grenoble, France), beamline ID23-I and ID14, for technical support during data collection and treatment.
This work was supported in part by the Region Bretagne and the CNRS.

This article contains supplemental Figs. S1–S6 and Tables S1 and S2.
The atomic coordinates and structure factors (codes 4asm, 4atf, and 4ate) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
T. Barbeyron, unpublished data.
P. Coutinho, personal communication.
- L6S
- α-l-galactose 6-sulfate ANTS, 8-aminonaphthalene-1,3,6-trisulfonate
- AMAC
- 2-aminoacridone.
REFERENCES
- 1. Popper Z. A., Michel G., Hervé C., Domozych D. S., Willats W. G., Tuohy M. G., Kloareg B., Stengel D. B. (2011) Evolution and diversity of plant cell walls. From algae to flowering plants. Annu. Rev. Plant Biol. 62, 567–590 [DOI] [PubMed] [Google Scholar]
- 2. Lechat H., Amat M., Mazoyer J., Buléon A., Lahaye M. (2000) Structure and distribution of glucomannan and sulfated glucan in the cell walls of the red alga Kappaphycus alvareszii (Gagartinasles, Rhodophyta). J. Phycol. 36, 891–902 [Google Scholar]
- 3. Lahaye M., Yaphe W., Phan Viet M. T., Rochas C. (1989) 13C NMR spectroscopic investigation of methylated and charged agarose oligosaccharides and polysaccharides. Carbohydr. Res. 190, 249–265 [Google Scholar]
- 4. van de Velde F., Knutsen S. H., Usov A. I., Rollema H. S., Cerezo A. S. (2002) 1H and 13C high resolution NMR spectroscopy of carrageenans: applications in research and industry. Trends Food Sci. Technol. 13, 73–92 [Google Scholar]
- 5. Knutsen S., Myslabodski D., Larsen B., Usov A. (1994) A modified system of nomenclature for red algal galactans. Botanica Marina 37, 163–169 [Google Scholar]
- 6. Anderson N. S., Rees D. A. (1965) Porphyran. A polysaccharide with a masked repeating structure. J. Chem. Soc. 5880–5887 [Google Scholar]
- 7. Hehemann J. H., Correc G., Barbeyron T., Helbert W., Czjzek M., Michel G. (2010) Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908–912 [DOI] [PubMed] [Google Scholar]
- 8. Correc G., Hehemann J. H., Czjzek M., Helbert W. (2011) Structural analysis of the degradation products of porphyran digested Zobellia galactanivorans β-porphyranase A. Carbohydr. Polym. 83, 277–283 [Google Scholar]
- 9. Michel G., Nyval-Collen P., Barbeyron T., Czjzek M., Helbert W. (2006) Bioconversion of red seaweed galactans. A focus on bacterial agarases and carrageenases. Appl. Microbiol. Biotechnol. 71, 23–33 [DOI] [PubMed] [Google Scholar]
- 10. Flament D., Barbeyron T., Jam M., Potin P., Czjzek M., Kloareg B., Michel G. (2007) α-Agarases define a new family of glycoside hydrolases, distinct from β-agarase families. Appl. Environ. Microbiol. 73, 4691–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dong J., Hashikawa S., Konishi T., Tamaru Y., Araki T. (2006) Cloning of the novel gene encoding β-agarase C from a marine bacterium, Vibrio sp. strain PO-303, and characterization of the gene product. Appl. Environ. Microbiol. 72, 6399–6401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ekborg N. A., Taylor L. E., Longmire A. G., Henrissat B., Weiner R. M., Hutcheson S. W. (2006) Genomic and proteomic analyses of the agarolytic system expressed by Saccharophagus degradans 2-40. Appl. Environ. Microbiol. 72, 3396–3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kirchman D. L. (2002) The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100 [DOI] [PubMed] [Google Scholar]
- 14. Thomas F., Hehemann J. H., Rebuffet E., Czjzek M., Michel G. (2011) Environmental and gut Bacteroidetes. The food connection. Front. Microbiol. 2, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barbeyron T., L'Haridon S., Corre E., Kloareg B., Potin P. (2001) Zobellia galactanovorans gen. nov., sp. nov., a marine species of Flavobacteriaceae isolated from a red alga, and classification of (Cytophaga) uliginosa (ZoBell and Upham 1944) Reichenbach 1989 as Zobellia uliginosa gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 51, 985–997 [DOI] [PubMed] [Google Scholar]
- 16. Jam M., Flament D., Allouch J., Potin P., Thion L., Kloareg B., Czjzek M., Helbert W., Michel G., Barbeyron T. (2005) The endo-β-agarases AgaA and AgaB from the marine bacterium Zobellia galactanivorans. Two paralogue enzymes with different molecular organizations and catalytic behaviors. Biochem. J. 385, 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Allouch J., Helbert W., Henrissat B., Czjzek M. (2004) Parallel substrate-binding sites in a β-agarase suggest a novel mode of action on double-helical agarose. Structure 12, 623–632 [DOI] [PubMed] [Google Scholar]
- 18. Allouch J., Jam M., Helbert W., Barbeyron T., Kloareg B., Henrissat B., Czjzek M. (2003) The three-dimensional structures of two β-agarases. J. Biol. Chem. 278, 47171–47180 [DOI] [PubMed] [Google Scholar]
- 19. Rebuffet E., Groisillier A., Thompson A., Jeudy A., Barbeyron T., Czjzek M., Michel G. (2011) Discovery and structural characterization of a novel glycosidase family of marine origin. Environ. Microbiol. 13, 1253–1270 [DOI] [PubMed] [Google Scholar]
- 20. Meyer F., Goesmann A., McHardy A. C., Bartels D., Bekel T., Clausen J., Kalinowski J., Linke B., Rupp O., Giegerich R., Pühler A. (2003) GenDB. An open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31, 2187–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bateman A., Coin L., Durbin R., Finn R. D., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E. L., Studholme D. J., Yeats C., Eddy S. R. (2004) The Pfam protein families database. Nucleic Acids Res. 32, 138–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nielsen H., Brunak S., von Heijne G. (1999) Machine learning approaches for the prediction of signal peptides and other protein sorting signals. Protein Eng. 12, 3–9 [DOI] [PubMed] [Google Scholar]
- 23. Sonnhammer E. L., von Heijne G., Krogh A. (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182 [PubMed] [Google Scholar]
- 24. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy). An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Katoh K., Misawa K., Kuma K., Miyata T. (2002) MAFFT. A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michel G., Chantalat L., Duee E., Barbeyron T., Henrissat B., Kloareg B., Dideberg O. (2001) The κ-carrageenase of P. carrageenovora features a tunnel-shaped active site. A novel insight in the evolution of Clan-B glycoside hydrolases. Structure 9, 513–525 [DOI] [PubMed] [Google Scholar]
- 27. Guindon S., Gascuel O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 [DOI] [PubMed] [Google Scholar]
- 28. Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011) MEGA5. Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Groisillier A., Hervé C., Jeudy A., Rebuffet E., Pluchon P. F., Chevolot Y., Flament D., Geslin C., Morgado I. M., Power D., Branno M., Moreau H., Michel G., Boyen C., Czjzek M. (2010) MARINE-EXPRESS. Taking advantage of high throughput cloning and expression strategies for the post-genomic analysis of marine organisms. Microb. Cell Fact. 9, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 31. Kidby D. K., Davidson D. J. (1973) A convenient ferricyanide estimation of reducing sugars in the nanomole range. Anal. Biochem. 55, 321–325 [DOI] [PubMed] [Google Scholar]
- 32. Starr C. M., Masada R. I. (1996) Fluorophore-assisted carbohydrate electrophoresis in the separation, analysis, and sequencing of carbohydrates. J. Chromatogr. A 720, 295–321 [DOI] [PubMed] [Google Scholar]
- 33. ZoBell C. (1941) Studies on marine bacteria. I. The cultural requirements of heterotrophic aerobes. J. Mar. Res. 4, 42–75 [Google Scholar]
- 34. Thomas F., Barbeyron T., Michel G. (2011) Evaluation of reference genes for real time quantitative PCR in the marine flavobacterium Zobellia galactanivorans. J. Microbiol. Methods 84, 61–66 [DOI] [PubMed] [Google Scholar]
- 35. Hehemann J. H., Michel G., Barbeyron T., Czjzek M. (2010) Expression, purification, and preliminary x-ray diffraction analysis of the catalytic module of a β-agarase from the flavobacterium Zobellia galactanivorans. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 413–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Navaza J. (2001) Implementation of molecular replacement in AMoRe. Acta Crystallogr. D Biol. Crystallogr. 57, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Collaborative Computational Project Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 40. Perrakis A., Sixma T. K., Wilson K. S., Lamzin V. S. (1997) wARP. Improvement and extension of crystallographic phases by weighted averaging of multiple-refined dummy atomic models. Acta Crystallogr. D Biol. Crystallogr. 53, 448–455 [DOI] [PubMed] [Google Scholar]
- 41. Perrakis A., Harkiolaki M., Wilson K. S., Lamzin V. S. (2001) ARP/wARP and molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 57, 1445–1450 [DOI] [PubMed] [Google Scholar]
- 42. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) SFCHECK. A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 26, 283–291 [DOI] [PubMed] [Google Scholar]
- 43. Karlsson E. N., Hachem M. A., Ramchuran S., Costa H., Holst O., Svenningsen S. F., Hreggvidsson G. O. (2004) The modular xylanase Xyn10A from Rhodothermus marinus is cell-attached, and its C-terminal domain has several putative homologues among cell-attached proteins within the phylum Bacteroidetes. FEMS Microbiol. Lett. 241, 233–242 [DOI] [PubMed] [Google Scholar]
- 44. Sato K., Naito M., Yukitake H., Hirakawa H., Shoji M., McBride M. J., Rhodes R. G., Nakayama K. (2010) A protein secretion system linked to Bacteroidete gliding motility and pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shoji M., Sato K., Yukitake H., Kondo Y., Narita Y., Kadowaki T., Naito M., Nakayama K. (2011) Por secretion system-dependent secretion and glycosylation of Porphyromonas gingivalis hemin-binding protein 35. PLoS One 6, e21372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomas F., Barbeyron T., Tonon T., Genicot S., Czjzek M., Michel G. (2012) Characterization of the first alginolytic operons in a marine bacterium. From their emergence in marine Flavobacteria to their independent transfers to marine Proteobacteria and human gut Bacteroides. Environ. Microbiol. 10.1111/j.1462-2920.2012.02751.x [DOI] [PubMed] [Google Scholar]
- 47. Anderson K. L., Salyers A. A. (1989) Genetic evidence that outer membrane binding of starch is required for starch utilization by Bacteroides thetaiotaomicron. J. Bacteriol. 171, 3199–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koropatkin N. M., Martens E. C., Gordon J. I., Smith T. J. (2008) Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16, 1105–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Davies G. J., Wilson K. S., Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vandenbussche S., Díaz D., Fernández-Alonso M. C., Pan W., Vincent S. P., Cuevas G., Cañada F. J., Jiménez-Barbero J., Bartik K. (2008) Aromatic-carbohydrate interactions. An NMR and computational study of model systems. Chemistry 14, 7570–7578 [DOI] [PubMed] [Google Scholar]
- 51. Davies G., Henrissat B. (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3, 853–859 [DOI] [PubMed] [Google Scholar]
- 52. Koropatkin N. M., Smith T. J. (2010) SusG. A unique cell-membrane-associated α-amylase from a prominent human gut symbiont targets complex starch molecules. Structure 18, 200–215 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.